Stereoselective Asymmetric Syntheses of Molecules with a 4,5-Dihydro-1H-[1,2,4]-Triazoline Core Possessing an Acetylated Carbohydrate Appendage: Crystal Structure, Spectroscopy, and Pharmacology

 , , , , and
, , , , and 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Chemical Synthesis
2.2. 1H NMR and 13C NMR Spectroscopic Characterization
2.3. Crystallographic Analysis of Derivative 8b
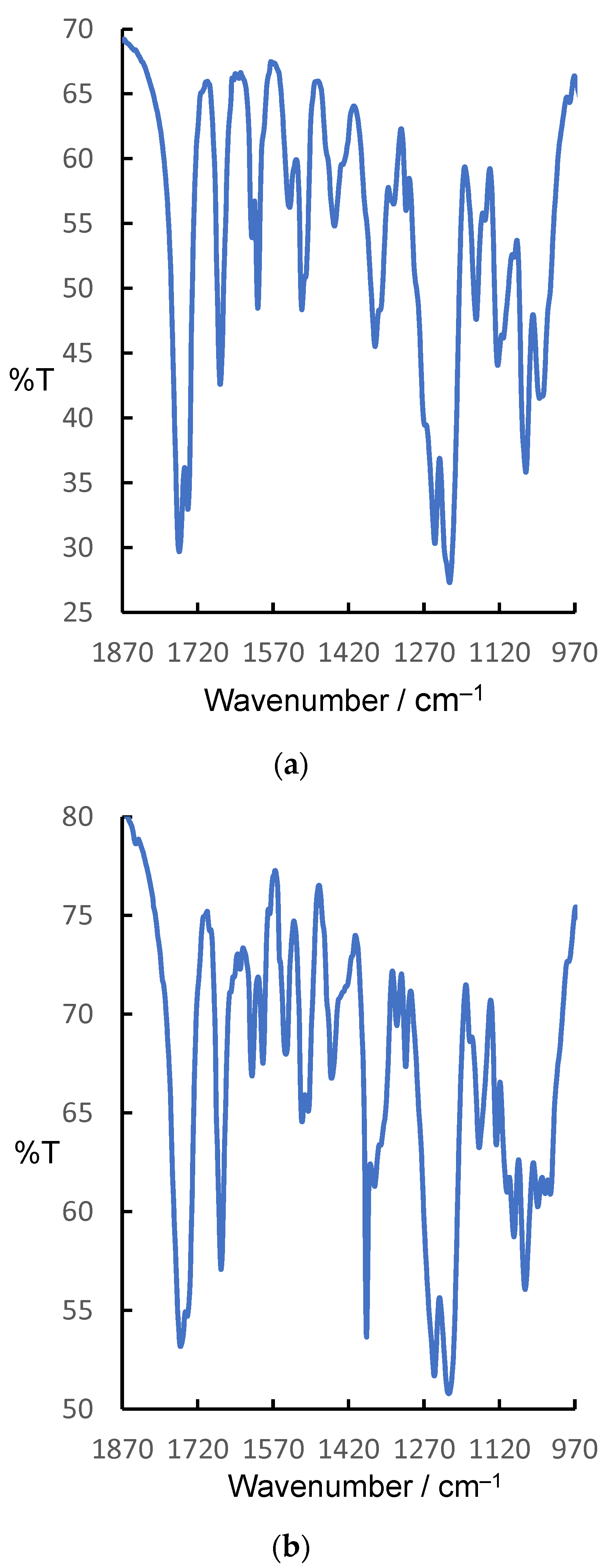
2.4. FTIR Spectroscopy
2.5. Biological Assay
2.5.1. Anti-Tumor Activity
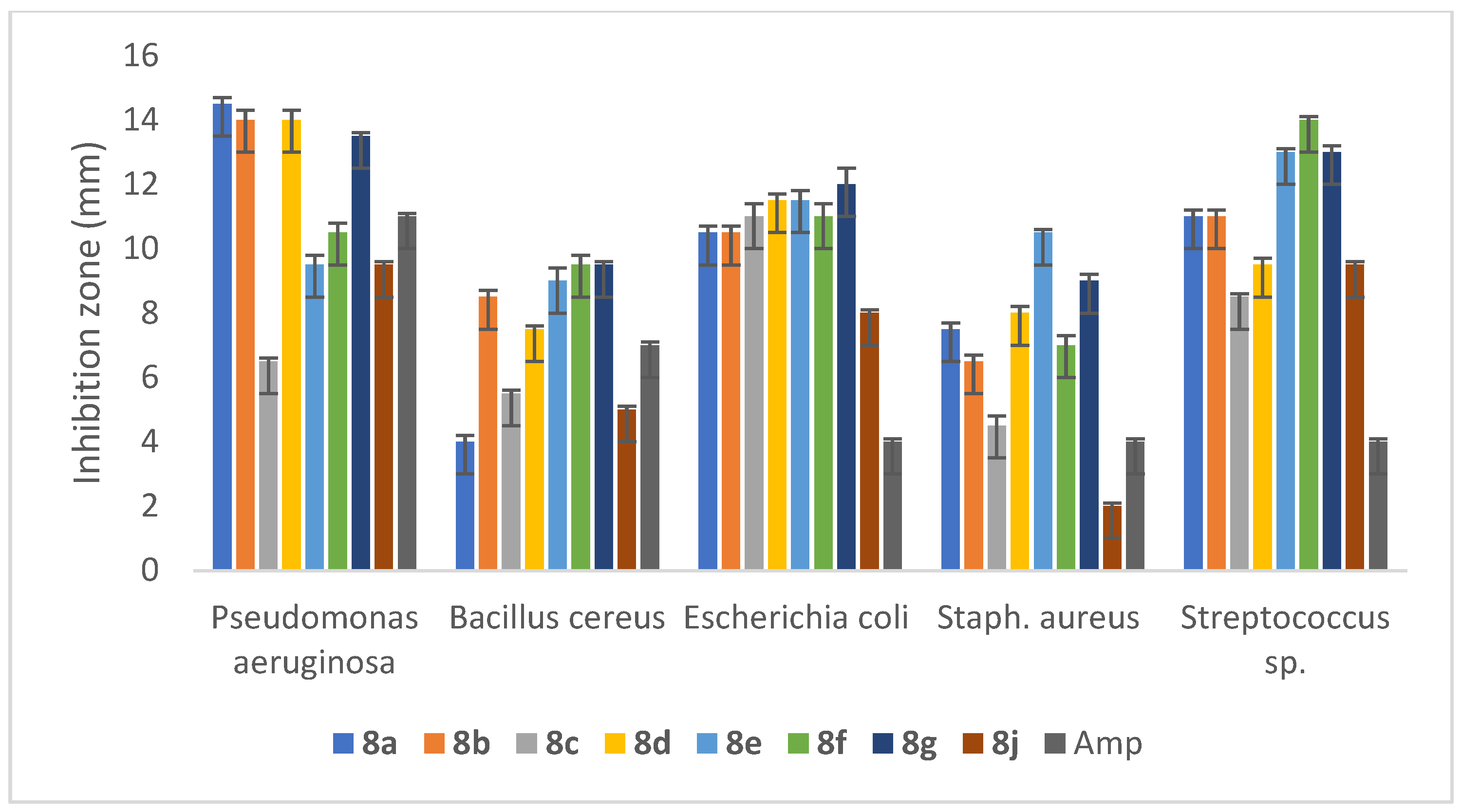
2.5.2. Anti-Bacterial Activity
2.5.3. Anti-Fungal Activity
3. Experimental Setup
3.1. General
3.2. Physical Techniques of Characterization
3.3. Single-Crystal X-ray Analysis
3.4. Anti-Tumor Activity
3.5. Anti-Bacterial Activity
3.6. Anti-Fungal Activity
3.7. General Synthesis of 4,5-Dihydro-1H-[1,2,4] Triazoline Derivatives
3.7.1. 2-(3-Acetyl-1-(phenyl)-5-((R)-4-methoxyphenyl)-1,2,4-triazolo-4-yl)-2-deoxy-1,3,4,6-tetraacetyl-β-ᴅ-glucose (8a)
3.7.2. 2-(3-Acetyl-1-(4-bromophenyl)-5-((S)-4-methoxyphenyl)-1,2,4-triazolo-4-yl)-2-deoxy-1,3,4,6-tetraacetyl-β-ᴅ-glucose (8b)
3.7.3. 2-(3-Acetyl-1-(4-bromophenyl)-5-((S)-4-methylphenyl)-1,2,4-triazolo-4-yl)-2-deoxy-1,3,4,6-tetraacetyl-β-ᴅ-glucose (8c)
3.7.4. 2-(3-Acetyl-1-(4-methylphenyl)-5-(4-methoxyphenyl)-1,2,4-triazolo-4-yl)-2-deoxy-1,3,4,6-tetraacetyl-β-ᴅ-glucose (8d)
3.7.5. 2-(3-Acetyl-1-(3-chlorophenyl)-5-(4-methoxyphenyl)-1,2,4-triazolo-4-yl)-2-deoxy-1,3,4,6-tetraacetyl-β-ᴅ-glucose (8e)
3.7.6. 2-(3-Acetyl-1-(4-bromophenyl)-5-((S)-4-chlorophenyl)-1,2,4-triazolo-4-yl)-2-deoxy-1,3,4,6-tetraacetyl-β-ᴅ-glucose (8f)
3.7.7. 2-(3-Acetyl-1-(p-tolyl)-5-((R)-4-chlorophenyl)-1,2,4-triazolo-4-yl)-2-deoxy-1,3,4,6-tetraacetyl-β-ᴅ-glucose (8g)
3.7.8. 2-(3-Acetyl-1-(naphthalen-1-yl)-5-(4-methylphenyl)-1,2,4-triazolo-4-yl)-2-deoxy-1,3,4,6-tetraacetyl-β-ᴅ-glucose (8h)
3.7.9. 2-(3-Acetyl-1-(phenyl)-5-((R)-p-tolyl)-1,2,4-triazolo-4-yl)-2-deoxy-1,3,4,6-tetraacetyl-β-ᴅ-glucose (8i)
3.7.10. 2-(3-Acetyl-1-(3,4-dichlorophenyl)-5-(3-methylphenyl)-1,2,4-triazolo-4-yl)-2-deoxy-1,3,4,6-tetraacetyl-β-ᴅ-glucose (8j)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fakhreddin, J. Stereochemically pure drugs: An overview. In Drug Stereochemistry: Analytical Methods and Pharmacology; Irving, W., Ed.; Marcel Dekker, Inc.: New York, NY, USA, 1993; pp. 375–382. [Google Scholar]
- Ötvös, S.B.; Kappe, C.O. Continuous flow asymmetric synthesis of chiral active pharmaceutical ingredients and their advanced intermediates. Green Chem. 2021, 23, 6117–6138. [Google Scholar] [CrossRef] [PubMed]
- Gawley, R.E.; Aubé, J. Principles of Asymmetric Synthesis; Elsevier: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Kunz, H.; Stoye, A. Carbohydrates as stereodifferentiating auxiliaries. In Heterocycles as Chiral Auxiliaries in Asymmetric Synthesis; Springer: Cham, Switzerland, 2017; pp. 1–72. [Google Scholar]
- Eswaran, S.; Adhikari, A.V.; Shetty, N.S. Synthesis and antimicrobial activities of novel quinoline derivatives carrying 1,2,4-triazole moiety. Eur. J. Med. Chem. 2009, 44, 4637–4647. [Google Scholar] [CrossRef] [PubMed]
- Saravolatz, L.D.; Johnson, L.B.; Kauffman, C.A. Voriconazole: A new triazole antifungal agent. Clin. Infect. Dis. 2003, 36, 630–637. [Google Scholar]
- Dheer, D.; Singh, V.; Shankar, R. Medicinal attributes of 1,2,3-triazoles: Current developments. Bioorg. Chem. 2017, 71, 30–54. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Zhang, S.; Gao, C.; Fan, J.; Zhao, F.; Lv, Z.-S.; Feng, L.-S. Isatin hybrids and their anti-tuberculosis activity. Chin. Chem. Lett. 2017, 28, 159–167. [Google Scholar] [CrossRef]
- Gilandoust, M.; Harsha, K.B.; Mohan, C.D.; Raquib, A.R.; Rangappa, S.; Pandey, V.; Lobie, P.E.; Rangappa, K.S. Synthesis, characterization and cytotoxicity studies of 1,2,3-triazoles and 1,2,4-triazolo [1,5-a] pyrimidines in human breast cancer cells. Bioorg. Med. Chem. Lett. 2018, 28, 2314–2319. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Chang, L.; Xu, Z.; Yan, X.-F.; Ding, C.; Zhao, F.; Wu, X.; Feng, L.-S. Recent advances of tetrazole derivatives as potential anti-tubercular and anti-malarial agents. Eur. J. Med. Chem. 2019, 163, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Christov, K.; Shilkaitis, A.; Green, A.; Mehta, R.G.; Grubbs, C.; Kelloff, G.; Lubet, R. Cellular responses of mammary carcinomas to aromatase inhibitors: Effects of vorozole. Breast Cancer Res. Treat. 2000, 60, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Roman, G.; Rahman, M.N.; Vukomanovic, D.; Jia, Z.; Nakatsu, K.; Szarek, W.A. Heme oxygenase inhibition by 2-Oxy-substituted 1-Azolyl-4-phenylbutanes: Effect of variation of the azole moiety. X-ray crystal structure of human heme oxygenase-1 in complex with 4-Phenyl-1-(1H-1,2,4-triazol-1-yl)-2-butanone. Chem. Biol. Drug Des. 2010, 75, 68–90. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, S.; Ba, Y.; Xu, Z. 1,2,4-Triazole-quinoline/quinolone hybrids as potential anti-bacterial agents. Eur. J. Med. Chem. 2019, 174, 1–8. [Google Scholar]
- Dai, J.; Tian, S.; Yang, X.; Liu, Z. Synthesis methods of 1,2,3-/1,2,4-triazoles: A review. Front. Chem. 2022, 10, 891484. [Google Scholar] [CrossRef] [PubMed]
- Bedekar, A.N.; Naik, A.N.; Pise, A.C. Schiff base derivatives of 2-amino-2-deoxy-1,3,4,6-tetra-O-acetyl-β-ᴅ-glucopyranose. Asian J. Chem. 2009, 21, 6661–6666. [Google Scholar]
- Eliseeva, A.I.; Nesterenko, O.O.; Slepukhin, P.A.; Benassi, E.; Belskaya, N.P. Synthesis and fluorescent behaviour of 2-Aryl-4,5-dihydro-1 H-1,2,4-triazoles. J. Org. Chem. 2017, 82, 86–100. [Google Scholar] [CrossRef]
- Karlsson, S.; Högberg, H.-E. Asymmetric 1,3-dipolar cycloadditions for the construction of enantiomerically pure heterocycles. A review. Org. Prep. Proced. Int. 2001, 33, 103–172. [Google Scholar] [CrossRef]
- El-Abadelah, M.M.; Hussein, A.Q.; Kamal, M.R.; Al-Adhami, K.H. Heterocycles from nitrile imines. I: 1,2,3,4-tetrahydro-1,2,4,5-Tetrazines. Heterocycles 1988, 27, 917–924. [Google Scholar] [CrossRef]
- Jian, F.; Bai, Z.; Xiao, H.; Li, K. 3-Benzyl-4-phenyl-1H-1,2,4-triazole-5 (4H)-thione. Acta Crystallogr. Sect. E Struct. Rep. Online 2005, 61, 557–558. [Google Scholar] [CrossRef]
- Kruszynski, R.; Trzesowska, A.; Przybycin, M.; Gil, K.; Dobosz, M. 2-(3-Methyl-5-thioxo-4,5-dihydro-1H-1,2,4-triazol-4-yl) acetic acid. Acta Crystallogr. Sect. E Struct. Rep. Online 2007, 63, 4378. [Google Scholar] [CrossRef]
- Wang, H.-Y.; Zhao, P.-S.; Li, R.-Q.; Zhou, S.-M. Synthesis, crystal structure and quantum chemical study on 3-phenylamino-4-phenyl-1,2,4-triazole-5-thione. Molecules 2009, 14, 608–620. [Google Scholar] [CrossRef] [PubMed]
- Saleem, R.S.Z.; Tepe, J.J. Synthesis of 1,2,4-triazolines and triazoles utilizing oxazolones. J. Org. Chem. 2010, 75, 4330–4332. [Google Scholar] [CrossRef]
- Karczmarzyk, Z.; Pitucha, M.; Wysocki, W.; Fruziński, A.; Olender, E. Ethyl 2-(3-methyl-5-sulfanylidene-4,5-dihydro-1H-1,2,4-triazol-4-yl) acetate. Acta Crystallogr. Sect. E Struct. Rep. Online 2012, 68, 3264–3265. [Google Scholar] [CrossRef]
- Boraei, A.T.; Soliman, S.M.; Haukka, M.; El Tamany, E.S.H.; Al-Majid, A.M.; Barakat, A. X-ray single crystal structure, tautomerism aspect, DFT, NBO, and Hirshfeld surface analysis of new Schiff bases based on 4-amino-5-indol-2-yl-1,2,4-triazole-3-thione hybrid. Crystals 2021, 11, 1041. [Google Scholar] [CrossRef]
- Altowyan, M.S.; Haukka, M.; Soliman, S.M.; Barakat, A.; Boraei, A.T.; Aboelmagd, A. Stereoselective synthesis of new 4-aryl-5-indolyl-1,2,4-triazole S-and N-β-galactosides: Characterizations, X-ray crystal structure and hirshfeld surface analysis. Crystals 2023, 13, 797. [Google Scholar] [CrossRef]
- Boraei, A.T.; Eltamany, E.H.; Haukka, M.; Soliman, S.M.; Barakat, A.; Sopaih, M. Synthesis and X-ray crystal structure analysis of substituted 1,2,4-triazolo [4′,3′:2,3] pyridazino [4,5-b] indole and its precursor. Crystals 2023, 13, 1036. [Google Scholar] [CrossRef]
- Liu, J.-Q.; Ji, L. Crystal structure of 4-(acetoxymethyl)-6-(3-acetyl-3-(4-fluorophenyl) thioureido) cyclohex-4-ene-1,2,3-triyl triacetate. Z. Für Krist. New Cryst. Struct. 2018, 234, 189–190. [Google Scholar] [CrossRef]
- Lopyrev, V.; Chipanina, N.; Rozinova, L.; Sarapulova, G.; Sultangareev, R.; Voronkov, M. Synthesis and absorption spectra of 3,5-diaryl-1,2,4-triazoles. Chem. Heterocycl. Compd. 1977, 13, 1346–1349. [Google Scholar] [CrossRef]
- Kahveci, B.; Ikizler, A.A. A study on some 4,5-dihydro-1H-1,2,4-triazol-5-one dervatives. Acta Pol. Pharm. 2000, 57, 119–122. [Google Scholar] [PubMed]
- Sharma, P.; Chen, A.; Wang, D.; Wang, G. Synthesis and self-assembling properties of peracetylated β-1-triazolyl alkyl ᴅ-glucosides and D-galactosides. Chemistry 2021, 3, 935–958. [Google Scholar] [CrossRef]
- Al-Ajely, M.; Al-Ajely, H.; Al-Naib, A. Synthesis of some substituted 1,2,3-triazole derivatives via 1,3-cycloaddition reaction of phenacylazides and some substituted propargyl compounds. Tikrit J. Pure Sci. 2008, 13, 100–106. [Google Scholar]
- Abdel-Jalil, R.J.; Arafeh, M.M.; Shongwe, M.S.; Maichle-Mößmer, C.; Kociok-Köhn, G.; Voelter, W. 1-(Naphthylamino)-1-(p-chlorophenylhydrazono)-2-propanone and 2-(p-tolyldiazenyl)-[1H]-3-methylbenzo [g] indole: Crystallographic and spectroscopic elucidation of the cyclisation of an arylamidrazone. J. Mol. Struct. 2015, 1079, 307–314. [Google Scholar] [CrossRef]
- Sert, Y.; El-Emam, A.A.; Al-Abdullah, E.S.; Al-Tamimi, A.-M.S.; Çırak, Ç.; Ucun, F. Use of vibrational spectroscopy to study 4-benzyl-3-(thiophen-2-yl)-4,5-dihydro-1H-1,2,4-triazole-5-thione: A combined theoretical and experimental approach. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2014, 126, 280–290. [Google Scholar] [CrossRef]
- El-Sakhawy, M.; Kamel, S.; Salama, A.; Tohamy, H.-A.S. Preparation and infrared study of cellulose based amphiphilic materials. Cellul. Chem. Technol. 2018, 52, 193–200. [Google Scholar]
- Rajasekaran, A.; Sivakumar, K.K.; Sureshkumar, K.; Manjushree, M. Design, synthesis, characterisation and in-vitro antimicrobial activity of some hybridized triazole scaffolds. Future J. Pharm. Sci. 2017, 3, 1–10. [Google Scholar] [CrossRef]
- Strzelecka, M.; Świątek, P. 1,2,4-triazoles as important antibacterial agents. Pharmaceuticals 2021, 14, 224. [Google Scholar] [CrossRef] [PubMed]
- Groll, A.; Piscitelli, S.; Walsh, T. Clinical pharmacology of systemic antifungal agents in clinical use, current investigational compounds and putative targets for antifungal drug development. Adv. Pharmacol. 1998, 44, 343–500. [Google Scholar] [PubMed]
- Ji, H.; Zhang, W.; Zhou, Y.; Zhang, M.; Zhu, J.; Song, Y.; Lü, J.; Zhu, J. A three-dimensional model of lanosterol 14α-demethylase of Candida albicans and its interaction with azole antifungals. J. Med. Chem. 2000, 43, 2493–2505. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-H.; Sheng, C.-Q.; Xu, X.-H.; Jiang, Y.-Y.; Zhang, W.-N.; He, C. Identification of Y118 amino acid residue in Candida albicans sterol 14α-demethylase associated with the enzyme activity and selective antifungal activity of azole analogues. Biol. Pharm. Bull. 2007, 30, 1246–1253. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Madison, V.; Chau, A.S.; Loebenberg, D.; Palermo, R.E.; McNicholas, P.M. Three-dimensional models of wild-type and mutated forms of cytochrome P450 14α-sterol demethylases from aspergillus fumigatus and candida albicans provide insights into posaconazole binding. Antimicrob. Agents Chemother. 2004, 48, 568–574. [Google Scholar] [CrossRef] [PubMed]
- Shalini, K.; Kumar, N.; Drabu, S.; Sharma, P.K. Advances in synthetic approach to and antifungal activity of triazoles. Beilstein J. Org. Chem. 2011, 7, 668–677. [Google Scholar] [CrossRef]
- Aggarwal, R.; Sumran, G. An insight on medicinal attributes of 1,2,4-triazoles. Eur. J. Med. Chem. 2020, 205, 112652. [Google Scholar] [CrossRef]
- Kazeminejad, Z.; Marzi, M.; Shiroudi, A.; Kouhpayeh, S.A.; Farjam, M.; Zarenezhad, E. Novel 1,2,4-triazoles as antifungal agents. BioMed Res. Int. 2022, 2022, 4584846. [Google Scholar] [CrossRef]
- Moghadam, E.S.; Mireskandari, K.; Abdel-Jalil, R.; Amini, M. An approach to pharmacological targets of pyrrole family from medicinal chemistry viewpoint. Mini Rev. Med. Chem. 2022, 22, 2486–2561. [Google Scholar] [PubMed]
- Stoe, C. X–AREA: Program for the Acquisition and Analysis of Data, Version 1.30; Stoe & Cie GmbH: Darmatadt, Germany, 2005. [Google Scholar]
- Stoe, C. X–RED: Program for Data Reduction and Absorption Correction, Version 1.28 b; Stoe & Cie GmbH: Darmatadt, Germany, 2005. [Google Scholar]
- Sheldrick, G. SHELXL-97. In Program for Crystal Structure Refinement; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Farrugia, L.J. WinGX suite for small-molecule single-crystal crystallography. J. Appl. Crystallogr. 1999, 32, 837–838. [Google Scholar] [CrossRef]
- Spek, A.L. PLATON SQUEEZE: A tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Holbeck, S.L.; Camalier, R.; Crowell, J.A.; Govindharajulu, J.P.; Hollingshead, M.; Anderson, L.W.; Polley, E.; Rubinstein, L.; Srivastava, A.; Wilsker, D. The National Cancer Institute ALMANAC: A comprehensive screening resource for the detection of anticancer drug pairs with enhanced therapeutic activity NCI ALMANAC of approved cancer drug combinations. Cancer Res. 2017, 77, 3564–3576. [Google Scholar] [CrossRef]
- Uenver, Y.; Düğdü, E.; Sancak, K.; Er, M.; Karaoğlu, S.A. Synthesis and antimicrobial and antitumor activity of some new [1,2,4] triazole-5-one derivatives. Turk. J. Chem. 2009, 33, 135–147. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Empirical formula | C31H34BrN3O11 |
| Molar mass (g/mol) | 704.52 |
| Temperature (K) | 298.15 |
| Crystal system | monoclinic |
| Space group | P21 |
| a (Å) | 11.850(2) |
| b (Å) | 10.170(2) |
| c (Å) | 16.090(3) |
| α (°) | 90 |
| β (°) | 104.40(3) |
| γ (°) | 90 |
| Volume (Å3) | 1878.2(7) |
| Z | 2 |
| ρcalc (g/cm3) | 1.246 |
| μ (mm–1) | 1.149 |
| F (000) | 728.0 |
| Crystal size (mm) | 0.10 × 0.10 × 0.05 |
| Radiation | Mo-Kα (λ = 0.71073 Å) |
| 2Θ range for data collection (°) | 4.904–49.414 |
| Index ranges | −13 ≤ h ≤ 13, −11 ≤ k ≤ 11, −18 ≤ l ≤ 18 |
| Reflections collected | 12,658 |
| Independent reflections | 6077 [Rint = 0.0798, Rsigma = 0.0869] |
| Data/restraints/parameters | 6077/355/418 |
| Goodness of fit on F2 | 1.015 |
| Final R indexes [I ≥ 2σ (I)] | R1 = 0.0722, wR2 = 0.1646 |
| Final R indexes [all data] | R1 = 0.1433, wR2 = 0.2008 |
| Largest diff. peak/hole (e Å–3) | 0.24/−0.35 |
| D–H···A | D–H | H···A | D–A | D–H···A | Symmetry Codes |
|---|---|---|---|---|---|
| C(3)–H(3)···O(10) | 0.98 | 2.48 | 3.163(12) | 126.7 | |
| C(5)–H(5)···O(10) | 0.98 | 2.30 | 3.046(12) | 132.3 | |
| C(8)–H(8C)···O(10) | 0.96 | 2.66 | 3.48(2) | 143.1 | −x + 1, y + ½, −z + 1 |
| C(10)–H(10B)···O(9) | 0.96 | 2.59 | 3.404(14) | 142.2 | x + 1, y, z |
| C(20)–H(20)···O(3) | 0.93 | 2.59 | 3.481(16) | 160.3 | −x + 1, y + ½, −z + 1 |
| C(12)–H(12B)···Br(1) | 0.96 | 2.94 | 3.89(2) | 170.0 | −x + 1, y−½, −z |
| C(29)–Br(1) | 1.887(13) |
| C(16)–O(10) | 1.189(13) |
| C(15)–N(1) | 1.415(13) |
| C(15)–N(2) | 1.299(12) |
| N(2)–N(3) | 1.361(11) |
| C(18)–N(1) | 1.490(12) |
| C(18)–N(3) | 1.505(13) |
| C(18)–C(19) | 1.485(14) |
| C(7)–O(2) | 1.340(14) |
| C(7)–O(3) | 1.165(17) |
| C(9)–O(4) | 1.358(11) |
| C(9)–O(5) | 1.174(13) |
| C(11)–O(6) | 1.343(14) |
| C(11)–O(7) | 1.186(15) |
| C(13)–O(8) | 1.341(13) |
| C(13)–O(9) | 1.217(14) |
| Compound ID | NCI Code | Mean G% |
|---|---|---|
| 8a | 835808 | 92.73 |
| 8b | 835811 | 70.77 |
| 8c | 835812 | 58.86 |
| 8d | 835813 | 78.32 |
| 8e | 835815 | 73.52 |
| 8f | 835816 | 61.24 |
| Cancer Panel Type | Cancer Cell Type | G% | |
|---|---|---|---|
| 8c NCI Code 835812 | 8f NCI Code 835816 | ||
| Leukemia | HL-60(TB) | 14.42 | 34.14 |
| RPMI-8226 | 35.38 | 38.03 | |
| MOLT-4 | 41.79 | 44.34 | |
| CNS Cancer | SF-268 | 58.40 | 58.57 |
| SF-295 | 29.43 | 36.96 | |
| Renal Cancer | 786-0 | 68.84 | 58.46 |
| A498 | 59.58 | 131.61 | |
| ACHN | 52.23 | 50.09 | |
| UO-31 | 32.19 | 27.59 | |
| Breast Cancer | MCF7 | 43.90 | 34.59 |
| T-47D | 26.78 | 34.57 | |
| MDA-MB-468 | 49.38 | 26.20 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al Maqbali, A.S.; Al Rasbi, N.K.; Zoghaib, W.M.; Sivakumar, N.; Robertson, C.C.; Shongwe, M.S.; Grzegorzek, N.; Abdel-Jalil, R.J. Stereoselective Asymmetric Syntheses of Molecules with a 4,5-Dihydro-1H-[1,2,4]-Triazoline Core Possessing an Acetylated Carbohydrate Appendage: Crystal Structure, Spectroscopy, and Pharmacology. Molecules 2024, 29, 2839. https://doi.org/10.3390/molecules29122839
Al Maqbali AS, Al Rasbi NK, Zoghaib WM, Sivakumar N, Robertson CC, Shongwe MS, Grzegorzek N, Abdel-Jalil RJ. Stereoselective Asymmetric Syntheses of Molecules with a 4,5-Dihydro-1H-[1,2,4]-Triazoline Core Possessing an Acetylated Carbohydrate Appendage: Crystal Structure, Spectroscopy, and Pharmacology. Molecules. 2024; 29(12):2839. https://doi.org/10.3390/molecules29122839
Chicago/Turabian StyleAl Maqbali, Anwaar S., Nawal K. Al Rasbi, Wajdi M. Zoghaib, Nallusamy Sivakumar, Craig C. Robertson, Musa S. Shongwe, Norbert Grzegorzek, and Raid J. Abdel-Jalil. 2024. "Stereoselective Asymmetric Syntheses of Molecules with a 4,5-Dihydro-1H-[1,2,4]-Triazoline Core Possessing an Acetylated Carbohydrate Appendage: Crystal Structure, Spectroscopy, and Pharmacology" Molecules 29, no. 12: 2839. https://doi.org/10.3390/molecules29122839