


The Development of Visible-Light Organic Photocatalysts for Atom Transfer Radical Polymerization via Conjugation Extension


Abstract
1. Introduction
2. Results and Discussion
2.1. Photocatalyst Development
2.2. Catalyst Evaluation
2.3. Kinetic Study, Temporal Control, and Polymerization Extension
3. Materials and Methods
3.1. Materials and Instruments
3.2. Synthesis and Characterization of Catalyst PSeH
3.3. Typical Procedures for Photoinduced O-ATRP
3.4. Polymerization Procedure for Chain Extension from PMMA Macroinitiator
3.5. General Methods for Analysis of Kinetics and Molecular Weight Growth
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Corrigan, N.; Jung, K.; Moad, G.; Hawker, C.J.; Matyjaszewski, K.; Boyer, C. Reversible-Deactivation Radical Polymerization (Controlled/Living Radical Polymerization): From Discovery to Materials Design and Applications. Prog. Polym. Sci. 2020, 111, 101311. [Google Scholar] [CrossRef]
- Corrigan, N.; Yeow, J.; Judzewitsch, P.; Xu, J.; Boyer, C. Seeing the Light: Advancing Materials Chemistry through Photopolymerization. Angew. Chem. Int. Ed. 2019, 58, 5170–5189. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.J.; Hyatt, M.G.; Miller, S.A.; Guironnet, D. Recent Trends in Catalytic Polymerizations. ACS Catal. 2019, 9, 11153–11188. [Google Scholar] [CrossRef]
- de Ávila Gonçalves, S.; Rodrigues, P.R.; Pioli Vieira, R. Metal-Free Organocatalyzed Atom Transfer Radical Polymerization: Synthesis, Applications, and Future Perspectives. Macromol. Rapid Commun. 2021, 42, 2100221. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-S.; Ejeta, D.D.; Kuo, S.-W.; Nakamura, Y.; Huang, C.-F. Combinations (Є) among Controlled/Living Polymerizations and Utilizations of Efficient Chemical Reactions for the Synthesis of Novel Polymeric Materials. Polym. Chem. 2023, 14, 4783–4803. [Google Scholar] [CrossRef]
- Wang, J.-S.; Matyjaszewski, K. Controlled/”living” Radical Polymerization. Atom Transfer Radical Polymerization in the Presence of Transition-Metal Complexes. J. Am. Chem. Soc. 1995, 117, 5614–5615. [Google Scholar] [CrossRef]
- Kato, M.; Kamigaito, M.; Sawamoto, M.; Higashimura, T. Polymerization of Methyl Methacrylate with the Carbon Tetrachloride/Dichlorotris-(Triphenylphosphine)Ruthenium(II)/Methylaluminum Bis(2,6-di-tert-butylphenoxide) Initiating System: Possibility of Living Radical Polymerization. Macromolecules 1995, 28, 1721–1723. [Google Scholar] [CrossRef]
- Pan, X.; Tasdelen, M.A.; Laun, J.; Junkers, T.; Yagci, Y.; Matyjaszewski, K. Photomediated Controlled Radical Polymerization. Prog. Polym. Sci. 2016, 62, 73–125. [Google Scholar] [CrossRef]
- Corrigan, N.; Shanmugam, S.; Xu, J.; Boyer, C. Photocatalysis in Organic and Polymer Synthesis. Chem. Soc. Rev. 2016, 45, 6165–6212. [Google Scholar] [CrossRef]
- Dworakowska, S.; Lorandi, F.; Gorczyński, A.; Matyjaszewski, K. Toward Green Atom Transfer Radical Polymerization: Current Status and Future Challenges. Adv. Sci. 2022, 9, 2106076. [Google Scholar] [CrossRef]
- Treat, N.J.; Sprafke, H.; Kramer, J.W.; Clark, P.G.; Barton, B.E.; Read de Alaniz, J.; Fors, B.P.; Hawker, C.J. Metal-Free Atom Transfer Radical Polymerization. J. Am. Chem. Soc. 2014, 136, 16096–16101. [Google Scholar] [CrossRef]
- Pan, X.; Fang, C.; Fantin, M.; Malhotra, N.; So, W.Y.; Peteanu, L.A.; Isse, A.A.; Gennaro, A.; Liu, P.; Matyjaszewski, K. Mechanism of Photoinduced Metal-Free Atom Transfer Radical Polymerization: Experimental and Computational Studies. J. Am. Chem. Soc. 2016, 138, 2411–2425. [Google Scholar] [CrossRef]
- Theriot, J.; Lim, C.-H.; Yang, H.; Ryan, M.; Musgrave, C.; Miyake, G. Organocatalyzed Atom Transfer Radical Polymerization Driven by Visible Light. Science 2016, 352, 1082–1086. [Google Scholar] [CrossRef]
- Pearson, R.M.; Lim, C.-H.; McCarthy, B.G.; Musgrave, C.B.; Miyake, G.M. Organocatalyzed Atom Transfer Radical Polymerization Using N -Aryl Phenoxazines as Photoredox Catalysts. J. Am. Chem. Soc. 2016, 138, 11399–11407. [Google Scholar] [CrossRef]
- McCarthy, B.; Miyake, G.M. Organocatalyzed Atom Transfer Radical Polymerization Catalyzed by Core Modified N-Aryl Phenoxazines Performed under Air. ACS Macro Lett. 2018, 7, 1016–1021. [Google Scholar] [CrossRef] [PubMed]
- Christensen, J.A.; Phelan, B.T.; Chaudhuri, S.; Acharya, A.; Batista, V.S.; Wasielewski, M.R. Phenothiazine Radical Cation Excited States as Super-Oxidants for Energy-Demanding Reactions. J. Am. Chem. Soc. 2018, 140, 5290–5299. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, Q.; Tong, Y.; Ma, Y. 9,9-Dimethyl Dihydroacridine-Based Organic Photocatalyst for Atom Transfer Radical Polymerization from Modifying “Unstable” Electron Donor. Macromolecules 2020, 53, 7053–7062. [Google Scholar] [CrossRef]
- Sartor, S.M.; Lattke, Y.M.; McCarthy, B.G.; Miyake, G.M.; Damrauer, N.H. Effects of Naphthyl Connectivity on the Photophysics of Compact Organic Charge-Transfer Photoredox Catalysts. J. Phys. Chem. A 2019, 123, 4727–4736. [Google Scholar] [CrossRef] [PubMed]
- Buss, B.L.; Lim, C.-H.; Miyake, G.M. Dimethyl Dihydroacridines as Photocatalysts in Organocatalyzed Atom Transfer Radical Polymerization of Acrylate Monomers. Angew. Chem. Int. Ed. 2020, 59, 3209–3217. [Google Scholar] [CrossRef]
- Lattke, Y.M.; Corbin, D.A.; Sartor, S.M.; McCarthy, B.G.; Miyake, G.M.; Damrauer, N.H. Interrogation of O-ATRP Activation Conducted by Singlet and Triplet Excited States of Phenoxazine Photocatalysts. J. Phys. Chem. A 2021, 125, 3109–3121. [Google Scholar] [CrossRef]
- Swisher, N.A.; Corbin, D.A.; Miyake, G.M. Synthesis, Characterization, and Reactivity of N-Alkyl Phenoxazines in Organocatalyzed Atom Transfer Radical Polymerization. ACS Macro Lett. 2021, 10, 453–459. [Google Scholar] [CrossRef]
- Corbin, D.A.; Cremer, C.; Puffer, K.O.; Newell, B.S.; Patureau, F.W.; Miyake, G.M. Effects of the Chalcogenide Identity in N-Aryl Phenochalcogenazine Photoredox Catalysts. ChemCatChem 2022, 14, e202200485. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Shao, H.; Ma, J.; Liao, S. Chalcogenide-Doped Anthracenes as Organophotocatalysts for Metal-Free Atom Transfer Radical Polymerization. Macromol. Chem. Phys. 2023, 224, 2200382. [Google Scholar] [CrossRef]
- Ma, Q.; Song, J.; Zhang, X.; Jiang, Y.; Ji, L.; Liao, S. Metal-free Atom Transfer Radical Polymerization with ppm Catalyst Loading under Sunlight. Nat. Commun. 2021, 12, 429. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Zhang, X.; Jiang, Y.; Lin, J.; Graff, B.; Hu, S.; Lalevée, J.; Liao, S. Organocatalytic PET-RAFT Polymerization with a Low Ppm of Organic Photocatalyst under Visible Light. Polym. Chem. 2022, 13, 209–219. [Google Scholar] [CrossRef]
- Zhang, X.; Jiang, Y.; Ma, Q.; Hu, S.; Liao, S. Metal-Free Cation-ic Polymerization of Vinyl Ethers with Strict Temporal Control by Employing an Organophotocatalyst. J. Am. Chem. Soc. 2021, 143, 6357–6362. [Google Scholar] [CrossRef] [PubMed]
- Dadashi-Silab, S.; Pan, X.; Matyjaszewski, K. Phenyl Benzo[b]Phenothiazine as a Visible Light Photoredox Catalyst for Metal-Free Atom Transfer Radical Polymerization. Chem. A Eur. J. 2017, 23, 5972–5977. [Google Scholar] [CrossRef] [PubMed]
- Discekici, E.H.; Anastasaki, A.; Read de Alaniz, J.; Hawker, C.J. Evolution and Future Directions of Metal-Free Atom Transfer Radical Polymerization. Macromolecules 2018, 51, 7421–7434. [Google Scholar] [CrossRef]
- Peters, A.T.; Behesti, Y.S.S. Benzo[k, l]xanthene-3, 4-dicarboximides and Benzimidazoxanthenoisoquinolinones—Yellow and Orange Dyes for Synthetic-polymer Fibres. J. Soc. Dye. Colour. 1989, 105, 29–35. [Google Scholar] [CrossRef]
- Cremer, C.; Eltester, M.A.; Bourakhouadar, H.; Atodiresei, I.L.; Patureau, F.W. Dehydrogenative C–H Phenochalcogenazination. Org. Lett. 2021, 23, 3243–3247. [Google Scholar] [CrossRef]
- Zagranyarski, Y.; Skabeev, A.; Ma, Y.; Müllen, K.; Li, C. Facile Synthesis of Annulated Heterocyclic Benzo[Kl]Acridine Derivatives via One-Pot N–H/C–H Coupling. Org. Chem. Front. 2016, 3, 1520–1523. [Google Scholar] [CrossRef]
- Singh, V.K.; Yu, C.; Badgujar, S.; Kim, Y.; Kwon, Y.; Kim, D.; Lee, J.; Akhter, T.; Thangavel, G.; Park, L.S.; et al. Highly Efficient Organic Photocatalysts Discovered via a Computer-Aided-Design Strategy for Visible-Light-Driven Atom Transfer Radical Polymerization. Nat. Catal. 2018, 1, 794–804. [Google Scholar] [CrossRef]
- Wu, C.; Corrigan, N.; Lim, C.-H.; Liu, W.; Miyake, G.; Boyer, C. Rational Design of Photocatalysts for Controlled Polymerization: Effect of Structures on Photocatalytic Activities. Chem. Rev. 2022, 122, 5476–5518. [Google Scholar] [CrossRef] [PubMed]
- Winget, P.; Cramer, C.J.; Truhlar, D.G. Computation of Equilibrium Oxidation and Reduction Potentials for Reversible and Dissociative Electron-Transfer Reactions in Solution. Theor. Chem. Acc. 2004, 112, 217–227. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- He, H.; Zapol, P.; Curtiss, L.A. A Theoretical Study of CO2 Anions on Anatase (101) Surface. J. Phys. Chem. C 2010, 114, 21474–21481. [Google Scholar] [CrossRef]
- Roth, H.; Romero, N.; Nicewicz, D. Experimental and Calculated Electrochemical Potentials of Common Organic Molecules for Applications to Single-Electron Redox Chemistry. Synlett 2015, 27, 714–723. [Google Scholar]
- Lee, K.; Serdiuk, I.E.; Kwon, G.; Min, D.J.; Kang, K.; Park, S.Y.; Kwon, J.E. Phenoxazine as a High-Voltage p-Type Redox Center for Organic Battery Cathode Materials: Small Structural Reorganization for Faster Charging and Narrow Operating Voltage. Energy Environ. Sci. 2020, 13, 4142–4156. [Google Scholar] [CrossRef]
- Weiss, R.; VanOrman, Z.A.; Sullivan, C.M.; Nienhaus, L. A Sensitizer of Purpose: Generating Triplet Excitons with Semiconductor Nanocrystals. ACS Mater. Au 2022, 2, 641–654. [Google Scholar] [CrossRef]
- Malinge, A.; Kumar, S.; Chen, D.; Zysman-Colman, E.; Kéna-Cohen, S. Heavy Atom Effect in Halogenated mCP and Its Influence on the Efficiency of the Thermally Activated Delayed Fluorescence of Dopant Molecules. J. Phys. Chem. C 2024, 128, 1122–1130. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
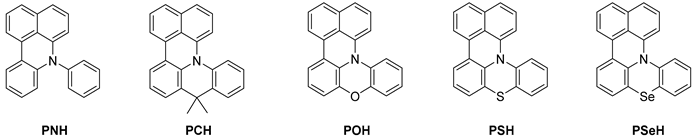 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| PC | λmax (nm) a | ε (M−1cm−1) | λem (nm) b | Τ (ns) c | Φ d | ES1,exp (eV) e | E1/2 (PC•+/PC) (V vs. SCE) | E0 (PC•+/1PC*) (V vs. SCE) e | E0calc (PC•+/3PC*) (V vs. SCE) f |
| PNH | 414 | 10,209 | 460 | 7.5 | 0.431 | 2.774 | 0.57 | −2.20 | −1.65 |
| PCH | 410 | 25,597 | 461 | 11.3 | 0.561 | 2.805 | 0.56 | −2.25 | −1.78 |
| POH | 435 | 13,405 | 470 | 10.8 | 0.505 | 2.805 | 0.74 | −2.06 | −1.51 |
| PSH | 412 | 13,111 | 474 | 6.0 | 0.368 | 2.774 | 0.77 | −2.00 | −1.43 |
| PSeH | 408 | 13,407 | 473 | 2.9 | 0.069 | 2.790 | 0.76 | −2.03 | −1.59/−2.04 g |
| Entry | Photocatalyst | Conv. b | Mn,theo (kg/mol) c | Mn,GPC (kg/mol) d | Đ d | I* e (%) |
|---|---|---|---|---|---|---|
| 1 | PNH | 64% | 6.8 | 11.6 | 1.49 | 58 |
| 2 | POH | 80% | 8.3 | 8.9 | 1.49 | 93 |
| 3 | PCH | 78% | 8.1 | 12.3 | 1.27 | 66 |
| 4 | PSH | 67% | 7.0 | 9.5 | 1.58 | 74 |
| 5 f | 45% | 4.8 | 8.1 | 1.26 | 59 | |
| 6 f | Nap-PTZ | 40% | 4.2 | 7.0 | 1.31 | 60 |
| 7 | PSeH | 85% | 8.7 | 8.0 | 1.24 | 111 |
| 8 g | 81% | 8.4 | 9.8 | 1.32 | 85 | |
| 9 | Nap-PSeZ | 85% | 8.8 | 11.3 | 1.61 | 61 |
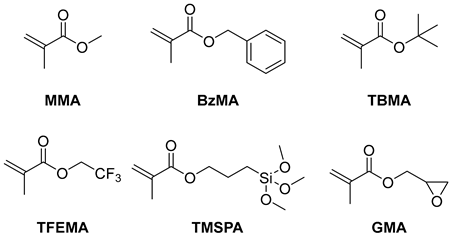 | ||||||
|---|---|---|---|---|---|---|
| Entry | Monomer | Solvent | Conv. | Mn,GPC (kg/mol) | Đ | I* (%) |
| 1 | BzMA | DMAc | 84% | 18.2 | 1.27 | 82 |
| 2 | TFEMA | DCM | 72% | 14.8 | 1.33 | 84 |
| 3 | TBMA | DCM | 85% | 17.8 | 1.51 | 70 |
| 4 b | TMSPA | anisole | 84% | 12.0 | 1.39 | 80 |
| 5 | GMA | DCM | 57% | 13.1 | 1.35 | 92 |
| 6 | MMA | DCM | 85% | 8.0 | 1.24 | 111 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shao, H.; Long, R.; Xu, H.; Sun, P.; Wang, G.; Li, Y.; Liao, S. The Development of Visible-Light Organic Photocatalysts for Atom Transfer Radical Polymerization via Conjugation Extension. Molecules 2024, 29, 2763. https://doi.org/10.3390/molecules29122763
Shao H, Long R, Xu H, Sun P, Wang G, Li Y, Liao S. The Development of Visible-Light Organic Photocatalysts for Atom Transfer Radical Polymerization via Conjugation Extension. Molecules. 2024; 29(12):2763. https://doi.org/10.3390/molecules29122763
Chicago/Turabian StyleShao, Hui, Runzhi Long, Hui Xu, Pan Sun, Guangrong Wang, Yuanming Li, and Saihu Liao. 2024. "The Development of Visible-Light Organic Photocatalysts for Atom Transfer Radical Polymerization via Conjugation Extension" Molecules 29, no. 12: 2763. https://doi.org/10.3390/molecules29122763
APA StyleShao, H., Long, R., Xu, H., Sun, P., Wang, G., Li, Y., & Liao, S. (2024). The Development of Visible-Light Organic Photocatalysts for Atom Transfer Radical Polymerization via Conjugation Extension. Molecules, 29(12), 2763. https://doi.org/10.3390/molecules29122763