
Structural, Biophysical, and Computational Studies of a Murine Light Chain Dimer
 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
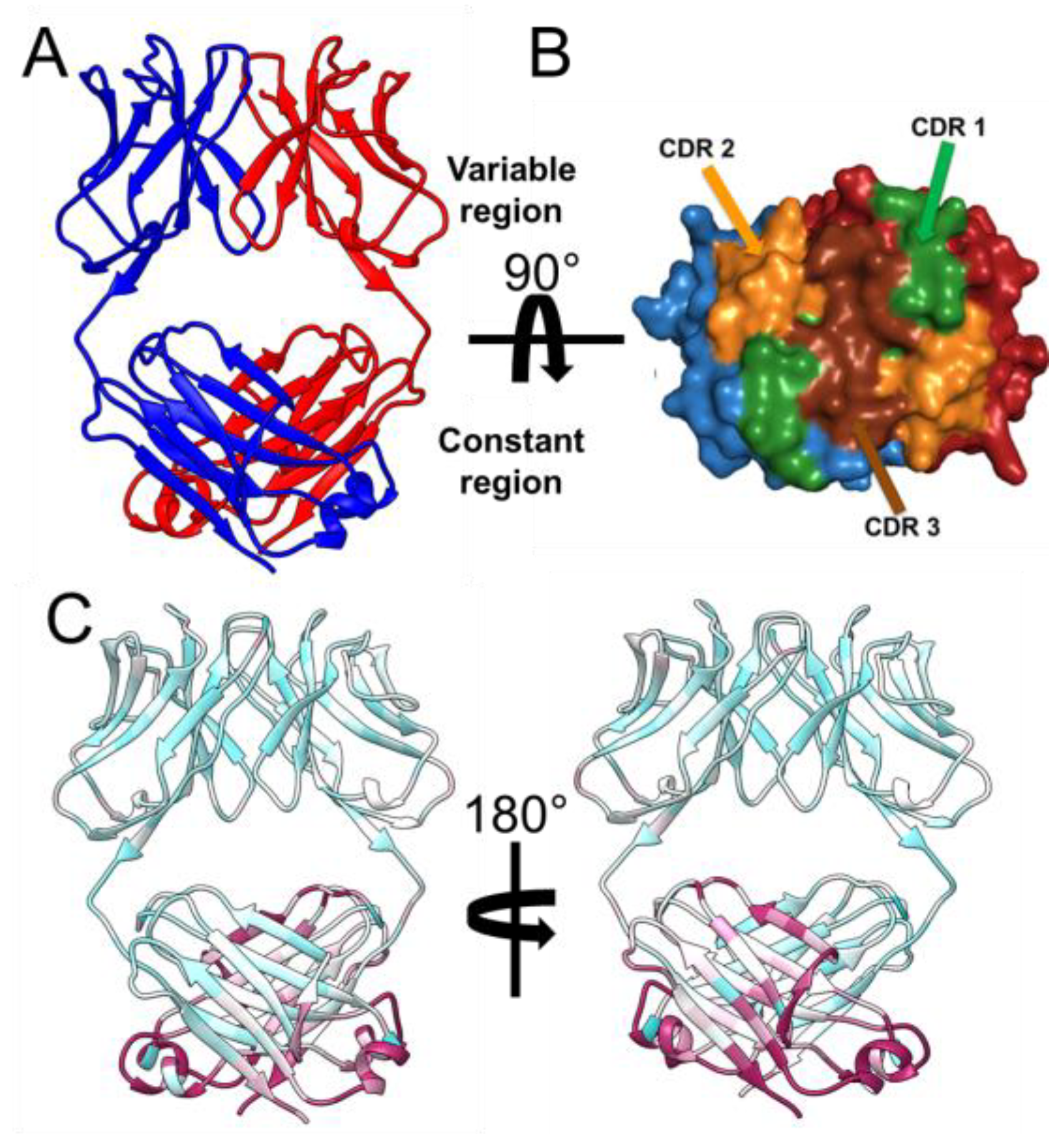
2.1. Crystallization, Structural, and Biochemical Analysis of 6A8 LCD
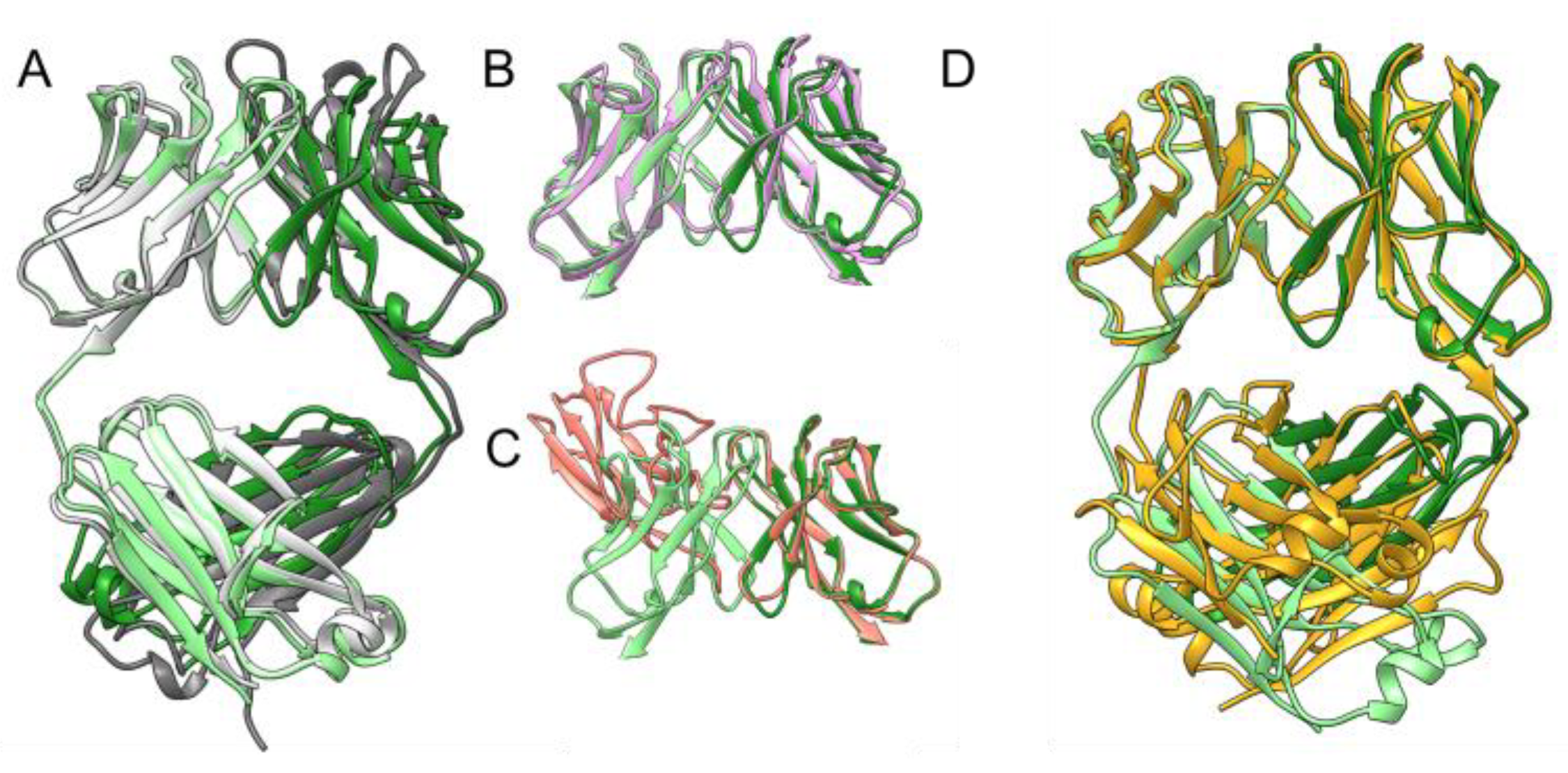
2.2. Comparision of the Quaternary Structure of 6A8 LCD with a 6A8 Fab AlphaFold2 Model and Homologous Light Chain Crystal Structures
2.3. Investigating the Specificity and Relative Binding Affinity Properties of 6A8 Light Chain versus the Full 6A8 IgG
2.4. Computational Studies to Explore the Difference in Specificity between 6A8 scFv and 6A8 LCD towards Der f 1
3. Discussion
4. Materials and Methods
4.1. Expression and Purification of 6A8 Fab and Light Chain
4.2. Crystallization, Data Collection, and Structure Determination of 6A8 LCD
4.3. Thermal Stability via DSF
4.4. ELISA Experiments: Specificity and Binding Affinities of mAb 6A8 LCD and Full IgG Ab
4.5. Computational Studies on Protein–Protein Interactions
4.6. Additional Computational Tools
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Kapingidza, A.B.; Kowal, K.; Chruszcz, M. Antigen-Antibody Complexes. Subcell. Biochem. 2020, 94, 465–497. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, H.W., Jr.; Cavacini, L. Structure and function of immunoglobulins. J. Allergy Clin. Immunol. 2010, 125, S41–S52. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, B.; Livneh, A.; Sela, B.-A. Immunoglobulin free light chain dimers in human diseases. Sci. World J. 2011, 11, 726–735. [Google Scholar] [CrossRef] [PubMed]
- Katzmann, J.A.; Clark, R.J.; Abraham, R.S.; Bryant, S.; Lymp, J.F.; Bradwell, A.R.; Kyle, R.A. Serum reference intervals and diagnostic ranges for free κ and free λ immunoglobulin light chains: Relative sensitivity for detection of monoclonal light chains. Clin. Chem. 2002, 48, 1437–1444. [Google Scholar] [CrossRef] [PubMed]
- Abe, M.; Goto, T.; Kosaka, M.; Wolfenbarger, D.; Weiss, D.; Solomon, A. Differences in kappa to lambda (κ: λ) ratios of serum and urinary free light chains. Clin. Exp. Immunol. 1998, 111, 457. [Google Scholar] [CrossRef] [PubMed]
- Berggård, I.; Peterson, P. Polymeric forms of free normal κ and λ chains of human immunoglobulin. J. Biol. Chem. 1969, 244, 4299–4307. [Google Scholar] [CrossRef] [PubMed]
- Powe, D.G.; Kormelink, T.G.; Sisson, M.; Blokhuis, B.J.; Kramer, M.F.; Jones, N.S.; Redegeld, F.A. Evidence for the involvement of free light chain immunoglobulins in allergic and nonallergic rhinitis. J. Allergy Clin. Immunol. 2010, 125, 139–145.e133. [Google Scholar] [CrossRef] [PubMed]
- Redegeld, F.A.; Nijkamp, F.P. Immunoglobulin free light chains and mast cells: Pivotal role in T-cell-mediated immune reactions? Trends Immunol. 2003, 24, 181–185. [Google Scholar] [CrossRef]
- Redegeld, F.A.; van der Heijden, M.W.; Kool, M.; Heijdra, B.M.; Garssen, J.; Kraneveld, A.D.; Van Loveren, H.; Roholl, P.; Saito, T.; Verbeek, J.S. Immunoglobulin-free light chains elicit immediate hypersensitivity-like responses. Nat. Med. 2002, 8, 694. [Google Scholar] [CrossRef]
- Baden, E.M.; Owen, B.A.; Peterson, F.C.; Volkman, B.F.; Ramirez-Alvarado, M.; Thompson, J.R. Altered dimer interface decreases stability in an amyloidogenic protein. J. Biol. Chem. 2008, 283, 15853–15860. [Google Scholar] [CrossRef]
- Gupta, N.; Sharma, A. Emerging biomarkers in Multiple Myeloma: A review. Clin. Chim. Acta 2020, 503, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Appella, E.; McIntire, K.R.; Perham, R.N. Lambda Bence Jones proteins of the mouse: Chemical and immunological characterization. J. Mol. Biol. 1967, 27, 391–394. [Google Scholar] [CrossRef] [PubMed]
- Fahey, J.L.; Potter, M. Bence Jones proteinuria associated with a transplantable mouse plasma-cell neoplasm. Nature 1959, 184 (Suppl. S9), 654–655. [Google Scholar] [CrossRef] [PubMed]
- Khatri, K.; Richardson, C.M.; Glesner, J.; Kapingidza, A.B.; Mueller, G.A.; Zhang, J.; Dolamore, C.; Vailes, L.D.; Wünschmann, S.; Peebles, R.S.; et al. Human IgE monoclonal antibody recognition of mite allergen Der p 2 defines structural basis of an epitope for IgE cross-linking and anaphylaxis. PNAS Nexus 2022, 1, pgac054. [Google Scholar] [CrossRef] [PubMed]
- Chruszcz, M.; Pomés, A.; Glesner, J.; Vailes, L.D.; Osinski, T.; Porebski, P.J.; Majorek, K.A.; Heymann, P.W.; Platts-Mills, T.A.; Minor, W.; et al. Molecular determinants for antibody binding on group 1 house dust mite allergens. J. Biol. Chem. 2012, 287, 7388–7398. [Google Scholar] [CrossRef] [PubMed]
- Osinski, T.; Pomés, A.; Majorek, K.A.; Glesner, J.; Offermann, L.R.; Vailes, L.D.; Chapman, M.D.; Minor, W.; Chruszcz, M. Structural Analysis of Der p 1-Antibody Complexes and Comparison with Complexes of Proteins or Peptides with Monoclonal Antibodies. J. Immunol. 2015, 195, 307–316. [Google Scholar] [CrossRef]
- Glesner, J.; Kapingidza, A.B.; Godzwon, M.; Offermann, L.R.; Mueller, G.A.; DeRose, E.F.; Wright, P.; Richardson, C.M.; Woodfolk, J.A.; Vailes, L.D.; et al. A Human IgE Antibody Binding Site on Der p 2 for the Design of a Recombinant Allergen for Immunotherapy. J. Immunol. 2019, 203, 2545–2556. [Google Scholar] [CrossRef]
- Glesner, J.; Vailes, L.D.; Schlachter, C.; Mank, N.; Minor, W.; Osinski, T.; Chruszcz, M.; Chapman, M.D.; Pomés, A. Antigenic Determinants of Der p 1: Specificity and Cross-Reactivity Associated with IgE Antibody Recognition. J. Immunol. 2017, 198, 1334–1344. [Google Scholar] [CrossRef]
- Chruszcz, M.; Chapman, M.D.; Vailes, L.D.; Stura, E.A.; Saint-Remy, J.M.; Minor, W.; Pomés, A. Crystal Structures of Mite Allergens Der f 1 and Der p 1 Reveal Differences in Surface-Exposed Residues that May Influence Antibody Binding. J. Mol. Biol. 2009, 386, 520–530. [Google Scholar] [CrossRef]
- Chapman, M.; Heymann, P.; Platts-Mills, T. Epitope mapping of two major inhalant allergens, Der p I and Der f I, from mites of the genus Dermatophagoides. J. Immunol. 1987, 139, 1479–1484. [Google Scholar] [CrossRef]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef]
- Kunik, V.; Ashkenazi, S.; Ofran, Y. Paratome: An online tool for systematic identification of antigen-binding regions in antibodies based on sequence or structure. Nucleic Acids Res. 2012, 40, W521–W524. [Google Scholar] [CrossRef]
- Blancas-Mejia, L.M.; Misra, P.; Dick, C.J.; Cooper, S.A.; Redhage, K.R.; Bergman, M.R.; Jordan, T.L.; Maar, K.; Ramirez-Alvarado, M. Immunoglobulin light chain amyloid aggregation. Chem. Commun. 2018, 54, 10664–10674. [Google Scholar] [CrossRef] [PubMed]
- Buxbaum, J. Mechanisms of disease: Monoclonal immunoglobulin deposition. Amyloidosis, light chain deposition disease, and light and heavy chain deposition disease. Hematol. Oncol. Clin. N. Am. 1992, 6, 323–346. [Google Scholar] [CrossRef]
- Bodi, K.; Prokaeva, T.; Spencer, B.; Eberhard, M.; Connors, L.H.; Seldin, D.C. AL-Base: A visual platform analysis tool for the study of amyloidogenic immunoglobulin light chain sequences. Amyloid 2009, 16, 1–8. [Google Scholar] [CrossRef]
- Olsen, K.E.; Sletten, K.; Westermark, P. Fragments of the constant region of immunoglobulin light chains are constituents of AL-amyloid proteins. Biochem. Biophys. Res. Commun. 1998, 251, 642–647. [Google Scholar] [CrossRef]
- Brumshtein, B.; Esswein, S.R.; Landau, M.; Ryan, C.M.; Whitelegge, J.P.; Phillips, M.L.; Cascio, D.; Sawaya, M.R.; Eisenberg, D.S. Formation of amyloid fibers by monomeric light chain variable domains. J. Biol. Chem. 2014, 289, 27513–27525. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Nymalm, Y.; Kravchuk, Z.; Salminen, T.; Chumanevich, A.A.; Dubnovitsky, A.P.; Kankare, J.; Pentikäinen, O.; Lehtonen, J.; Arosio, P.; Martsev, S.; et al. Antiferritin VL homodimer binds human spleen ferritin with high specificity. J. Struct. Biol. 2002, 138, 171–186. [Google Scholar] [CrossRef]
- Li, Z.; Jaroszewski, L.; Iyer, M.; Sedova, M.; Godzik, A. FATCAT 2.0: Towards a better understanding of the structural diversity of proteins. Nucleic Acids Res. 2020, 48, W60–W64. [Google Scholar] [CrossRef]
- Roussel, A.; Spinelli, S.; Déret, S.; Navaza, J.; Aucouturier, P.; Cambillau, C. The structure of an entire noncovalent immunoglobulin kappa light-chain dimer (Bence-Jones protein) reveals a weak and unusual constant domains association. Eur. J. Biochem. 1999, 260, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.L.; Davis, M.M. Diversity in the CDR3 region of V(H) is sufficient for most antibody specificities. Immunity 2000, 13, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Gavish, M.; Dwek, R.A.; Givol, D. Comparison of the fine specificity of anti-dinitrophenyl-combining site composed of either VL dimer or VL and VH of protein 315. Biochemistry 1977, 16, 3154–3159. [Google Scholar] [CrossRef] [PubMed]
- Dunbar, J.; Krawczyk, K.; Leem, J.; Marks, C.; Nowak, J.; Regep, C.; Georges, G.; Kelm, S.; Popovic, B.; Deane, C.M. SAbPred: A structure-based antibody prediction server. Nucleic Acids Res. 2016, 44, W474–W478. [Google Scholar] [CrossRef]
- Krawczyk, K.; Baker, T.; Shi, J.; Deane, C.M. Antibody i-Patch prediction of the antibody binding site improves rigid local antibody–antigen docking. Protein Eng. Des. Sel. 2013, 26, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Kozakov, D.; Beglov, D.; Bohnuud, T.; Mottarella, S.E.; Xia, B.; Hall, D.R.; Vajda, S. How good is automated protein docking? Proteins 2013, 81, 2159–2166. [Google Scholar] [CrossRef] [PubMed]
- Vajda, S.; Yueh, C.; Beglov, D.; Bohnuud, T.; Mottarella, S.E.; Xia, B.; Hall, D.R.; Kozakov, D. New additions to the ClusPro server motivated by CAPRI. Proteins 2017, 85, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Brenke, R.; Hall, D.R.; Chuang, G.Y.; Comeau, S.R.; Bohnuud, T.; Beglov, D.; Schueler-Furman, O.; Vajda, S.; Kozakov, D. Application of asymmetric statistical potentials to antibody-protein docking. Bioinformatics 2012, 28, 2608–2614. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, D.; Gray, J.J. Shape complementarity and hydrogen bond preferences in protein-protein interfaces: Implications for antibody modeling and protein-protein docking. Bioinformatics 2016, 32, 2451–2456. [Google Scholar] [CrossRef]
- Peng, H.P.; Lee, K.H.; Jian, J.W.; Yang, A.S. Origins of specificity and affinity in antibody-protein interactions. Proc. Natl. Acad. Sci. USA 2014, 111, E2656–E2665. [Google Scholar] [CrossRef]
- Marshall, J.S.; Warrington, R.; Watson, W.; Kim, H.L. An introduction to immunology and immunopathology. Allergy Asthma Clin. Immunol. 2018, 14, 49. [Google Scholar] [CrossRef] [PubMed]
- Lipman, N.S.; Jackson, L.R.; Trudel, L.J.; Weis-Garcia, F. Monoclonal versus polyclonal antibodies: Distinguishing characteristics, applications, and information resources. ILAR J. 2005, 46, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Ayoubi, R.; Ryan, J.; Biddle, M.S.; Alshafie, W.; Fotouhi, M.; Bolivar, S.G.; Moleon, V.R.; Eckmann, P.; Worrall, D.; McDowell, I.; et al. Scaling of an antibody validation procedure enables quantification of antibody performance in major research applications. eLife 2023, 12, RP91645. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, A.; Plückthun, A. Reproducibility: Standardize antibodies used in research. Nature 2015, 518, 27–29. [Google Scholar] [CrossRef] [PubMed]
- Voskuil, J.L.A.; Bandrowski, A.; Begley, C.G.; Bradbury, A.R.M.; Chalmers, A.D.; Gomes, A.V.; Hardcastle, T.; Lund-Johansen, F.; Plückthun, A.; Roncador, G.; et al. The Antibody Society’s antibody validation webinar series. MAbs 2020, 12, 1794421. [Google Scholar] [CrossRef] [PubMed]
- Baker, M. When antibodies mislead: The quest for validation. Nature 2020, 585, 313–314. [Google Scholar] [CrossRef] [PubMed]
- Oberti, L.; Rognoni, P.; Barbiroli, A.; Lavatelli, F.; Russo, R.; Maritan, M.; Palladini, G.; Bolognesi, M.; Merlini, G.; Ricagno, S. Concurrent structural and biophysical traits link with immunoglobulin light chains amyloid propensity. Sci. Rep. 2017, 7, 16809. [Google Scholar] [CrossRef] [PubMed]
- Hurle, M.R.; Helms, L.R.; Li, L.; Chan, W.; Wetzel, R. A role for destabilizing amino acid replacements in light-chain amyloidosis. Proc. Natl. Acad. Sci. USA 1994, 91, 5446–5450. [Google Scholar] [CrossRef] [PubMed]
- Ausserwöger, H.; Schneider, M.M.; Herling, T.W.; Arosio, P.; Invernizzi, G.; Knowles, T.P.J.; Lorenzen, N. Non-specificity as the sticky problem in therapeutic antibody development. Nat. Rev. Chem. 2022, 6, 844–861. [Google Scholar] [CrossRef]
- Minor, W.; Cymborowski, M.; Otwinowski, Z.; Chruszcz, M. HKL-3000: The integration of data reduction and structure solution–from diffraction images to an initial model in minutes. Acta Crystallogr. Sect. D Biol. Crystallogr. 2006, 62, 859–866. [Google Scholar] [CrossRef]
- Vagin, A.; Teplyakov, A. MOLREP: An automated program for molecular replacement. J. Appl. Crystallogr. 1997, 30, 1022–1025. [Google Scholar] [CrossRef]
- Cowtan, K. The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr. Sect. D Biol. Crystallogr. 2006, 62, 1002–1011. [Google Scholar] [CrossRef]
- Langer, G.; Cohen, S.X.; Lamzin, V.S.; Perrakis, A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 2008, 3, 1171. [Google Scholar] [CrossRef]
- Krissinel, E.; Lebedev, A.A.; Uski, V.; Ballard, C.B.; Keegan, R.M.; Kovalevskiy, O.; Nicholls, R.A.; Pannu, N.S.; Skubák, P.; Berrisford, J.; et al. CCP4 Cloud for structure determination and project management in macromolecular crystallography. Acta Crystallogr. D Struct. Biol. 2022, 78 Pt 9, 1079–1089. [Google Scholar] [CrossRef]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed]
- Davis, I.W.; Leaver-Fay, A.; Chen, V.B.; Block, J.N.; Kapral, G.J.; Wang, X.; Murray, L.W.; Arendall, W.B., III; Snoeyink, J.; Richardson, J.S. MolProbity: All-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007, 35 (Suppl. S2), W375–W383. [Google Scholar] [CrossRef]
- Painter, J.; Merritt, E.A. TLSMD web server for the generation of multi-group TLS models. J. Appl. Crystallogr. 2006, 39, 109–111. [Google Scholar] [CrossRef]
- Daneshian, L.; Renggli, I.; Hanaway, R.; Offermann, L.R.; Schlachter, C.R.; Hernandez Arriaza, R.; Henry, S.; Prakash, R.; Wybouw, N.; Dermauw, W.; et al. Structural and functional characterization of β-cyanoalanine synthase from Tetranychus urticae. Insect Biochem. Mol. Biol. 2022, 142, 103722. [Google Scholar] [CrossRef]
- Krawczyk, K.; Liu, X.; Baker, T.; Shi, J.; Deane, C.M. Improving B-cell epitope prediction and its application to global antibody-antigen docking. Bioinformatics 2014, 30, 2288–2294. [Google Scholar] [CrossRef]
- PyMOL. The PyMOL Molecular Graphics System, Version 2.0; Schrödinger, LLC: New York, NY, USA, 2016. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arriaza, R.H.; Kapingidza, A.B.; Dolamore, C.; Khatri, K.; O’Malley, A.; Glesner, J.; Wuenschmann, S.; Hyduke, N.P.; Easley, W.; Chhiv, C.; et al. Structural, Biophysical, and Computational Studies of a Murine Light Chain Dimer. Molecules 2024, 29, 2885. https://doi.org/10.3390/molecules29122885
Arriaza RH, Kapingidza AB, Dolamore C, Khatri K, O’Malley A, Glesner J, Wuenschmann S, Hyduke NP, Easley W, Chhiv C, et al. Structural, Biophysical, and Computational Studies of a Murine Light Chain Dimer. Molecules. 2024; 29(12):2885. https://doi.org/10.3390/molecules29122885
Chicago/Turabian StyleArriaza, Ricardo H., A. Brenda Kapingidza, Coleman Dolamore, Kriti Khatri, Andrea O’Malley, Jill Glesner, Sabina Wuenschmann, Noah P. Hyduke, William Easley, Charline Chhiv, and et al. 2024. "Structural, Biophysical, and Computational Studies of a Murine Light Chain Dimer" Molecules 29, no. 12: 2885. https://doi.org/10.3390/molecules29122885