
C2-Symmetrical Terphenyl Derivatives as Small Molecule Inhibitors of Programmed Cell Death 1/Programmed Death Ligand 1 Protein–Protein Interaction
 , , , , , , , , , and
, , , , , , , , , and
Abstract
:1. Introduction
2. Results and Discussion
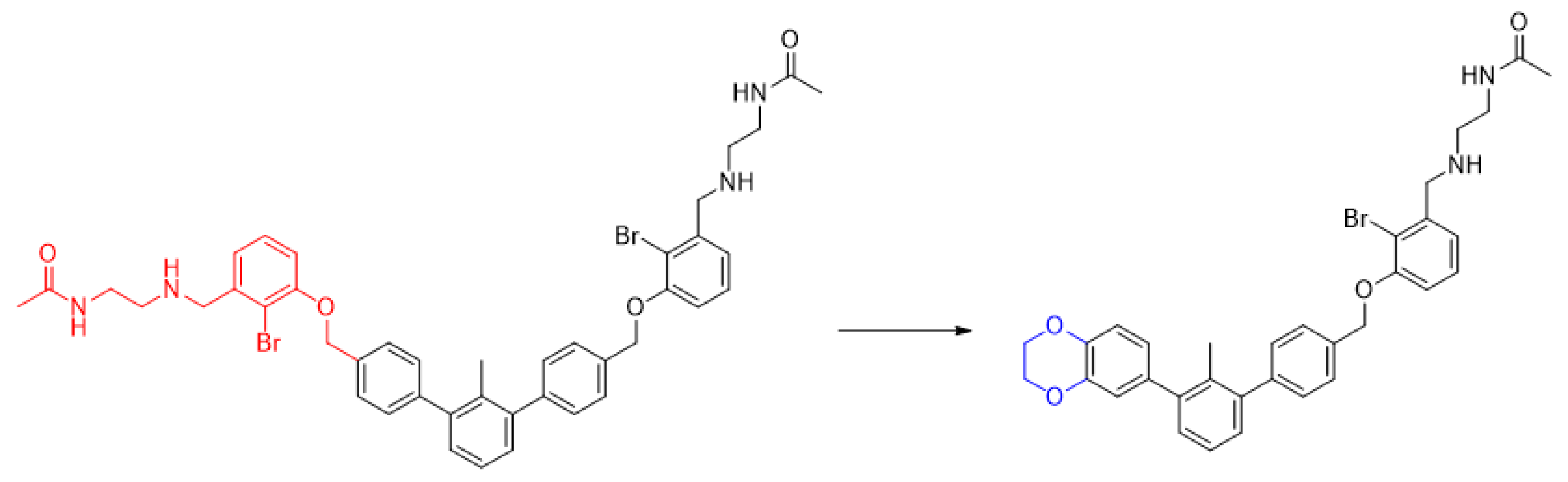
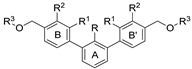
2.1. Chemical Synthesis of C2-Symmetrical Inhibitors of PD-1/PD-L1 Interactions
2.2. HTRF-Based Structure–Activity Relationship (SAR) Results and Correlations
2.3. NMR Binding Assay
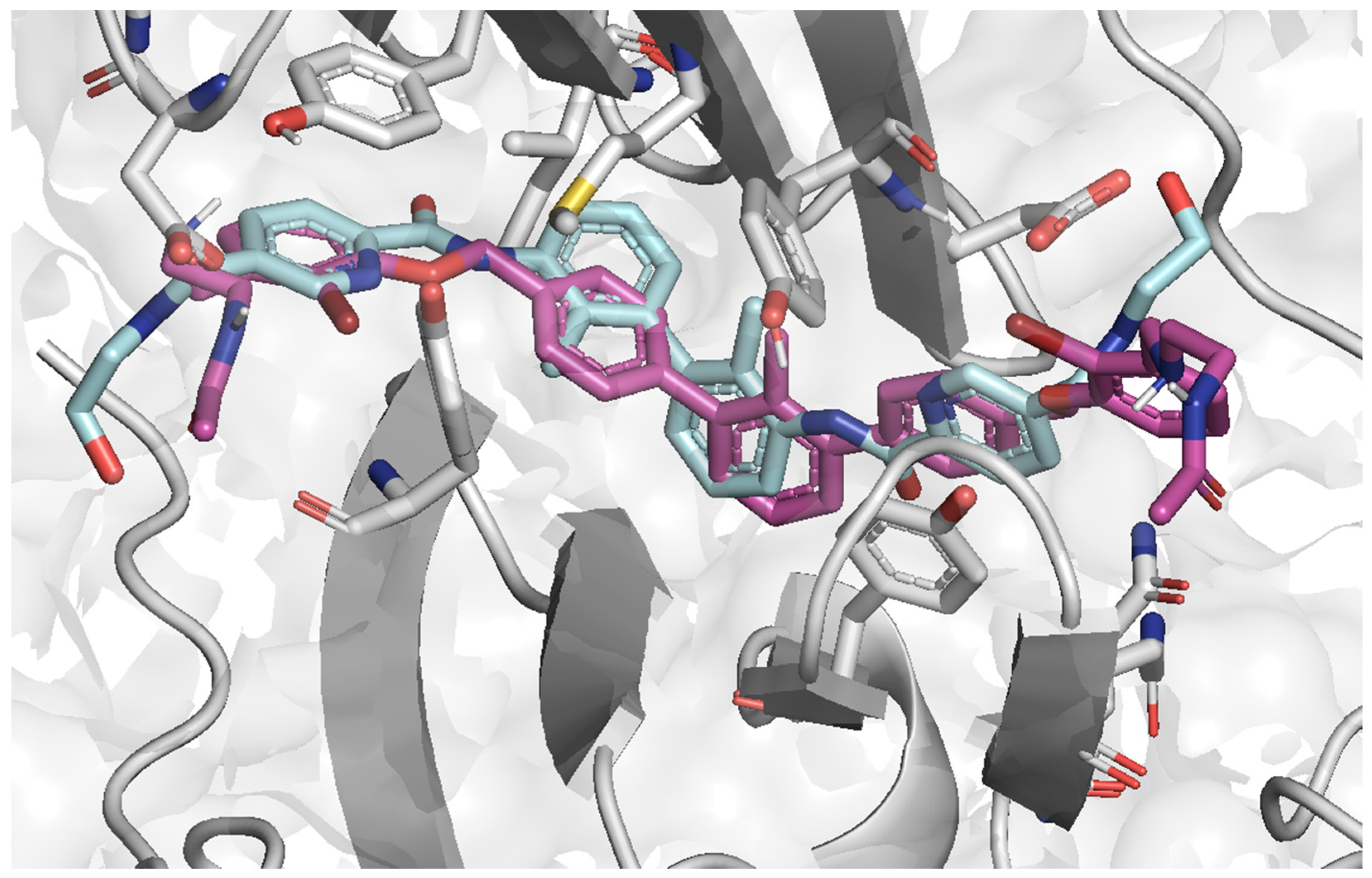
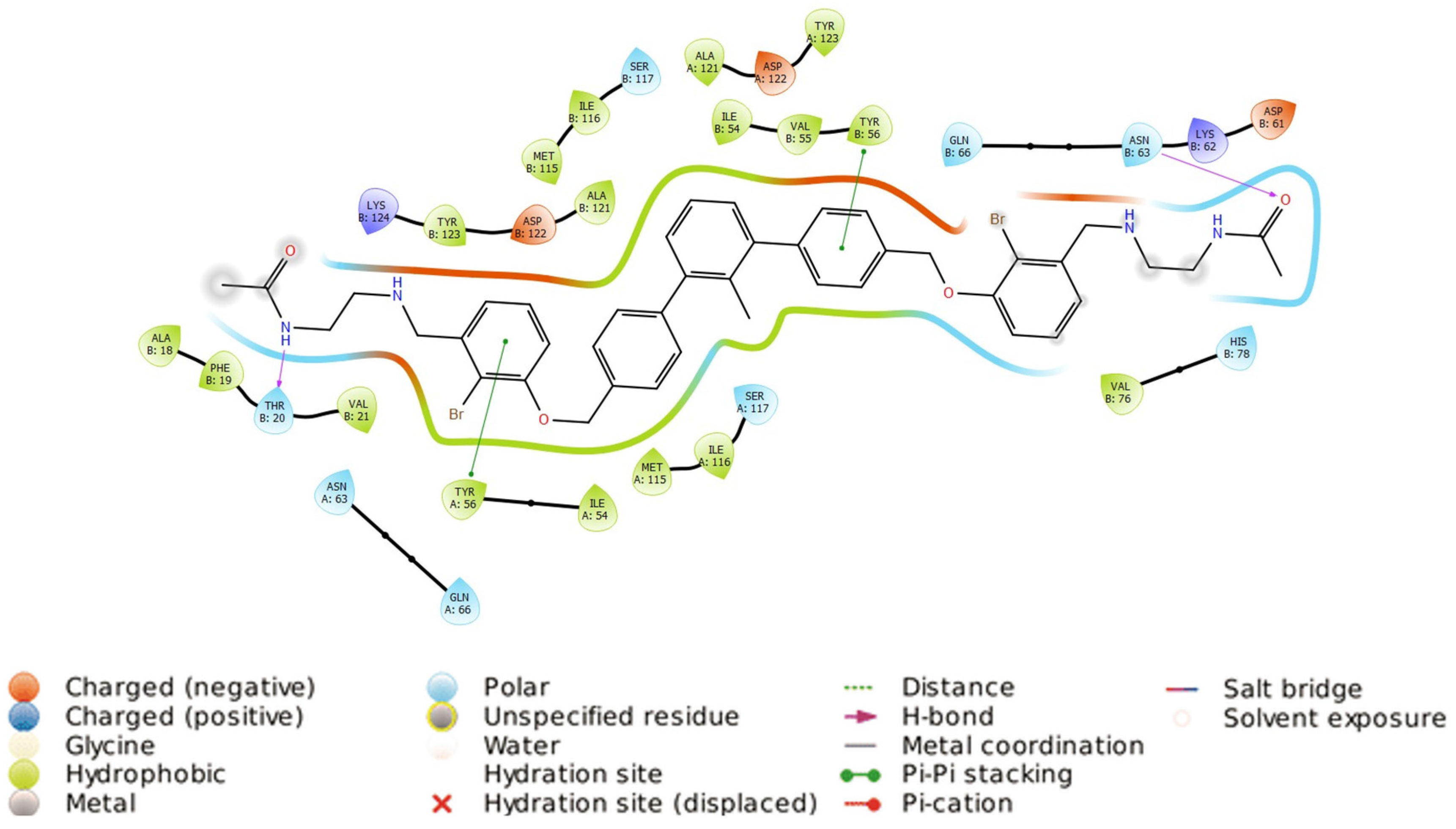
2.4. Molecular Docking
2.5. Ligands’ Pose Geometry Analysis
3. Materials and Methods
3.1. Chemical Synthesis
3.1.1. General Procedure for Final Product Synthesis: Method A. Reductive Amination
3.1.2. General Procedure for Final Product Synthesis: Method B. Nucleophilic Substitution
- N,N’-(((((((5′-methyl-[1,1′:3′,1′′-terphenyl]-3,3′′-diyl)bis(methylene))bis(oxy))bis(3,1-phenylene))bis(methylene))bis(azanediyl))bis(ethane-2,1-diyl))diacetamide (4a)
- N,N’-(((((((5′-methyl-[1,1′:3′,1′′-terphenyl]-3,3′′-diyl)bis(methylene))bis(oxy))bis(2-methoxy-4,1-phenylene))bis(methylene))bis(azanediyl))bis(ethane-2,1-diyl))diacetamide (4b)
- N,N’-(((((((5′-methyl-[1,1′:3′,1′′-terphenyl]-3,3′′-diyl)bis(methylene))bis(oxy))bis(2-bromo-3,1-phenylene))bis(methylene))bis(azanediyl))bis(ethane-2,1-diyl))diacetamide (4c)
- N,N’-(((((((2,2′′-difluoro-5′-methyl-[1,1′:3′,1′′-terphenyl]-3,3′′-diyl)bis(methylene))bis(oxy))bis(3,1-phenylene))bis(methylene))bis(azanediyl))bis(ethane-2,1-diyl))diacetamide (4d)
- N,N’-(((((((2,2′′,5′-trimethyl-[1,1′:3′,1′′-terphenyl]-3,3′′-diyl)bis(methylene))bis(oxy))bis(2-bromo-3,1-phenylene))bis(methylene))bis(azanediyl))bis(ethane-2,1-diyl))diacetamide (4e)
- N,N’-(((((((2,2′′,5′-trimethyl-[1,1′:3′,1′′-terphenyl]-3,3′′-diyl)bis(methylene))bis(oxy))bis(2,5-difluoro-4,1-phenylene))bis(methylene))bis(azanediyl))bis(ethane-2,1-diyl))diacetamide (4f)
- N,N’-(((((((5′-methyl-[1,1′:3′,1′′-terphenyl]-2,2′′-diyl)bis(methylene))bis(oxy))bis(3-methoxy-2,1-phenylene))bis(methylene))bis(azanediyl))bis(ethane-2,1-diyl))diacetamide (5a)
- N,N’-(((((((3,3′′-dimethoxy-5′-methyl-[1,1′:3′,1′′-terphenyl]-4,4′′-diyl)bis(methylene))bis(oxy))bis(3-methoxy-2,1-phenylene))bis(methylene))bis(azanediyl))bis(ethane-2,1-diyl))diacetamide (6a)
- N,N’-(((((((3,3′′-dimethoxy-2′-methyl-[1,1′:3′,1′′-terphenyl]-4,4′′-diyl)bis(methylene))bis(oxy))bis(3-methoxy-4,1-phenylene))bis(methylene))bis(azanediyl))bis(ethane-2,1-diyl))diacetamide (7a)
- 2,2′-((((((3,3′′-dimethoxy-2′-methyl-[1,1′:3′,1′′-terphenyl]-4,4′′-diyl)bis(methylene))bis(oxy))bis(3-methoxy-4,1-phenylene))bis(methylene))bis(azanediyl))bis(ethan-1-ol) (7b)
- N,N’-(((((((2,2′,2′′-trimethyl-[1,1′:3′,1′′-terphenyl]-4,4′′-diyl)bis(methylene))bis(oxy))bis(3-methoxy-4,1-phenylene))bis(methylene))bis(azanediyl))bis(ethane-2,1-diyl))diacetamide (7c)
- 2,2′-((((((2,2′,2′′-trimethyl-[1,1′:3′,1′′-terphenyl]-4,4′′-diyl)bis(methylene))bis(oxy))bis(3-methoxy-4,1-phenylene))bis(methylene))bis(azanediyl))bis(ethan-1-ol) (7d)
- N,N’-(((((((2′,3,3′′-trimethyl-[1,1′:3′,1′′-terphenyl]-4,4′′-diyl)bis(methylene))bis(oxy))bis(3-methoxy-4,1-phenylene))bis(methylene))bis(azanediyl))bis(ethane-2,1-diyl))diacetamide (7e)
- 2,2′-((((((2′,3,3′′-trimethyl-[1,1′:3′,1′′-terphenyl]-4,4′′-diyl)bis(methylene))bis(oxy))bis(3-methoxy-4,1-phenylene))bis(methylene))bis(azanediyl))bis(propane-1,3-diol) (7g)
- N,N’-(((((((2′,3,3′′-trimethyl-[1,1′:3′,1′′-terphenyl]-4,4′′-diyl)bis(methylene))bis(oxy))bis(2,5-difluoro-4,1-phenylene))bis(methylene))bis(azanediyl))bis(ethane-2,1-diyl))diacetamide (7h)
- 2,2′-((((((2′,3,3′′-trimethyl-[1,1′:3′,1′′-terphenyl]-4,4′′-diyl)bis(methylene))bis(oxy))bis(2,5-difluoro-4,1-phenylene))bis(methylene))bis(azanediyl))bis(ethan-1-ol) (7i)
- N,N’-(((((((2′-methyl-[1,1′:3′,1′′-terphenyl]-4,4′′-diyl)bis(methylene))bis(oxy))bis(2-bromo-3,1-phenylene))bis(methylene))bis(azanediyl))bis(ethane-2,1-diyl))diacetamide (7j)
- N,N’-(((((((2′-methyl-[1,1′:3′,1′′-terphenyl]-4,4′′-diyl)bis(methylene))bis(oxy))bis(2-methoxy-4,1-phenylene))bis(methylene))bis(azanediyl))bis(ethane-2,1-diyl))diacetamide (7k)
- 2,2′-((((((2′-methyl-[1,1′:3′,1′′-terphenyl]-4,4′′-diyl)bis(methylene))bis(oxy))bis(2-methoxy-4,1-phenylene))bis(methylene))bis(azanediyl))bis(ethan-1-ol) (7l)
- N,N’-(((((((2′-chloro-[1,1′:3′,1′′-terphenyl]-4,4′′-diyl)bis(methylene))bis(oxy))bis(2-methoxy-4,1-phenylene))bis(methylene))bis(azanediyl))bis(ethane-2,1-diyl))diacetamide (7m)
- 2,2′-((((((2′-chloro-[1,1′:3′,1′′-terphenyl]-4,4′′-diyl)bis(methylene))bis(oxy))bis(2-methoxy-4,1-phenylene))bis(methylene))bis(azanediyl))bis(ethan-1-ol) (7n)
- 2,2′-((((((2′-methyl-[1,1′:3′,1′′-terphenyl]-3,3′′-diyl)bis(methylene))bis(oxy))bis(2-bromo-3,1-phenylene))bis(methylene))bis(azanediyl))bis(ethan-1-ol) (8a)
- N,N’-(((((((2′-methyl-[1,1′:3′,1′′-terphenyl]-3,3′′-diyl)bis(methylene))bis(oxy))bis(3-methoxy-2,1-phenylene))bis(methylene))bis(azanediyl))bis(ethane-2,1-diyl))diacetamide (8b)
- 2,2′-((((((2′-methyl-[1,1′:3′,1′′-terphenyl]-3,3′′-diyl)bis(methylene))bis(oxy))bis(3-methoxy-2,1-phenylene))bis(methylene))bis(azanediyl))bis(ethan-1-ol) (8c)
3.2. Homogenous Time-Resolved Fluorescence
3.3. Protein Expression and Purification
3.4. NMR Binding Assay
3.5. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer Statistics, 2024. CA. Cancer J. Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef] [PubMed]
- Das, C.K.; Singh, S.K. Immune Checkpoint Inhibitors in Cancer Therapy: A Ray of Hope. In Biomedical Translational Research from Disease Diagnosis to Treatment; Springer Nature: Singapore, 2022; pp. 393–411. [Google Scholar] [CrossRef]
- Sharma, P.; Goswami, S.; Raychaudhuri, D.; Siddiqui, B.A.; Singh, P.; Nagarajan, A.; Liu, J.; Subudhi, S.K.; Poon, C.; Gant, K.L.; et al. Immune Checkpoint Therapy—Current Perspectives and Future Directions. Cell 2023, 186, 1652–1669. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Wolchok, J.D. Cancer Immunotherapy Using Checkpoint Blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Huang, Q.; Xie, Y.; Wu, X.; Ma, H.; Zhang, Y.; Xia, Y. Improvement of the Anticancer Efficacy of PD-1/PD-L1 Blockade via Combination Therapy and PD-L1 Regulation. BioMed Cent. 2022, 15, 24. [Google Scholar] [CrossRef] [PubMed]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and Its Ligands in Tolerance and Immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, T.; Honjo, T. PD-1 and PD-1 Ligands: From Discovery to Clinical Application. Int. Immunol. 2007, 19, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Mezzadra, R.; Schumacher, T.N. Regulation and Function of the PD-L1 Checkpoint. Immunity 2018, 48, 434–452. [Google Scholar] [CrossRef] [PubMed]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced Expression of PD-1, a Novel Member of the Immunoglobulin Gene Superfamily, upon Programmed Cell Death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef]
- Patel, S.P.; Kurzrock, R. PD-L1 Expression as a Predictive Biomarker in Cancer Immunotherapy. Mol. Cancer Ther. 2015, 14, 847–856. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Ge, J.; Xiang, B.; Wu, X.; Ma, J.; Zhou, M.; Li, X.; et al. Role of the Tumor Microenvironment in PD-L1/PD-1-Mediated Tumor Immune Escape. Mol. Cancer 2019, 18, 10. [Google Scholar] [CrossRef]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 Pathway: Current Researches in Cancer. Am. J. Cancer Res. 2020, 10, 727–742. [Google Scholar] [PubMed]
- Zhang, J.; Zhang, Y.; Qu, B.; Yang, H.; Hu, S.; Dong, X. If Small Molecules Immunotherapy Comes, Can the Prime Be Far Behind? Eur. J. Med. Chem. 2021, 218, 113356. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.T.; Lee, S.H.; Heo, Y.S. Molecular Interactions of Antibody Drugs Targeting PD-1, PD-L1, and CTLA-4 in Immuno-Oncology. Molecules 2019, 24, 1190. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Cai, S.; Cheng, Y.; Zhang, W.; Wang, M.; Sun, H.; Guo, B.; Li, Z.; Xiao, Y.; Jiang, S. Discovery of Small-Molecule Inhibitors of the PD-1/PD-L1 Axis That Promote PD-L1 Internalization and Degradation. J. Med. Chem. 2022, 65, 3879–3893. [Google Scholar] [CrossRef] [PubMed]
- Martins, F.; Sofiya, L.; Sykiotis, G.P.; Lamine, F.; Maillard, M.; Fraga, M.; Shabafrouz, K.; Ribi, C.; Cairoli, A.; Guex-Crosier, Y.; et al. Adverse Effects of Immune-Checkpoint Inhibitors: Epidemiology, Management and Surveillance. Nat. Rev. Clin. Oncol. 2019, 16, 563–580. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Roudi, R.; Dai, T.; Chen, S.; Fan, B.; Li, H.; Zhou, Y.; Zhou, M.; Zhu, B.; Yin, C.; et al. Immune-Related Adverse Events Associated with Programmed Cell Death Protein-1 and Programmed Cell Death Ligand 1 Inhibitors for Non-Small Cell Lung Cancer: A PRISMA Systematic Review and Meta-Analysis. BMC Cancer 2019, 19, 558. [Google Scholar] [CrossRef] [PubMed]
- Imai, K.; Takaoka, A. Comparing Antibody and Small-Molecule Therapies for Cancer. Nat. Rev. Cancer 2006, 6, 714–727. [Google Scholar] [CrossRef] [PubMed]
- Zak, K.M.; Kitel, R.; Przetocka, S.; Golik, P.; Guzik, K.; Musielak, B.; Dömling, A.; Dubin, G.; Holak, T.A. Structure of the Complex of Human Programmed Death 1, PD-1, and Its Ligand PD-L1. Structure 2015, 23, 2341–2348. [Google Scholar] [CrossRef] [PubMed]
- Chupak, L.S.; Zheng, X. Bistol-Myers Squibb Company Compounds Useful a S Immunomodulatory. WO 2015/034820 A1, 12 March 2015. [Google Scholar]
- Chupak, L.S.; Ding, M.; Martin, S.W.; Zheng, X.; Hewawasam, P.; Conolly, T.P.; Xu, N.; Yeung, K.-S.; Zhu, J.; Langley, D.R.; et al. Compounds Useful as Immunomodulatory. WO Patent WO 2015/160641 A2, 22 October 2015. [Google Scholar]
- Liang, J.; Wang, B.; Yang, Y.; Liu, B.; Jin, Y. Approaching the Dimerization Mechanism of Small Molecule Inhibitors Targeting PD-L1 with Molecular Simulation. Int. J. Mol. Sci. 2023, 24, 1280. [Google Scholar] [CrossRef]
- Zak, K.M.; Grudnik, P.; Guzik, K.; Zieba, B.J.; Musielak, B.; Dömling, A.; Dubin, G.; Holak, T.A. Structural Basis for Small Molecule Targeting of the Programmed Death Ligand 1 (PD-L1). Oncotarget 2016, 7, 30323–30335. [Google Scholar] [CrossRef]
- Perry, E.; Mills, J.J.; Zhao, B.; Wang, F.; Sun, Q.; Christov, P.P.; Tarr, J.C.; Rietz, T.A.; Olejniczak, E.T.; Lee, T.; et al. Fragment-Based Screening of Programmed Death Ligand 1 (PD-L1). Bioorganic Med. Chem. Lett. 2019, 29, 786–790. [Google Scholar] [CrossRef]
- Wang, K.; Zhang, X.; Cheng, Y.; Qi, Z.; Ye, K.; Zhang, K.; Jiang, S.; Liu, Y.; Xiao, Y.; Wang, T. Discovery of Novel PD-L1 Inhibitors That Induce the Dimerization, Internalization, and Degradation of PD-L1 Based on the Fragment Coupling Strategy. J. Med. Chem. 2023, 66, 16807–16827. [Google Scholar] [CrossRef]
- Basu, S.; Yang, J.; Xu, B.; Magiera-Mularz, K.; Skalniak, L.; Musielak, B.; Kholodovych, V.; Holak, T.A.; Hu, L. Design, Synthesis, Evaluation, and Structural Studies of C2-Symmetric Small Molecule Inhibitors of Programmed Cell Death-1/Programmed Death-Ligand 1 Protein-Protein Interaction. J. Med. Chem. 2019, 62, 7250–7263. [Google Scholar] [CrossRef]
- Kawashita, S.; Aoyagi, K.; Yamanaka, H.; Hantani, R.; Naruoka, S.; Tanimoto, A.; Hori, Y.; Toyonaga, Y.; Fukushima, K.; Miyazaki, S.; et al. Symmetry-Based Ligand Design and Evaluation of Small Molecule Inhibitors of Programmed Cell Death-1/Programmed Death-Ligand 1 Interaction. Bioorganic Med. Chem. Lett. 2019, 29, 2464–2467. [Google Scholar] [CrossRef]
- Park, J.J.; Thi, E.P.; Carpio, V.H.; Bi, Y.; Cole, A.G.; Dorsey, B.D.; Fan, K.; Harasym, T.; Iott, C.L.; Kadhim, S.; et al. Checkpoint Inhibition through Small Molecule-Induced Internalization of Programmed Death-Ligand 1. Nat. Commun. 2021, 12, 1222. [Google Scholar] [CrossRef]
- Deng, J.; Cheng, Z.; Long, J.; Dömling, A.; Tortorella, M.; Wang, Y. Small Molecule Inhibitors of Programmed Cell Death Ligand 1 (PD-L1): A Patent Review (2019–2021). Expert Opin. Ther. Pat. 2022, 32, 575–589. [Google Scholar] [CrossRef]
- Sasikumar, P.G.; Sudarshan, N.S.; Adurthi, S.; Ramachandra, R.K.; Samiulla, D.S.; Lakshminarasimhan, A.; Ramanathan, A.; Chandrasekhar, T.; Dhudashiya, A.A.; Talapati, S.R.; et al. PD-1 Derived CA-170 Is an Oral Immune Checkpoint Inhibitor That Exhibits Preclinical Anti-Tumor Efficacy. Commun. Biol. 2021, 4, 699. [Google Scholar] [CrossRef]
- Aktoudianakis, E.; Cho, A.; Du, Z.; Graupe, M.; LAD, L.T.; Machicao Tello, P.; Medley, J.W.; Metobo, S.; Mukherjee, P.K. Gilead Sciences Incorporated PD-1/PD-L1 Inhibitors. WO Patent WO 2019/160882 A1, 22 August 2019. [Google Scholar]
- Burris, H.; Kotecki, N.; Kristeleit, R.; Pinato, D.; Sahebjam, S. Phase 1 Study of INCB06550, an Oral PD-L1 Inhibitor, in Immune-Chceckpoint Naive Patients with Advanced Solid Tumors. J. Immuno Ther. Cancer 2021, 9, 559–560. [Google Scholar]
- Koblish, H.K.; Wu, L.; Wang, L.C.S.; Liu, P.C.C.; Wynn, R.; Rios-Doria, J.; Spitz, S.; Liu, H.; Volgina, A.; Zolotarjova, N.; et al. Characterization of INCB086550: A Potent and Novel Small-Molecule PD-L1 Inhibitor. Cancer Discov. 2022, 12, 1482–1499. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, Z.; Wu, T.; He, M.; Zhang, N. Aromatic Acetylene or Aromatic Ethylene Compound, Intermediate, Preparation Method, Pharmaceutical Composition and Use Thereof. WO Patent WO 2018/006795 A1, 11 January 2018. [Google Scholar]
- Zwergel, C.; Fioravanti, R.; Mai, A. PD-L1 Small-Molecule Modulators: A New Hope in Epigenetic-Based Multidrug Cancer Therapy? Drug Discov. Today 2023, 28, 103435. [Google Scholar] [CrossRef]
- Muszak, D.; Surmiak, E.; Plewka, J.; Magiera-Mularz, K.; Kocik-Krol, J.; Musielak, B.; Sala, D.; Kitel, R.; Stec, M.; Weglarczyk, K.; et al. Terphenyl-Based Small-Molecule Inhibitors of Programmed Cell Death-1/Programmed Death-Ligand 1 Protein-Protein Interaction. J. Med. Chem. 2021, 64, 11614–11636. [Google Scholar] [CrossRef]
- Surmiak, E.; Ząber, J.; Plewka, J.; Wojtanowicz, G.; Kocik-Krol, J.; Kruc, O.; Muszak, D.; Rodríguez, I.; Musielak, B.; Viviano, M.; et al. Solubilizer Tag Effect on PD-L1/Inhibitor Binding Properties for m-Terphenyl Derivatives. ACS Med. Chem. Lett. 2024, 15, 36–44. [Google Scholar] [CrossRef]
- Wang, T.; Cai, S.; Wang, M.; Zhang, W.; Zhang, K.; Chen, D.; Li, Z.; Jiang, S. Novel Biphenyl Pyridines as Potent Small-Molecule Inhibitors Targeting the Programmed Cell Death-1/Programmed Cell Death-Ligand 1 Interaction. J. Med. Chem. 2021, 64, 7390–7403. [Google Scholar] [CrossRef]
- Wang, S.; Wang, Y.; Yan, H. Progress on Biphenyl Derivatives as PD-1/PD-L1 Inhibitors. Med. Chem. Res. 2023, 32, 2089–2115. [Google Scholar] [CrossRef]
- Konieczny, M.; Musielak, B.; Kocik, J.; Skalniak, L.; Sala, D.; Czub, M.; Magiera-Mularz, K.; Rodriguez, I.; Myrcha, M.; Stec, M.; et al. Di-Bromo-Based Small-Molecule Inhibitors of the PD-1/PD-L1 Immune Checkpoint. J. Med. Chem. 2020, 63, 11271–11285. [Google Scholar] [CrossRef]
- Kitel, R.; Rodríguez, I.; Del Corte, X.; Atmaj, J.; Żarnik, M.; Surmiak, E.; Muszak, D.; Magiera-Mularz, K.; Popowicz, G.M.; Holak, T.A.; et al. Exploring the Surface of the Ectodomain of the PD-L1 Immune Checkpoint with Small-Molecule Fragments. ACS Chem. Biol. 2022, 17, 2655–2663. [Google Scholar] [CrossRef]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved Protein–Ligand Docking Using GOLD Marcel. Proteins 2003, 52, 609–623. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Adasme, M.F.; Linnemann, K.L.; Bolz, S.N.; Kaiser, F.; Salentin, S.; Haupt, V.J.; Schroeder, M. PLIP 2021: Expanding the Scope of the Protein-Ligand Interaction Profiler to DNA and RNA. Nucleic Acids Res. 2021, 49, W530–W534. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
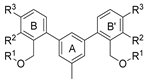
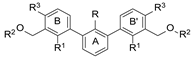
| Name | R1 | R2 | R3 | HTRF ([%) 1 | IC50 Estimated (µM) | |
|---|---|---|---|---|---|---|
| 5 µM | 0.5 µM | |||||
 | ||||||
| 4a | H | 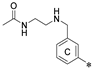 | H | 56.8 ± 15.4 | 80.4 ± 18.8 | 7.72 |
| 4b | H |  | H | 62.1 ± 8.4 | 97.5 ± 6.0 | 7.41 |
| 4c | H |  | H | 70.8 ± 18.8 | 88.6 ± 14.0 | 20.52 |
| 4d | F |  | H | 22.5 ± 9.17 | 89.9 ± 19.8 | 2.08 |
| 4e | CH3 |  | H | 94.2 ± 2.3 | 93.9 ± 5.5 | not active |
| 4f | CH3 | 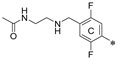 | H | 28.2 ± 1.5 | 85.7 ± 10.9 | 2.18 |
 | ||||||
| 5a | 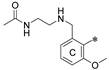 | H | H | 78.4 ± 6.4 | 84.3 ± 13.1 | 36.61 |
 | ||||||
| 6a | H | OCH3 |  | 59.2 ± 2.6 | 86.1 ± 4.5 | 9.01 |
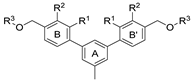
| Name | R | R1 | R2 | R3 | HTRF [%] 1 | IC50 Estimated [µM] | |
|---|---|---|---|---|---|---|---|
| 5 µM | 0.5 µM | ||||||
 | |||||||
| 7a | CH3 | H | OCH3 | 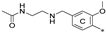 | 23.8 ± 0.3 | 72.5 ± 8.1 | 1.36 |
| 7b 2 | CH3 | H | OCH3 | 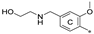 | 11.9 ± 1.0 | 60.9 ± 3.9 | 0.74 |
| 7c | CH3 | CH3 | H | 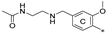 | 77.1 ± 0.7 | 82.1 ± 8.0 | 33.13 |
| 7d | CH3 | CH3 | H | 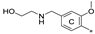 | 35.7 ± 3.3 | 86.1 ± 12.7 | 2.75 |
| 7e | CH3 | H | CH3 | 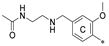 | 55.3 ± 13.4 | 93.8 ± 3.6 | 6.19 |
| 7f | CH3 | H | CH3 | 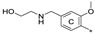 | 46.4 ± 9.6 | 93.1 ± 17.4 | 4.39 |
| 7g | CH3 | H | CH3 |  | 53.7 ± 1.7 | 94.5 ± 18.1 | 5.74 |
| 7h | CH3 | H | CH3 | 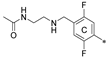 | 69.0 ± 0.9 | 84.1 ± 26.4 | 18.80 |
| 7i | CH3 | H | CH3 |  | 42.2 ± 7.2 | 78.5 ± 7.3 | 3.09 |
| 7j 2 | CH3 | H | H |  | 11.1 ± 0.3 | 40.4 ± 1.5 | 0.31 |
| 7k | CH3 | H | H | 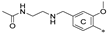 | 75.1 ± 11.0 | 95.5 ± 15.2 | 20.02 |
| 7l | CH3 | H | H | 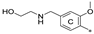 | 59.8 ± 13.5 | 97.9 ± 11.2 | 6.68 |
| 7m 2 | Cl | H | H | 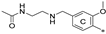 | 35.0 ± 11.7 | 51.6 ± 1.6 | 0.69 |
| 7n | Cl | H | H |  | 40.0 ± 9.9 | 67.1 ± 14.1 | 2.04 |
| Name | R | R1 | R2 | R3 | HTRF [%] 1 | IC50 Estimated [µM] | |
|---|---|---|---|---|---|---|---|
| 5 µM | 0.5 µM | ||||||
 | |||||||
| 8a | CH3 | H | 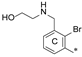 | H | 23.0 ± 0.1 | 94.6 ± 0.1 | 2.43 |
| 8b | CH3 | H | 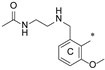 | H | 73.6 ± 5.3 | 88.9 ± 3.8 | 25.1 |
| 8c | CH3 | H |  | H | 61.1 ± 4.5 | 103.9 ± 3.3 | 5.96 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klimek, J.; Kruc, O.; Ceklarz, J.; Kamińska, B.; Musielak, B.; van der Straat, R.; Dӧmling, A.; Holak, T.A.; Muszak, D.; Kalinowska-Tłuścik, J.; et al. C2-Symmetrical Terphenyl Derivatives as Small Molecule Inhibitors of Programmed Cell Death 1/Programmed Death Ligand 1 Protein–Protein Interaction. Molecules 2024, 29, 2646. https://doi.org/10.3390/molecules29112646
Klimek J, Kruc O, Ceklarz J, Kamińska B, Musielak B, van der Straat R, Dӧmling A, Holak TA, Muszak D, Kalinowska-Tłuścik J, et al. C2-Symmetrical Terphenyl Derivatives as Small Molecule Inhibitors of Programmed Cell Death 1/Programmed Death Ligand 1 Protein–Protein Interaction. Molecules. 2024; 29(11):2646. https://doi.org/10.3390/molecules29112646
Chicago/Turabian StyleKlimek, Joanna, Oskar Kruc, Joanna Ceklarz, Beata Kamińska, Bogdan Musielak, Robin van der Straat, Alexander Dӧmling, Tad A. Holak, Damian Muszak, Justyna Kalinowska-Tłuścik, and et al. 2024. "C2-Symmetrical Terphenyl Derivatives as Small Molecule Inhibitors of Programmed Cell Death 1/Programmed Death Ligand 1 Protein–Protein Interaction" Molecules 29, no. 11: 2646. https://doi.org/10.3390/molecules29112646