Abstract
The genome—the source of life and platform of evolution—is continuously exposed to harmful factors, both extra- and intra-cellular. Their activity causes different types of DNA damage, with approximately 80 different types of lesions having been identified so far. In this paper, the influence of a clustered DNA damage site containing imidazolone (Iz) or oxazolone (Oz) and 7,8-dihydro-8-oxo-2′-deoxyguanosine (OXOdG) on the charge transfer through the double helix as well as their electronic properties were investigated. To this end, the structures of oligo-Iz, d[A1Iz2A3OXOG4A5]*d[T5C4T3C2T1], and oligo-Oz, d[A1Oz2A3OXOG4A5]*d[T5C4T3C2T1], were optimized at the M06-2X/6-D95**//M06-2X/sto-3G level of theory in the aqueous phase using the ONIOM methodology; all the discussed energies were obtained at the M06-2X/6-31++G** level of theory. The non-equilibrated and equilibrated solvent–solute interactions were taken into consideration. The following results were found: (A) In all the discussed cases, OXOdG showed a higher predisposition to radical cation formation, and B) the excess electron migration toward Iz and Oz was preferred. However, in the case of oligo-Oz, the electron transfer from Oz2 to complementary C4 was noted during vertical to adiabatic anion relaxation, while for oligo-Iz, it was settled exclusively on the Iz2 moiety. The above was reflected in the charge transfer rate constant, vertical/adiabatic ionization potential, and electron affinity energy values, as well as the charge and spin distribution. It can be postulated that imidazolone moiety formation within the CDL ds-oligo structure and its conversion to oxazolone can significantly influence the charge migration process, depending on the C2 carbon hybridization sp2 or sp3. The above can confuse the single DNA damage recognition and removal processes, cause an increase in mutagenesis, and harm the effectiveness of anticancer therapy.
1. Introduction
Across all species, each and every cell contains genetic information encoded in the sequence of nucleobases. The human genome comprises approximately 3.2 × 109 base pairs (BPs) arranged in two complementary counter-rotating oligonucleotide chains settled as chromatin in the nucleus [1]. The mitochondrial circular genome, on the other hand, consists of only 16 569 BP units [2]. The above should be multiplied by the number of human body cells, i.e., 1014 [3]. Each of the cells, including those located within nucleic acids, is continuously exposed to harmful chemical and physical external factors, such as ionization radiation (X-ray, gamma, and beta), ultraviolet, photosynthesis, xenobiotics, environmental pollution, and reactive oxygen or nitrogen species (ROS or RNS). Additionally, other classes of potentially toxic agents exist within cells, including metabolites, products of the respiratory cycle, and estrogens [4]. The activity of all these factors causes the formation of various types of DNA damage. To date, more than 70 different kinds of DNA lesions have been identified, including apurinic-apyrimidinic sites (AP-sites), DNA-DNA or protein–DNA cross-links, single/double-strand breaks (S/DSB), sugar/base modifications, and tandem and clustered types [5]. Such DNA damage can constitute serious threats to genetic information. As a consequence, throughout evolution, prokaryotic and eukaryotic life forms have developed DNA damage response (DDR) systems that exploit base and nucleotide excision repair (BER and NER), mismatch repair, homologous recombination, and non-homologous end-joining systems, all of which remove lesions in a substrate-dependent manner [6]. Due to the abundance of sugar and base lesions which are removed from the genome by BER, this protective system has become the most active and common, which is present in the nucleus and mitochondria [7]. The protein deficit/defect involved in this repair process causes changes in genetic information (i.e., mutation), which can lead to cancers or an acceleration in the aging process [8]. Activation of the BER enzyme cascade is triggered when a DNA lesion is recognized by mono- or bifunctional glycosylase [9]. Of all the nucleosides, 2′-deoxyguaosine (dG) is especially sensitive to harmful effects because of its particularly low reduction potential among canonical nucleosides (E7(dG(-H)•/dG = 1.29 V) [10,11,12]. The lesions derived from guanosine have been identified as the most abundant. Their distribution depends on the endocellular environment (hypoxic or normoxic), the presence of a 2-deoxyribose substituent, and the ways in which ROS are generated, especially hydroxyl radicals (●OH) [13,14]. In a recent study using the high-salt DNA extraction method, 7,8-dihydro-8-oxo-2′-deoxyguanosine (OXOdG)—the most abundant DNA lesion—was measured to be 14.62 ± 1.45 per 106 DNA bases [15]. The redox potential of OXOdG was found to be 0.58–0.75 V [16]. This lesion is recognized and removed from the genome by the bifunctional glycosylase OGG1 [17]. A lack or defect in this enzyme leads to GC→TA transversion, because OXOdG, after adopting an anti-conformation, can form a base pair with 2′-dexyadenosine (dA) [18]. Fortunately, the glycosylase MutYh is able to recognize and remove the badly paired adenine [19]. All the above leads to the low mutagenicity of the abundant 7,8-dihydro-8-oxo-2′-deoxyguanosine. The above-mentioned OXOdG can be produced in the genome not only by ●OH activity but also as the product of the two-electron dG oxidation process [20]. The dG and OXOdG also undergo conversion to 2,5-Diamino-4H-imidazol-4-one (Iz), which, after hydrolysis, rearranges itself as 2,2,4-triamino-5(2H)-oxazolone (Oz) (Figure 1) [16,21,22].
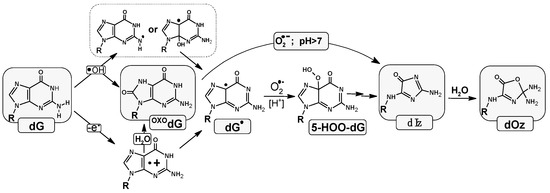
Figure 1.
Graphical representation of imidazolone formation in the physiological condition and its conversion into oxazolone as the final product of four-electron guanine oxidation.
It should be pointed out that Iz and therefore Oz are the results of the 2′-deoxyguanosine four-electron oxidation process. Moreover, Oz, and therefore Iz, leads to GC→CG or GC→TA transversion [23]. Careful analytical studies have revealed that there are 2–6 oxazolone molecules per 107 of guanines in human hepatocytes [16,24].
It should be pointed out that Oz is at least one magnitude more promutagenic than OXOG [25,26]. Fortunately, this lesion is removed from the genome by Nei, Nth prokaryotic glycosylases, and by eukaryotic NEIL1, NTH1 glycosylases [27,28].
Recently, a theory has been proposed that some proteins utilize a charge transfer to scan the double helix to detect their sites’ action in a highly effective way [29]. Proteins containing [4Fe-4S] clusters, such as glycosylases, polymerases, helicases, and primases, are communicated by charge transfer through ds-DNA [30]. Hence, the formation of clustered DNA lesions containing OXOdG and Iz/Oz can pose a challenge for the activity of such an enzyme in the sea of canonical nucleobases. In light of the influence of clustered DNA lesions (CDLs) on the ds-DNA structure, their electronic properties and role in charge transfer are worth investigation. (A CDL is defined as two or more DNA damage events per one or two double helix turns [31,32].) The above has led to a better understanding of the roles of CDLs in genetic information repair and replication [33,34]. Furthermore, the effectiveness of radiotherapy, photodynamics, chemotherapy, etc., relies on inducing DNA damage in the targeted genome. Therefore, studying the charge transfer process through the double helix should result in the increased safety of anticancer therapies (by reducing genotoxicity in normal cells) [31,34,35].
2. Results
Nucleobases present in ds-DNA interacted mutually by the hydrogen bond and stacking interaction equally [36]. These gentle interactions stabilized the ds-DNA structure and, therefore, genetic information. Moreover, the presence of the π-π interaction facilitated the protein (containing a [4Fe-4S] cluster) communication by charge transfer (CT), e.g., glycosylases [37]. The proposed mechanism is evidence of the high effectiveness of the ds-DNA scanning/verification process by dedicated proteins [38]. Spatial geometry disruption, triggered by DNA damage, infected the CT and “activated” the glycosylase action. For these studies, short ds-oligonucleotides (ds-oligos) containing a multi-damage site consisting of OXOdG and Iz or its degradation product, Oz, were investigated (Figure 2).
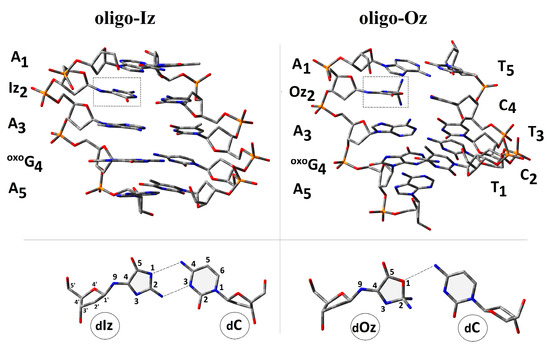
Figure 2.
A graphical representation of the d[A1Iz2A3OXOG4A5]*d[T1C2T3C4T5] and d[A1Iz2A3OXOG4A5]*d[T1C2T3C4T5] spatial geometry optimized at the M06-2x/D95** level of theory in the aqueous phase using the PCM solvation model. Iz: 2-amino-5-[2-deoxy-β-d-erythro-pentofuranosyl)amino]-4H-imidazol-4-one; Oz: (2-deoxy-β-d-erythro-pentofuranosyl)amino]-5(2H)-oxazolone; and OXOG: 7,8-dihydro-8-oxo-2’-deoxyguanosine. Extracted from the double helix base pair formed by dIz/dOz and 2’-deoxycytidine (dC) as well as the atom numbering and hydrogen bond indicated by the dash lines.
The following notation of the ds-oligonucleotides has been used within this article: oligo-Oz: d[A1Iz2A3OXOG4A5]*d[T5C4T3C2T1], and oligo-Oz: d[A1Oz2A3OXOG4A5]*d[T5C4T3C2T1]. As a reference for the presented studies, the unmodified ds-oligonucleotide d[A1G2A3G4A5]*d[T5C4T3C2T1] (oligo-N) was chosen [39].
2.1. Influence of Iz and Oz on Double Helix Spatial Geometry
The influence of a CDL containing OXOdG and Iz or Oz on the double helix three-dimensional geometry was taken into consideration. The spatial structure of both oligo-Iz and oligo-Oz were optimized using the ONIOM (Our Own N-layered Integrated Molecular Orbital and Molecular Mechanics) methodology [40]. The central/pivotal part of the ds-oligo containing the complementary nucleobases was described as a high layer, while the sugar-phosphate backbone was noted as low and optimized at the M06-2x/D95** and M06-2x/sto-3G levels of theory, respectively. All the theoretical experiments were performed in the aqueous phase using the integral equation formalism variant of the polarizable continuum model (IEF-PCM) [41,42]. According to the standard reference frame for the description of DNA double helix geometry, an analysis of the mutual base arrangement was carried out for oligo-Iz and oligo-Oz [43]. For the proposal under discussion, the hydrogen bond length (HB), the distance between C1′ (dC1…C1′) of the complementary BP and rotation of each base around the C1′ atom, i.e., λ1 and λ2 as angles determined by N9-C1′ (pyrimidine)-C1′(purine) and N1-C1′(purine)-C1′(pyrimidine) (Table S1), have been discussed.
The analysis of the spatial geometry obtained for oligo-Iz and oligo-Oz reveals that Iz can form two hydrogen bonds with complementary cytidine, while oxazolone forms only one in its cyclic form, as presented in Figure 1. For the remainder of the BPs, the scheme of the formed HB was found to be unchanged; moreover, the lengths of the HBs were at a similar level to those noted in the standard reference frame [43]. The mutual base orientation within the BP was defined by the λ1, λ2, and dC1′…C1′ parameters. A significant divergence was noted in the part of ds-oligo in which Iz and Oz appear. For oligo-Iz, the decreases in λ1 for the Iz2C4 and the neighboring A3T3 were noted as follows: 42.5[O] and 48.7[O], respectively; subsequently, a distance dC1…C1 increase by 0.7 Å in the case of Iz2C4 was observed (Table S1 in the Supplementary Materials). The subsequent imidazolone hydrolysis led to oxazolone formation and caused further λ1 decreases (39.6[O]); surprisingly, the λ1 of A3T3 reconstructs close to the value of the native AT base pair (52.8[O]). Additionally, the changes in dC1′…C1′ of A1T5 and Oz2C4 were noted as follows: decreases by 1 Å and 0.2 Å, in comparison to those assigned for the precursor (oligo-Iz). In all the discussed cases, λ2 calculated for the pyrimidine strand was found to be close to the reference value of 54.5[O].
The above-discussed parameter changes influence the distance within the base-pair dimer, noted as Rise (Table S1) [43]. The calculated Rise parameters reveal slight decreases in comparison to the reference (oligo-N), with the average values assigned as 2.96 Å and 3.12 Å for oligo-Iz and oligo-Oz, respectively. It should be pointed out that in the wake of Iz hydrolysis, the distance between Oz2T4 and A3T3 increases from 2.93 Å up to 3.46 Å, which can be the result of sp3 hybridization adoption by carbon C2 and the appearance of an additional exocyclic NH2 group. All this forces a higher spatial hindrance than that observed in the case of a flat Iz heterocycle moiety (Figure 1).
2.2. Influence of Iz and Oz on Double Hydrogen Bonds and Stacking Energies
Two main factors determine the stability of the double helix, i.e., the stacking and hydrogen bonds [36]. These noncovalent interactions are sensitive to the changes within the nucleoside/tide subunit structure. Imidozolone and oxazolone, in fact, are the products of the four-electron guanine oxidation process [22]. As shown in Figure 1, in both cases, the rearrangement process spreads over the whole bicyclic purine moiety. This structural difference from the initial guanine caused the change in the HB and Rise parameters as discussed previously. These geometrical changes were reflected in the values of the HB (EHB) and stacking (EST) energies. An analysis of the results, presented in Table S2, shows that the flat structure of Iz (oligo-Iz) left the stacking interaction between A1T5/A3T3 and Iz2C4 equal to 14.61 and 14.97 kcal, respectively. Decreases in the HB energy versus native G:::C system (17.23 kcal) were observed for the Iz2C4 base pair, i.e., 11.47 kcal, as a result of the reduction in HB numbers. However, it was still at the level of the EHB of the AT base pair. Hydrolysis of the Iz moiety led to significant EST decreases in the Oz neighborhood, as denoted by the results obtained for oligo-Oz, as follows: 12.41 kcal and 12.77 kcal for A1T5||Oz2C4 and Oz2C4||A3T3, respectively. Moreover, the Iz rearrangement toward Oz led to only one hydrogen bond remaining between Oz and the complementary cytidine. The above was manifested by an EHB decrease of up to 7.69 kcal, which is lower by approximately 10 kcal, than that observed in the cases of the GC or OXOGC pair (Table S2).
2.3. Influence of Iz and Oz on Double Helix Electronic Properties
Because of the π-π interaction of the nucleobase (heterocycles), the DNA double helix can be perceived as a nanowire [44]. Moreover, investigations have revealed that charge transfer is exploited by proteins to scan the genome for different activity [37,45]. With the above in mind, the electronic properties of oligo-Iz and oligo-Oz were investigated. As shown above, Iz and Oz change the 3D geometries of the double helix. Therefore, complete ds-DNA structures consisting of a sugar–phosphate skeleton and nucleobase pairs, and solely base pair ladders, were taken into investigation. All the energy calculations were performed at the M06-2x/6-31++G** level of theory in the aqueous phase, using the IEF-PCM model. Both non-equilibrated (NE) and equilibrated (EQ) solvent–solute interactions were used. Firstly, the spatial structure of oligo-Iz and oligo-Oz in anionic and cation forms was optimized to the ground state using the ONIOM strategy at the same level as that for the non-charged ds-oligo.
The global structural changes, forced by electron attachment or electron loss, were assigned as the differences between the atomic positions between the neutral versus anionic or cationic forms of the discussed oligo, i.e., oligo-Iz and oligo-Oz, and expressed as an RMSD (Root Mean Square Deviation) in [Å2] (Table 1) [46]. As expected, the molecule charge changes were compensated mainly by the phosphate-sugar backbone’s flexibility, while the internal part containing base pairs was less affected, as presented in Table 1. Furthermore, the appearance of an extra electron in the oligo-Iz system resulted in a higher RMSD value in all the discussed cases.

Table 1.
The electronic properties, in [eV], of oligo-Iz and oligo-Oz: the vertical (VIP) and adiabatic ionization potential (AIP) and the vertical (VEA) and adiabatic (AEA) electron affinity calculated at the M062x/6-31++G** level of theory in the aqueous phase. (a) Complete double helix and (b) base-pair skeleton, NE—non-equilibrated solvent–solute interaction, EQ—equilibrated solvent–solute interaction, and * data calculated for oligo-N [39]. The Root Mean Square Deviation (RMSD) of the atomic positions in [Å2], calculated for the neutral, anionic, and cationic forms of oligo-Iz and oligo-Oz. BP—base pair, PS—phospho-sugar backbone. The raw data have been given in Table S3.
Contrary to this, the electron lost by oligo-Iz did not force a significant structural perturbation and left the RMSD close to 0.18 Å. Comparing the oligo-Oz structure in the anionic and cationic modes with the structure in a neutral mode revealed a similar geometry sensitivity to the electron adoption or ejection by the ds-DNA as follows in [Å2]: 0.46 and 0.69 for the oligo-Oz anion and cation versus neutral form, respectively (Table 1).
The observed structural changes in the double helix should influence the global electronic properties. To this end, the NE and EQ interaction between the solvent and solute was investigated. It should be pointed out here that whole short fragments of ds-oligos were submerged in the aqueous phase. The results obtained for the vertical ionization potential (VIP) calculated in the non-equilibrated mode were found, respectively, to be oligo-N>oligo-Oz>oligo-Iz, irrespective of the investigated system, i.e., whether a complete double helix or base-pair skeleton. Moreover, the observed order of the VIPNE remained unchanged for the equilibrated VIP and adiabatic ionization potential (AIP) (Table 1) [39]. However, the values of the discussed parameters found for oligo-Iz and oligo-Oz were at the same level in comparison to the native oligo-N. These indicated that the presence of OXOG in ds-DNA exerts a stronger influence on radical cation formation/stabilization than Iz or Oz. The ionization potential decreases with the progress of the discussed ds-oligo system relaxations, i.e., VIPNE > VIPEQ > AIP.
The situation changes when the extra electron appears in the ds-DNA structure. The oligo-Iz in all the discussed cases had a significantly higher electron affinity than oligo-Oz and the reference oligo-N, as shown in Table 1. After structural relaxation, the following order of AEA (adiabatic electron affinity) was found for the complete ds-DNA and BP skeleton as follows: oligo-Iz > oligo-N > oligo-Oz. Interestingly, the AEA value established for oligo-Oz was lower than that obtained for oligo-N in both modes. Moreover, the VEAEQ and VEANE were found to be completely incoherent and depend on the investigated system, i.e., a complete double helix or base-pair ladder (Table 1). These observations indicate that the presence of Oz in the ds-oligo structure, as a part of a CDL, causes significant “electro-negativity” perturbation and “covers” the influence of OXOG.
As shown above, the CDL appears in the ds-DNA structure, forcing different results depending on the four-electron oxidation product, i.e., imidazolone or oxazolone in the global context. To shed light on this phenomenon, the electronic properties of isolated base pairs were taken into consideration. The above allows for the AIP, VIP, VEA, and AEA values to be obtained for the BPs in their geometry adopted in the neutral and charged (anionic and cationic) oligo-Iz and oligo-Oz, calculated at the M06-2x/6-31++G** level of theory in the condensed phase, using the equilibrated mode of solvent–solute interaction. In all the discussed cases, the lower vertical/adiabatic ionization potentials in [eV] were found for OXOG4C2 as follows: 5.94/5.52 (oligo-Iz) and 5.91/5.56 (oligo-Oz), respectively (Table 2). On the other hand, a higher VIP and AIP were noted for the Iz2/C4 and Oz2/C4 base pairs at a level of 7 eV, with a negligible difference between the vertical and adiabatic states.
Table 2.
The electronic properties of the isolated base pairs from oligo-Iz and oligo-Oz: the vertical (VIP) and adiabatic ionization potential (AIP) and the vertical (VEA) and the adiabatic (AEA) electron affinity calculated at the M062x/6-31++G** level of theory in the condensed phase. The raw data have been given in Table S4.
For the remaining A1T5, A3T3, and A5T1 base pairs present as previously in the oligo-Iz and oligo-Oz structures, the difference between the VIP and AIP was found to be negligible in a range of between 6.63 and 6.73 [eV]. The appearance of an extra electron within the ds-DNA structure leads to anion formation. The ability of electron adoption by the isolated nucleobase was described by the electron affinity parameters calculated in the vertical and adiabatic modes at the same level of theory as the ionization potential (Table 2). The highest VEA and AEA was found for the Iz2C4 moiety (oligo-Iz), i.e., 2.45 and 2.97 [eV], respectively, while for the Oz2C4 base pair located in oligo-Oz, these values were as follows: 1.82 and 1.89 [eV]. For the remaining A1T5, A3T3, A5T1, and OXOG4C2, the difference between the VIP and AIP was found to be scant for both the discussed ds-oligos. The average value for the AT base pairs was found to be, in [eV], 1.42 and 1.40, respectively, while for OXOGCs this value was measured as 1.51 [eV] for both the VEA and AEA (Table 2).
The above results indicate the differentiation in the spin and charge distribution within the oligo-Iz and oligo-Oz structures. To investigate this phenomenon, all the calculations were carried out at the M06-2x/6-31++G** level of theory using the Hirshfeld population analysis of the density functional electronic charge distribution [47]. As could be expected, the electron lost by oligo-Iz or oligo-Oz leads to a positive charge and spin accumulation, mainly on the OXOG4C2 base pairs, of ~80% and ~90%, respectively, in both the discussed vertical and adiabatic modes. The above charge pattern found in the case of equilibrated and non-equilibrated solvent–solute interaction was affected too.
In both ds-oligo cases, OXOG was found as the main beneficiary of the positive charge and spin localization/accumulation, as presented in Table S3. In contrast, the electron adoption by the discussed systems (oligo-Iz and oligo-Oz) caused a different negative charge and spin distribution depending on whether an imidazoline or oxazolone moiety was present in the structure of the lesioned ds-oligo. In both cases (oligo-Iz and oligo-Oz), the base pairs Iz2C4 and Oz2C4 turned out to be the settling point of the extra electron (Table S3). Hardly any spread of negative charge and spin was found on the A1T5 moiety. However, a more precise analysis revealed that the product of Iz heterocycle hydrolysis, i.e., Oz, exerted a significant influence on the negative charge and spin location within the double helix. Firstly, in the case of Iz2C4, the charge and spin were found in ~80% and 90% on the Iz2 moiety, respectively, for the vertical anion: the non-equilibrated and equilibrated state as well as for the adiabatic one (Figure 3). Secondly, the imidazolone heterocycle transformation into oxazolone caused the negative charge to settle initially (a non-equilibrated solvent–solute interaction) on the Oz (74%) and with subsequent increases up to 82% after system equilibration. A similar observation was noted in the case of the spin analysis within oligo-Oz, with 84% and 91% located on the Oz2 moiety in the vertical anion state, non-equilibrated and equilibrated, respectively, and barely anything at all on C4. A structural relaxation and achievement of the adiabatic form of the negative charge was found on the C4 subunit of Oz2C4 in 67% of the remaining part of the charge dispersed along the purine strand. The above is in good agreement with the spin distribution, which was located in 85% on the C4 base and only in 10% on T5, as shown in Figure 3.
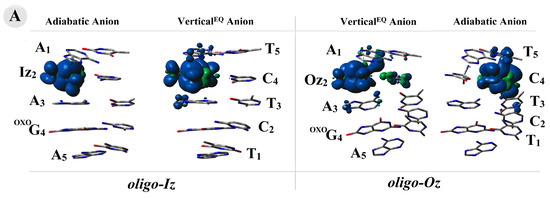
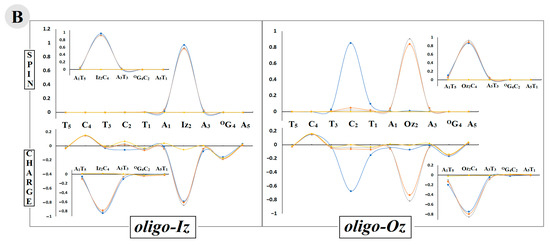
Figure 3.
(A) The spin and charge distribution within oligo-Iz and oligo-Oz calculated at the M062x/6-31++G** level of theory in the condensed phase, with the base-pair skeleton taken into consideration.  adiabatic radical anion,
adiabatic radical anion,  vertical radical anion, non-equilibrated solvent–solute interaction,
vertical radical anion, non-equilibrated solvent–solute interaction,  vertical radical cation/anion, equilibrated solvent–solute interaction, and
vertical radical cation/anion, equilibrated solvent–solute interaction, and  neutral form. The raw data of the charge and spin distribution have been given in Tables S3 and S4 (Supplementary Materials). (B) A graphical visualization of the spin distribution within oligo-Iz and oligo-Oz. The raw data have been given in Table S5.
neutral form. The raw data of the charge and spin distribution have been given in Tables S3 and S4 (Supplementary Materials). (B) A graphical visualization of the spin distribution within oligo-Iz and oligo-Oz. The raw data have been given in Table S5.
adiabatic radical anion, vertical radical anion, non-equilibrated solvent–solute interaction, vertical radical cation/anion, equilibrated solvent–solute interaction, and neutral form. The raw data of the charge and spin distribution have been given in Tables S3 and S4 (Supplementary Materials). (B) A graphical visualization of the spin distribution within oligo-Iz and oligo-Oz. The raw data have been given in Table S5.
2.4. Influence of Iz or Oz on Charge Transfer through oligo-Iz and oligo-Oz
The double helix’s ability to transfer charge follows directly from the base-pair stacking interaction [48]. Since the 1990s, ds-oligo has been perceived as a nanowire [49]. As discussed previously, the ds-DNA structure and electronic properties are influenced by the presence of Iz and Oz as part of a CDL. The additional electron or electron-hole migration process can be perceived according to three categories: single-step tunnelling, random-walk multistep, and polaron-like hopping [50]. It has been confirmed theoretically and experimentally that the charge can migrate over 200 Å within ds-DNA from the place of induction via an incoherent mechanism [51]. Contrary to this, a single-step super exchange process over a distance of a few BPs can be observed. In the case of both mechanisms, the base pairs’ spatial orientation and their individual electronic properties are essential. The theory proposed by Marcus takes all the above-discussed factors together with the rate constant (kET) equation, as presented below:
(kb—the Boltzmann constant, h—the Planck constant, T—the temperature [K], λ—the reorganization energy [eV], ΔG—the driving force [eV], and V12—the electron coupling) [52,53,54]. For details, please see Voityuk’s review [55].
The charge transfer rate depends on the following parameters: the driving force ΔG (the free energy difference between the initial and final molecule states involved in charge transfer) and the reorganization energy (λ) (the changes in adjacent structures, which occur during hole or electron migration). Both of the above are linked together in the activation energy (Ea), which depends on the intervening system [56]. Due to the nature of the charge transfer, only the process with negative ΔG was taken into consideration (Table 3). Higher |ΔG| electron-hole (positive charge) migration values were found for A3T3→OXOG4C2 and OXOG4C2←A5T1 in oligo-Iz and oligo-Oz at a level of close to 1.1 [eV]. Meanwhile, for A1T5←Iz2C4 and Iz2C4→A3T3 of oligo-Iz and A1T5←Oz2C4;Oz2C4→A3T3 of oligo-Oz, this value was found to be almost two and half times smaller, in the range of 0.3–0.4 [eV]. The calculated kET for A3T3→OXOG4C2 and OXOG4C2←A5T1 was found as follows, in [s−1]: 106; 1010 and 109 for oligo-Iz and oligo-Oz, respectively. Hence, negative reorganization values and activation energies were noted in the case of electron-hole transfer from the A1T5 toward the Iz2C4 and Oz2C4 base pair (A1T5→Oz2/Iz2C4) for both the discussed ds-oligos and Iz2C4→A3T3 for oligo-Iz. On the other hand, an analysis of the ΔG values indicates that the radical cation migration from Iz2C4 toward OXOG4C2 and Oz2C4 toward OXOG4C2 is privileged, i.e., kET = 104 and 103 [s−1] in the case of oligo-Iz and oligo-Oz, respectively (Table 3). The raw data have been given in Table S6a,b.
Table 3.
The charge transfer parameters. ΔG—driving force, λ—reorganization energy, Ea—activation energy, V12—electron coupling, and kHT—charge rate constant of permissible transfer between base pairs of oligo-Iz and oligo-Oz, calculated at the m062x/6-31++G** level of theory in the aqueous phase and given in [eV]. The arrows indicate the direction of the charge migration.
It should be pointed out that in the case of oligo-Oz, the hole transfer from A1T5 to A3T3 has the highest value, as expected. Additionally, the main spin density was found on the OXOG4C2 moiety of the discussed ds-oligo in the adiabatic cation state. Based on the above and because OXOG4C2 has the lowest ionization potential, this base pair can be assigned as the radical cation sink during electron-hole transfer.
This is in good agreement with previous observations. Because of its nature, the charge transfer through ds-DNA may occur in its oxidative and reduced state. The highest rate constant of the excess electron transfer was found for A3T3 → OXOG4C2 of oligo-Iz and OXOG4C2 ← A5T1 of oligo-Oz as follows: 7.1 × 1011 and 1.44 × 1014, respectively, in [s−1]. However, because of the highest absolute value of the ΔG and spin distribution results, it can be predicted that excess electron transfers are privileged toward Iz2C4 and Oz2C4 moieties within oligo-Iz and oligo-Oz. For the above, the high-rate constant value was calculated for excess electron migration toward the Iz2C4 or Oz2C4 base pair from the neighboring pairs, as presented in Table 3. Additionally, a difference in the transfer predisposition between Iz2C4/Oz2C4 and OXOG4C2 was observed. In the case of oligo-Iz, it was Iz2C2←OXOG4C4 (kET = 1.39 × 109 s−1), while in the case of oligo-Oz, it was noted as 8 orders of magnitude slower Oz2C4←OXOG4C2 (kET = 4.5 × 101 s−1). The above is supported by the results of the adiabatic electron affinity and the negative charge and spin distribution calculation.
3. Discussion
The genome (ds-DNA) is continuously exposed to various extra- and intra-cellular harms [4,57]. Under suitable conditions, their activity causes different types of DNA damage. Until now, more than 70 lesion types have been identified [5,58]. Therefore, the correct sequence of nucleobases in the genome is governed by different DNA repair systems, such as BER, NER, NHEJ, HR, etc. [59]. Any deficiency can lead to mutations and, as a consequence, to carcinogenesis or an accelerated aging process [60]. Most of the DNA damage is removed from ds-DNA by BER machinery [61]. The subsequent cascade of protein activity is initiated by glycosylases [62,63]. These specific enzymes identify and remove the designated lesion. However, the above process raises the important question of how these proteins are capable of scanning the genome in such an effective way so as to prevent genetic information from being permanently altered. Moreover, the accumulation of unrepaired units in the genome can cause multiple damage sites to form and subsequently affect further repairs to lesions [64]. Cappelli at al. have shown that the abundant glycosylases (105) present in the nucleus must scan approximately 7 × 104 base pairs per individual [65]. Additionally, in a single cell, 104 DNA lesions are induced daily [3]. Recently, Barton et al. have shown that glycosylases like MutY can recognize a DNA lesion by charge transfer, making this process extremely fast and effective in the case of isolated lesions [37,66,67]. However, the situation becomes more complicated when a multi-DNA damage site is taken into consideration. Given the above, the influence of CDLs containing Iz or Oz and OXOdG on the electron or hole transfer process was considered worthy of investigation. With this in mind, the following ds-oligos were chosen: oligo-Iz: d[A1Iz2A3OXOG4A5]*d[T5C4T3C2T1] and oligo-Oz: d[A1Oz2A3OXOG4A5]*d[T5C4T3C2T1]. The OXOdG is established as one of the more abundant one-electron oxidation lesions, while imidazolone or oxazolone are less propagated four-electron oxidation examples [68]. It should be pointed out that Oz can appear in the genome as the second product of 2′-deoxyguanosie or 7,8-dihydro-8-oxo-2′-deoxyguanosine degradation [23,25]. Meanwhile, Iz can be perceived as intermediate, which after slow hydrolysis (147 min) is converted to oxazolone [23] (Figure 1). Moreover, Ming et al. have shown that the number of both lesions, i.e., OXOdG and Oz, increases in the presence of 5-methycytosine when a CpG island of p53 is taken into consideration [69]. Therefore, the discussed type of CDL on charge transfer should not be omitted. The results presented in this manuscript reveal that imidazolone forms two hydrogen bonds with complementary cytidine, while within the Oz2:C4 base only one is found. The following EHBs were noted for Iz::C and Oz:C, respectively: 12.8 and 7.7 kcal. Moreover, the presence of the final guanine four-electron oxidizing product within oligo-Oz impaired the stacking interaction energy of the Oz2C4||A3T3 BP dimer to 12.77 kcal, which is in good agreement with the Rise parameter increases of up to 3.46 Å (Tables S1 and S2). Surprisingly, when oligo-Iz was taken into consideration, these parameters were found at the level assigned for native oligo-N as follows: Rise = 2.93 Å and EST = 14.97 kcal. The appearance of an additional electron within the oligo-Iz leads to greater structural changes than those noted for oligo-Oz, while the electron loss by the system leads to the opposite observation, with a greater geometry susceptibility to a positive charge being noted for oligo-Oz, which is shown in Table 1 as the results of RMSD value calculations. The analysis of the calculated ionization potential and electron affinity parameters of ds-oligo showed the highest AEA for oligo-Iz (2.83 eV), compared to oligo-Oz (2.06 eV), while the AIP was found at the same level (5.47 eV) in both cases. The above indicates a greater influence of Iz and Oz on electron affinity than OXOG if both lesions are part of a CDL structure. Conversely, OXOG determines the ionization properties of the discussed ds-oligo. The above is in good agreement with the results of the charge and spin distribution analysis performed in both the vertical and adiabatic modes. The radical cation is mainly located at the OXOdG4C2 base pair, irrespective of the presence of a second DNA lesion (Iz or Oz). Conversely, the adoption of an extra electron by oligo-Iz and oligo-Oz leads to similar outcomes. The negative charge and spin were mainly found on Oz2C4 and Iz2C4. However, an advanced analysis revealed that the extra charge is mainly located on the Iz moiety in the vertical and adiabatic anion states of oligo-Iz. On the other hand, in the case of oligo-Oz, the extra electron (vertical anion) is initially located on the Oz2 subunit and subsequently migrates to the complementary cytidine (C4) base moiety. These results correspond well with the assigned electronic properties for individual base pairs extracted from a ds-oligo. For both oligo-Iz and oligo-Oz, lower ionization potentials were found for OXOG4C2 (~5.5 eV), while the highest electron affinity was found for Oz2C4 and Iz2C4, i.e., 2.97 and 1.89, respectively, in eV. Taking this all together, it can be predicted that the guanosine four-electron oxidation product, constituting part of a clustered lesion together with OXOG, can affect the charge transfer through a double helix. An electron-hole transfer investigation, according to the Marcus [70] theory, revealed that in the case of oligo-Iz and oligo-Oz, the radical cation migration toward the OXOdG4C2 base pair was privileged. However, in the case of excess electron transfer, differences in preference were observed. In both cases, i.e., oligo-Iz and oligo-Oz, the electron transfer toward the Iz2C4 and Oz2C4 moiety was noted as privileged (Table 3). Therefore, it can be concluded that in both ds-oligo cases, the OXOG4C2 base pair should be privileged to a suitable radical cation formation, while the excess electron should settle at Iz2C4 of oligo-Iz and at Oz2C4 of oligo-Oz. The above observation indicates that four-electron oxidation products can significantly affect the charge migration process through the double helix. It should be pointed out that the initial imidazolone moiety is subsequently hydrolyzed to oxazolone. This can manifest itself by the slowing down of other DNA lesion recognition and removal processes, which can increase the probability of mutagenesis and subsequent pathological processes. However, with regard to anticancer therapy (radio/chemo), the presence of Iz and further Oz in the structure of clustered DNA damage can result in improved cancer treatment.
It should be noted that four-electron oxidation is a rare event under physiological conditions. However, during anticancer therapy (e.g., ionization radiation), four-electron oxidation can occur, promoting Iz and subsequently Oz formation. Identifying and quantifying secondary oxidation products of 2′-deoxyguanosine such as Iz and Oz poses serious challenges in terms of analytical techniques because of their instability and high hydrophilicity [71]. Even in low abundance, Iz and Oz (2–6 per 107 [23]) can play a significant role in mutagenesis/cancerogenesis. It has been found that unlike OXOdG, which yields 3% of G → T transversion, Oz results in G → C preferences [72]. Furthermore, the presence of Iz and/or Oz in the structure of the double helix can lead to a protein communication process via charge transfer [73]. It has been found that after electron loss, the binding of MutYh to ds-DNA increases a thousand-fold [19,74,75]. If no reduction takes place, it can hinder the movement of other proteins, such as OGG1. The above indicates that the cumulation of DNA damage over a short distance renders the damage repair process difficult with a longer repair time and reduced fidelity [76].
4. Materials and Methods
All the theoretical calculations were carried out according to previous descriptions, in brief [77]: the starting geometries of the bi-stranded oligo-Iz and oligo-Oz were built using BIOVIA Discovery Studio Visualizer v20.1.0.19295 software [78] and denoted accordingly as d[A1Iz2A3OXOG4A5]*d[T5C4T3C2T1] and d[A1Oz2A3OXOG4A5]*d[T5C4T3C2T1], respectively.
The negative charges of the phosphate groups were neutralized by the addition of protons, and the other atoms were saturated by additional hydrogen atoms. The structure optimizations of oligo-Iz and oligo-Oz were performed using the ONIOM (Our Own N-layered Integrated Molecular Orbital and Molecular Mechanics) strategy [40]. The structures of the ds-oligos were divided into high—HL (nucleobases, M06-2X/D95**)—and low—LL (sugar-phosphate backbone, M06-2X/sto-3G)—levels of calculation [79]. In the course of the ONIOM calculation, the atoms in the HL, which are bonded to atoms in the LL system, were modeled using link atoms when the computations were performed on the HL system. In the case of a carbon atom (CLL) from the low level being connected with a carbon atom (CHL) from the high-level part (CLL-CHL bond), the CLL atom was replaced by a hydrogen atom in the ensuing energy computations [80]. The graphical representation of the ONIOM layers has been presented in Figure S1, Supplementary Materials. Following Truhlar’s and Lin’s studies, the Z3 scheme was found to be the default for the charge manipulation in the link atom approach in the ONIOM calculations using Gaussian 16 software. For details of ONIOM, please see references [81,82]. All the calculations were performed in the aqueous phase. The M06-2X functional with the augmented polarized valence double-ζ basis set 6-31++G** was used for the energy calculations. For all the optimized geometries, the charge and spin analyses were achieved using the Hirshfeld methodology at the M06-2X/6-31++G** level of theory [83]. The electronic properties of the molecules were calculated as described previously [84,85]. The chosen level of theory was previously carefully investigated and compared with other DFT functionals such as M-11, ωB97-XD, and M06-L, for which the following basis sets were used: 6-31++G*, 6-31++G**, and D95** aug-cc-pVDZ. In all the tested cases, the radical anion represented the valence type, not the dipole, except for ωB97-XD/6-31++G** [86]. The influence of the basis set and DFT functionals was previously carefully investigated by Sevilla et al. for isolated nucleobases [87]. It should be pointed out that the electronic behavior of the base pairs was found to be different in comparison to the isolated base pairs due to the possibility of a charge-proton transfer. The transition dipole moment of excited states and the single point calculation at the M06-2X/6-31++G** level of theory were performed using time-dependent DFT (TD-DFT) methodology [88]. The electron coupling was calculated according to the Generalized Mulliken–Hush model [89]. The solvation–solute interaction was investigated in both non-equilibrium (NE) and equilibrated (EQ) modes [41]. All the calculations of the electronic properties, i.e., the VIPNE (vertical ionization potential in the NE state), VIPEQ (vertical ionization potential in the EQ state), AIP (adiabatic ionization potential), VEANE (vertical electron affinity in the NE state), VEAEQ (vertical electron affinity in the EQ state), and AEA (adiabatic electron affinity), were conducted in [eV], as described previously [85]. All the above calculations were performed using the Gaussian G16 (version C.01) software suite [90].
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules29122754/s1, Table S1. The structural local base-pair parameters: rise and dC1′…C1′ in [Å] and λ1 and λ2 in [O] according to the standard DNA reference frame of oligo-Iz and oligo-Oz. The hydrogen bond lengths are given in [Å] (HB-1: Ade(N1), Thy(N3) and Gua(O6), and Cyt(N4); HB-2: Ade(N6), Thy(O4) and Gua(N1), and Cyt(N3); HB-3: Gua(N2) and Cyt(O2); for Iz2C4: HB-1 (Cyt(N4), Iz(N1), HB-2 Cyt(N3), and Iz(N2); and for Oz2C4: HB-1 (Cyt(N4) and Oz(O1)). Table S2. The stacking and hydrogen bond energies in [kcal] calculated at the M062x/6-31++G** level of theory in the aqueous phase. The raw data are given in Tables S1 and S2 of the Supplementary Materials. Table S3. The energies (in Hartree) of the neural, vertical cation (VCNC) (NE—non-equilibrated), vertical cation (VCEQ) (EQ—equilibrated), vertical anion (VANE), vertical anion (VAEQ), adiabatic cation (AC), and adiabatic anion (AA) of a complete DNA double helix and base-pair skeleton extracted from ds-oligonucleotides calculated at the M06-2X/6-31++G** level of theory in the aqueous phase, respectively. Table S4. The energies (in Hartree) of the neural, vertical cation, and adiabatic cation forms of the base pairs extracted from the ds-oligonucleotides calculated at the M06-2x/6-31++G** level of theory in the aqueous phase. Table S5. The Hirshfeld charge and spin distribution in the shape of oligo-Iz, d[A1Iz2A3OXOG4A5]*d[T5C4T3C2T1], and oligo-Oz, d[A1Oz2A3OXOG4A5]*d[T5C4T3C2T1], where only the nucleoside bases were taken into consideration, calculated at the M06-2x/6-31++G** level of theory in the aqueous phase. Vertical cation (VCNC) (NE—non-equilibrated), vertical cation (VCEQ) (EQ—equilibrated), vertical anion (VANE), vertical anion (VAEQ), adiabatic cation (AC), and adiabatic anion (AA). Table S6a. The energies: The ground (EGR) and excitation (EEX) state energies and excitation and HOMO energies as well as the corresponding dipole moment ground, excitation, and transition (DMG, DMEX, and D12) in Debays of the neighboring base pair extracted from the selected dimers of the ds-oligonucleotides, calculated at the M06-2x/6-31++G** level of theory in the aqueous phase using the DFT or TD-DFT methodology. Table S6b. The energies: The ground (EGR) and excitation (EEX) state energies and excitation and HOMO energies as well as the corresponding dipole moment ground, excitation, and transition (DMG, DMEX, and D12) in Debays of the distal base pair extracted from the selected trimers of the ds-oligonucleotides, calculated at the M06-2x/6-31++G** level of theory in the aqueous phase using the DFT or TD-DFT methodology. Figure S1. The graphical representation of the ONIOM layer distribution used in these studies.
Funding
This study was supported by the low number of the above glycosylases by the Medical University of Lodz, 503/3-045-02/503-31-002, and in part by PL-Grid infrastructure (ACC Cyfronet AGH, PLG/2021/014771).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The author would like to thank Idalgo van der Berg for his valuable input during discussions and the mental support.
Conflicts of Interest
The author declares no conflicts of interest.
References
- Brown, T.A. Genomes, 2nd ed.; Wiley-Liss: Oxford, UK, 2002; ISBN 10: 0-471-25046-5. [Google Scholar]
- Nissanka, N.; Minczuk, M.; Moraes, C.T. Mechanisms of Mitochondrial DNA Deletion Formation. Trends Genet. 2019, 35, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Sudhir Ambekar, S. DNA: Damage and Repair Mechanisms in Humans. Glob. J. Pharm. Pharm. Sci. 2017, 3, 555613. [Google Scholar] [CrossRef][Green Version]
- Tubbs, A.; Nussenzweig, A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.D.; Dizdaroglu, M.; Cooke, M.S. Oxidative DNA damage and disease: Induction, repair and significance. Mutat. Res. /Rev. Mutat. Res. 2004, 567, 1–61. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef] [PubMed]
- Poletto, M.; Legrand, A.J.; Dianov, G.L. DNA Base Excision Repair: The Achilles’ Heel of Tumour Cells and their Microenvironment? Curr. Pharm. Des. 2020, 23, 4758–4772. [Google Scholar] [CrossRef]
- Farnell, D.A. Nucleotide Excision Repair in the Three Domains of Life. West. Undergrad. Res. J. Health Nat. Sci. 2011, 2, 1–6. [Google Scholar] [CrossRef]
- Jacobs, A.L.; Schär, P. DNA glycosylases: In DNA repair and beyond. Chromosoma 2012, 121, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Steenken, S.; Jovanovic, S.V. How easily oxidizable is DNA? One-electron reduction potentials of adenosine and guanosine radicals in aqueous solution. J. Am. Chem. Soc. 1997, 119, 617–618. [Google Scholar] [CrossRef]
- Khanduri, D.; Adhikary, A.; Sevilla, M.D. Highly Oxidizing Excited States of One-Electron-Oxidized Guanine in DNA: Wavelength and pH Dependence. J. Am. Chem. Soc. 2011, 12, 4527–4537. [Google Scholar] [CrossRef]
- Barry Halliwell, A.; Adhikary, M.; Dingfelder, M.D. Hydroxyl radical is a significant player in oxidative DNA damage in vivo. Chem. Soc. Rev. 2021, 50, 8355–8360. [Google Scholar] [CrossRef] [PubMed]
- Jing, X.; Yang, F.; Shao, C.; Wei, K.; Xie, M.; Shen, H.; Shu, Y. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol. Cancer 2019, 18, 157. [Google Scholar] [CrossRef] [PubMed]
- Keith, B.; Simon, M.C. Hypoxia-Inducible Factors, Stem Cells, and Cancer. Cell 2007, 129, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Scanlan, L.D.; Coskun, S.H.; Jaruga, P.; Hanna, S.K.; Sims, C.M.; Almeida, J.L.; Catoe, D.; Coskun, E.; Golan, R.; Dizdaroglu, M.; et al. Measurement of Oxidatively Induced DNA Damage in Caenorhabditis elegans with High-Salt DNA Extraction and Isotope-Dilution Mass Spectrometry. Anal. Chem. 2019, 91, 12149–12155. [Google Scholar] [CrossRef] [PubMed]
- Matter, B.; Malejka-giganti, D.; Csallany, A.S.; Tretyakova, N. Quantitative analysis of the oxidative DNA lesion, 2,2-diamino-4-(2-deoxy-b-D-erythro-in vitro and in vivo by isotope dilution-capillary HPLC-ESI-MS/MS. Nucleic Acids Res. 2006, 34, 5449–5460. [Google Scholar] [CrossRef] [PubMed]
- Dizdaroglu, M. Base-excision repair of oxidative DNA damage by DNA glycosylases. Mutat. Res.—Fundam. Mol. Mech. Mutagen. 2005, 591, 45–59. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Kamiya, H. Mutations induced by 8-hydroxyguanine (8-oxo-7,8-dihydroguanine), a representative oxidized base, in mammalian cells. Genes Environ. 2017, 39, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Kairupan, C.; Scott, R.J. Base excision repair and the role of MUTYH. Hered. Cancer Clin. Pract. 2007, 5, 199–209. [Google Scholar] [CrossRef]
- Choi, S.; Cooley, R.B.; Hakemian, A.S.; Larrabee, Y.C.; Bunt, R.C.; Maupas, S.D.; Muller, J.G.; Burrows, C.J. Mechanism of Two-Electron Oxidation of Deoxyguanosine 5′-Monophosphate by a Platinum(IV) Complex. J. Am. Chem. Soc. 2004, 126, 591–598. [Google Scholar] [CrossRef]
- Kino, K.; Kawada, T.; Hirao-Suzuki, M.; Morikawa, M.; Miyazawa, H. Products of oxidative guanine damage form base pairs with guanine. Int. J. Mol. Sci. 2020, 21, 7645. [Google Scholar] [CrossRef]
- Morikawa, M.; Kino, K.; Oyoshi, T.; Suzuki, M.; Kobayashi, T.; Miyazawa, H. Product analysis of photooxidation in isolated quadruplex DNA; 8-oxo-7,8-dihydroguanine and its oxidation product at 3′-G are formed instead of 2,5-diamino-4H-imidazol-4-one. RSC Adv. 2013, 3, 25694–25697. [Google Scholar] [CrossRef]
- Suzuki, M.; Kino, K.; Kawada, T.; Morikawa, M.; Kobayashi, T.; Miyazawa, H. Analysis of Nucleotide Insertion Opposite 2,2,4-Triamino-5(2H)-oxazolone by Eukaryotic B- and Y-Family DNA Polymerases. Chem. Res. Toxicol. 2015, 28, 1307–1316. [Google Scholar] [CrossRef]
- Cadet, J.; Angelov, D.; Wagner, J.R. Hydroxyl radical is predominantly involved in oxidatively generated base damage to cellular DNA exposed to ionizing radiation. Int. J. Radiat. Biol. 2022, 98, 1684–1690. [Google Scholar] [CrossRef]
- Duarte, V. In vitro DNA synthesis opposite oxazolone and repair of this DNA damage using modified oligonucleotides. Nucleic Acids Res. 2000, 28, 1555–1563. [Google Scholar] [CrossRef] [PubMed]
- Henderson, P.T.; Delaney, J.C.; Gu, F.; Tannenbaum, S.R.; Essigmann, J.M. Oxidation of 7,8-dihydro-8-oxoguanine affords lesions that are potent sources of replication errors in vivo. Biochemistry 2002, 41, 914–921. [Google Scholar] [CrossRef]
- Zhou, J.; Liu, M.; Fleming, A.M.; Burrows, C.J.; Wallace, S.S. Neil3 and NEIL1 DNA glycosylases remove oxidative damages from quadruplex DNA and exhibit preferences for lesions in the telomeric sequence context. J. Biol. Chem. 2013, 288, 27263–27272. [Google Scholar] [CrossRef]
- Karahalil, B.; Hogue, B.A.; Souza-Pinto, N.C.; Bohr, V.A. Base excision repair capacity in mitochondria and nuclei: Tissue-specific variations. FASEB J. 2002, 16, 1895–1902. [Google Scholar] [CrossRef] [PubMed]
- Romano, C.A. DNA-mediated charge transfer between [4Fe-4S] cluster glycosylases. Ph.D. Dissertation, California Institute of Technology, Pasadena, CA, USA, 2011. [Google Scholar]
- White, M.F.; Dillingham, M.S. Iron-sulphur clusters in nucleic acid processing enzymes. Curr. Opin. Struct. Biol. 2012, 22, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Lomax, M.E.; Folkes, L.K.; Neill, P.O. Biological Consequences of Radiation-induced DNA Damage: Relevance to Radiotherapy Statement of Search Strategies Used and Sources of Information Why Radiation Damage is More Effective than Endogenous Damage at Killing Cells Ionising Radiation-induced Do. Clin. Oncol. 2013, 25, 578–585. [Google Scholar] [CrossRef]
- Bukowska, B.; Karwowski, B.T. The Clustered DNA Lesions—Types, Pathways of Repair and Relevance to Human Health. Curr. Med. Chem. 2018, 25, 2722–2735. [Google Scholar] [CrossRef]
- Karwowski, B.T. The Influence of Single, Tandem, and Clustered DNA Damage on the Electronic Properties of the Double Helix: A Theoretical Study. Molecules 2020, 25, 3126. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Zhang, Y.; Liu, C.; Zhang, M.; Han, S. Application of radiosensitizers in cancer radiotherapy. Int. J. Nanomed. 2021, 16, 1083–1102. [Google Scholar] [CrossRef]
- Huang, R.; Zhou, P. DNA damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal Transduct. Target. Ther. 2020, 5, 60. [Google Scholar] [CrossRef]
- Mignon, P.; Loverix, S.; Steyaert, J.; Geerlings, P. Influence of the π–π interaction on the hydrogen bonding capacity of stacked DNA/RNA bases. Nucleic Acids Res. 2005, 33, 1779–1789. [Google Scholar] [CrossRef] [PubMed]
- Boal, A.K.; Yavin, E.; Lukianova, O.A.; O’Shea, V.L.; David, S.S.; Barton, J.K. DNA-bound redox activity of DNA repair glycosylases containing [4Fe-4S] clusters. Biochemistry 2005, 44, 8397–8407. [Google Scholar] [CrossRef]
- Boal, A.K.; Yavin, E.; Barton, J.K. DNA repair glycosylases with a [4Fe-4S] cluster: A redox cofactor for DNA-mediated charge transport? J. Inorg. Biochem. 2007, 101, 1913–1921. [Google Scholar] [CrossRef][Green Version]
- Karwowski, B. How Clustered DNA Damage Can Change the Electronic Properties of ds-DNA, Differences between GAG, GAOXOG, OXOGAOXOG. Biomolecules 2023, 13, 517. [Google Scholar] [CrossRef]
- Dapprich, S.; Komáromi, I.; Byun, K.S.; Morokuma, K.; Frisch, M.J. A new ONIOM implementation in Gaussian98. Part I. The calculation of energies, gradients, vibrational frequencies and electric field derivatives. J. Mol. Struct. THEOCHEM 1999, 461–462, 1–21. [Google Scholar] [CrossRef]
- Miertus, S.; Tomasi, J. Approximate evaluations of the electrostatic free energy and internal energy changes in solution processes. Chem. Phys. 1982, 65, 239–245. [Google Scholar] [CrossRef]
- Cancès, E.; Mennucci, B.; Tomasi, J. A new integral equation formalism for the polarizable continuum model: Theoretical background and applications to Isotropic and anisotropic dielectrics. J. Chem. Phys. 1997, 107, 3032–3041. [Google Scholar] [CrossRef]
- Olson, W.K.; Bansal, M.; Burley, S.K.; Dickerson, R.E.; Gerstein, M.; Harvey, S.C.; Heinemann, U.; Lu, X.J.; Neidle, S.; Shakked, Z.; et al. A standard reference frame for the description of nucleic acid base-pair geometry. J. Mol. Biol. 2001, 313, 229–237. [Google Scholar] [CrossRef]
- Kim, H.J.; Ro, Y.; Hong, B.; Ji, H.G. Formation of au nanowires by DNA molecules as a template. J. Korean Phys. Soc. 2009, 55, 1892–1895. [Google Scholar] [CrossRef]
- Sontz, P.A.; Mui, T.P.; Fuss, J.O.; Tainer, J.A.; Barton, J.K. DNA charge transport as a first step in coordinating the detection of lesions by repair proteins. Proc. Natl. Acad. Sci. USA 2012, 109, 1856–1861. [Google Scholar] [CrossRef] [PubMed]
- Arnittali, M.; Rissanou, A.N.; Harmandaris, V. Structure of Biomolecules Through Molecular Dynamics Simulations. Procedia Comput. Sci. 2019, 156, 69–78. [Google Scholar] [CrossRef]
- Marenich, A.V.; Jerome, S.V.; Cramer, C.J.; Truhlar, D.G. Charge Model 5: An Extension of Hirshfeld Population Analysis for the Accurate Description of Molecular Interactions in Gaseous and Condensed Phases. J. Chem. Theory Comput. 2012, 8, 527–541. [Google Scholar] [CrossRef]
- Voityuk, A.A. Estimates of electronic coupling for excess electron transfer in DNA. J. Chem. Phys. 2005, 123, 34903. [Google Scholar] [CrossRef]
- Ram Kumar Pandian, S.; Yuan, C.J.; Lin, C.C.; Wang, W.H.; Chang, C.C. DNA-based nanowires and nanodevices. Adv. Phys. X 2017, 2, 22–34. [Google Scholar] [CrossRef]
- Lewis, F.D.; Liu, J.; Weigel, W.; Rettig, W.; Kurnikov, I.V.; Beratan, D.N. Donor-bridge-acceptor energetics determine the distance dependence of electron tunneling in DNA. Proc. Natl. Acad. Sci. USA 2002, 99, 12536–12541. [Google Scholar] [CrossRef]
- Genereux, J.C.; Barton, J.K. Mechanisms for DNA charge transport. Chem. Rev. 2010, 110, 1642–1662. [Google Scholar] [CrossRef]
- Marcus, R.A. Electron transfer reactions in chemistry. Theory and experiment. Rev. Mod. Phys. 1993, 65, 599–610. [Google Scholar] [CrossRef]
- Rawtani, D.; Kuntmal, B.; Agrawal, Y. Charge transfer in DNA and its diverse modelling approaches. Front. Life Sci. 2016, 9, 214–225. [Google Scholar] [CrossRef][Green Version]
- Bolton, J.R.; Archer, M.D. Basic Electron-Transfer Theory. In Electron Transfer in Inorganic, Organic, and Biological Systems; Bolton, J.R., Mataga, N., McLendon, G., Eds.; American Chemical Society: Washington, DC, USA, 1991; Volume 228, pp. 7–23. [Google Scholar]
- Rösch, N.; Voityuk, A.A. Quantum Chemical Calculation of Donor–Acceptor Coupling for Charge Transfer in DNA. In Long-Range Charge Transfer in DNA II; Springer: Berlin/Heidelberg, Germany, 2012; pp. 37–72. [Google Scholar] [CrossRef]
- Khan, A. Reorganization energy, activation energy, and mechanism of hole transfer process in DNA: A theoretical study. J. Chem. Phys. 2008, 128, 075101. [Google Scholar] [CrossRef] [PubMed]
- Dizdaroglu, M. Oxidatively Induced DNA Damage and Its Repair. Ref. Modul. Life Sci. 2017, 1–5. [Google Scholar] [CrossRef]
- Olinski, R.; Siomek, A.; Rozalski, R.; Gackowski, D.; Foksinski, M.; Guz, J.; Dziaman, T.; Szpila, A.; Tudek, B. Oxidative damage to DNA and antioxidant status in aging and age-related diseases. Acta Biochim. Pol. 2007, 54, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Demple, B.; Harrison, L. Repair of oxidative damage to DNA: Enzymology and biology. Annu. Rev. Biochem. 1994, 63, 915–948. [Google Scholar] [CrossRef] [PubMed]
- Wallace, S.S. Biological consequences of free radical-damaged dna bases. Free Radic. Biol. Med. 2002, 33, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Hang, B. Base excision repair. In DNA Repair, Genetic Instability, and Cancer, 1st ed.; Wei, Q., Li, L., Chen, D.J., Eds.; World Scientific: Singapore, 2007; pp. 23–64. [Google Scholar] [CrossRef]
- Shikazono, N.; Pearson, C.; Neill, P.O.; Thacker, J. The roles of specific glycosylases in determining the mutagenic consequences of clustered DNA base damage. Nucleic Acids Res. 2006, 34, 3722–3730. [Google Scholar] [CrossRef] [PubMed]
- Wallace, S.S. DNA glycosylases search for and remove oxidized DNA bases. Environ. Mol. Mutagen. 2013, 54, 691–704. [Google Scholar] [CrossRef]
- Byrne, S.; Cunniffe, S.; O’Neill, P.; Lomax, M.E. 5,6-Dihydrothymine impairs the base excision repair pathway of a closely opposed AP site or single-strand break. Radiat. Res. 2009, 172, 537–549. [Google Scholar] [CrossRef]
- Friedman, J.I.; Stivers, J.T. Detection of damaged DNA bases by DNA glycosylase enzymes. Biochemistry 2010, 49, 4957–4967. [Google Scholar] [CrossRef]
- Tse, E.C.M.; Zwang, T.J.; Barton, J.K. The Oxidation State of [4Fe4S] Clusters Modulates the DNA-Binding Affinity of DNA Repair Proteins. J. Am. Chem. Soc. 2017, 136, 12784–12792. [Google Scholar] [CrossRef] [PubMed]
- Baranovskiy, A.G.; Babayeva, N.D.; Zhang, Y.; Blanco, L.; Pavlov, Y.I.; Tahirov, T.H. Comment on “The [4Fe4S] cluster of human DNA primase functions as a redox switch using DNA charge transport. Science 2017, 357, 2954. [Google Scholar] [CrossRef] [PubMed]
- Fleming, A.M.; Burrows, C.J. Formation and processing of DNA damage substrates for the hNEIL enzymes. Free Radic. Biol. Med. 2017, 107, 35–52. [Google Scholar] [CrossRef] [PubMed]
- Ming, X.; Matter, B.; Song, M.; Veliath, E.; Shanley, R.; Jones, R. Mapping Structurally Defined Guanine Oxidation Products along DNA Duplexes: Influence of Local Sequence Context and Endogenous Cytosine Methylation. J. Am. Chem. Soc. 2014, 136, 4223–4235. [Google Scholar] [CrossRef] [PubMed]
- Fujitsuka, M.; Majima, T. Charge transfer in DNA. Pure Appl. Chem. 2013, 85, 1367–1377. [Google Scholar] [CrossRef]
- Yin, J.; Zhang, N.; Wang, H. Liquid chromatography-mass spectrometry for analysis of DNA damages induced by environmental exposure. Trends in Analytical Chemistry 2019, 120, 1–11. [Google Scholar] [CrossRef]
- Neeley, W.L.; Delaney, S.; Alekseyev, Y.O.; Jarosz, D.F.; Delaney, J.C.; Walker, G.C.; Essigmann, J.M. DNA polymerase V allows bypass of toxic guanine oxidation products in vivo. J. Biol. Chem. 2007, 282, 12741–12748. [Google Scholar] [CrossRef] [PubMed]
- Syed, A.; Tainer, J.A. Charge Transport Communication through DNA by Protein Fe − S Clusters: How Far Is Not Too Far? ACS Cent. Sci. 2019, 5, 7–9. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, A.H.S.; da Silva, A.E.; de Oliveira, I.M.; Henriques, J.A.P.; Agnez-Lima, L.F. MutY-glycosylase: An overview on mutagenesis and activities beyond the GO system. Mutat. Res.—Fundam. Mol. Mech. Mutagen. 2014, 769, 119–131. [Google Scholar] [CrossRef]
- Arnold, A.R.; Grodick, M.A.; Barton, J.K. DNA Charge Transport: From Chemical Principles to the Cell. Cell Chem. Biol. 2016, 23, 183–197. [Google Scholar] [CrossRef]
- Eccles, L.J.; Neill, P.O.; Lomax, M.E. Delayed repair of radiation induced clustered DNA damage: Friend or foe? Mutat. Res. /Fundam. Mol. Mech. Mutagen. 2011, 711, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Karwowski, B.T. Charge Transfer Depends on Its Diastereomeric Form: A Theoretical Study. Antioxidants 2023, 12, 881. [Google Scholar] [CrossRef] [PubMed]
- BIOVIA. BIOVIA. Discovery Studio Visualizer; v16.1.0.15350; BIOVIA: San Diego, CA, USA, 2015. [Google Scholar]
- Zhao, Y.; Pu, J.; Lynch, B.J.; Truhlar, D.G. Tests of second-generation and third-generation density functionals for thermochemical kinetics. Phys. Chem. Chem. Phys. 2004, 6, 673–676. [Google Scholar] [CrossRef]
- Lin, H.; Truhlar, D.G. Redistributed charge and dipole schemes for combined quantum mechanical and molecular mechanical calculations. J. Phys. Chem. A 2005, 109, 3991–4004. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Truhlar, D.G. QM/MM: What have we learned, where are we, and where do we go from here? Theor. Chem. Acc. 2007, 117, 185–199. [Google Scholar] [CrossRef]
- Mayhall, N.J.; Raghavachari, K. Charge transfer across ONIOM QM:QM boundaries: The impact of model system preparation. J. Chem. Theory Comput. 2010, 6, 3131–3136. [Google Scholar] [CrossRef] [PubMed]
- Hirshfeld, F.L. Bonded-atom fragments for describing molecular charge densities. Theor. Chim. Acta 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Karwowski, B.T. Ionisation potential and electron affinity of free 5′,8-cyclopurine-2′-deoxynucleosides. DFT study in gaseous and aqueous phase. Cent. Eur. J. Chem. 2010, 8, 70–76. [Google Scholar] [CrossRef]
- Karwowski, B.T. The AT Interstrand Cross-Link: Structure, Electronic Properties, and Influence on Charge Transfer in dsDNA. Mol. Ther.—Nucleic Acids 2018, 13, 665–685. [Google Scholar] [CrossRef]
- Karwowski, B.T. The 2Ih and OXOG Proximity Consequences on Charge Transfer through ds -DNA: Theoretical Studies of Clustered DNA Damage. Molecules 2023, 28, 2180. [Google Scholar] [CrossRef]
- Li, X.; Cai, Z.; Sevilla, M.D. DFT calculations of the electron affinities of nucleic acid bases: Dealing with negative electron affinities. J. Phys. Chem. A 2002, 106, 1596–1603. [Google Scholar] [CrossRef]
- Kumar, A.; Sevilla, M.D. Photoexcitation of dinucleoside radical cations: A time-dependent density functional study. J. Phys. Chem. B 2006, 110, 24181–24188. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cave, R.J.; Newton, M.D. Generalization of the Mulliken-Hush treatment for the calculation of electron transfer matrix elements. Chem. Phys. Lett. 1996, 249, 15–19. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 16, Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2019. [Google Scholar]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).