
Investigation of the Efficacy of Benzylidene-3-methyl-2-thioxothiazolidin-4-one Analogs with Antioxidant Activities on the Inhibition of Mushroom and Mammal Tyrosinases

 , , , , , , , and
, , , , , , , and
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Synthesis of Benzylidene-3-Methyl-2-Thioxothiazolidin-4-One (BMTTZD) Analogs 1–8
2.2. Mushroom Tyrosinase Inhibitory Activities of BMTTZD Analogs 1–8
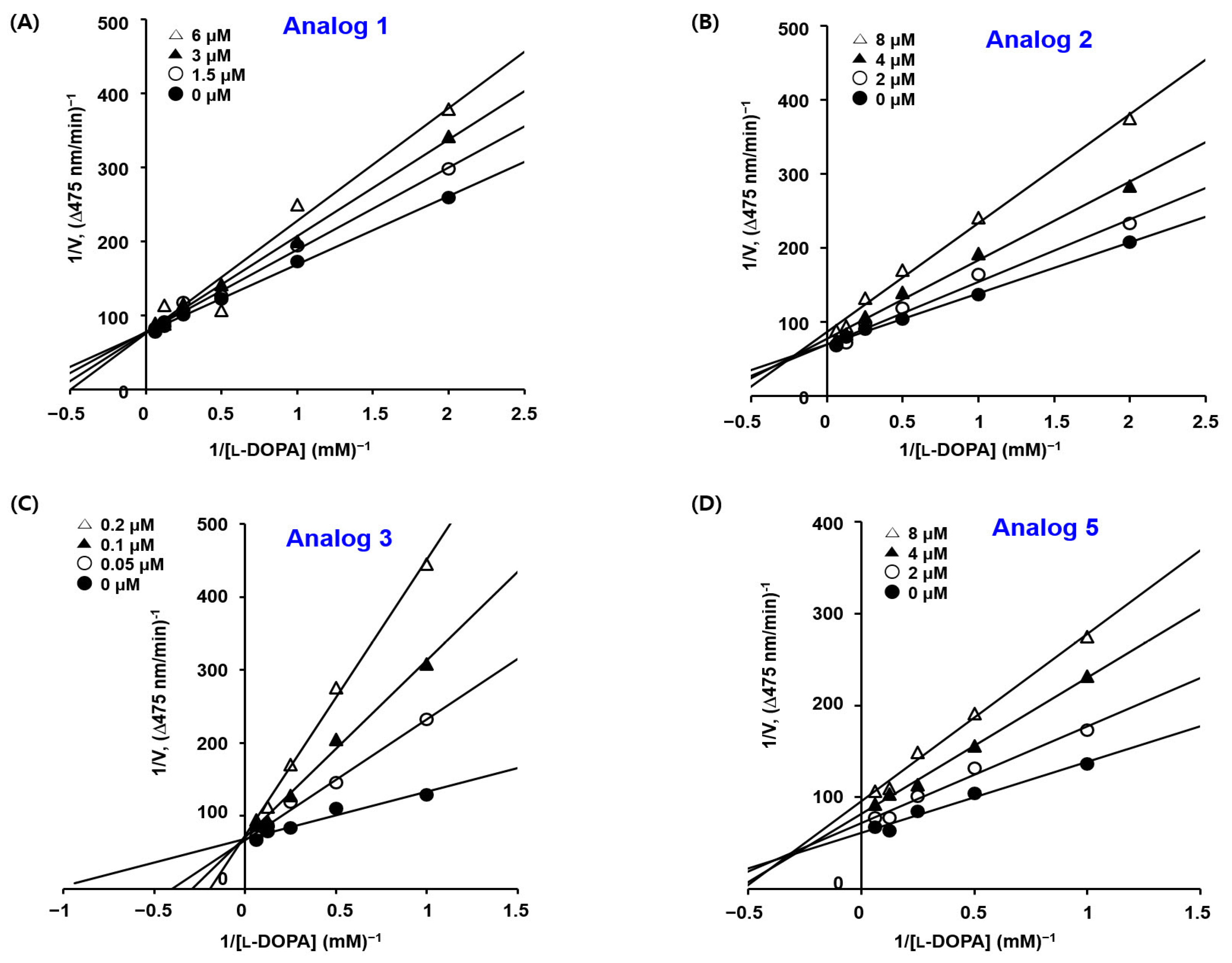
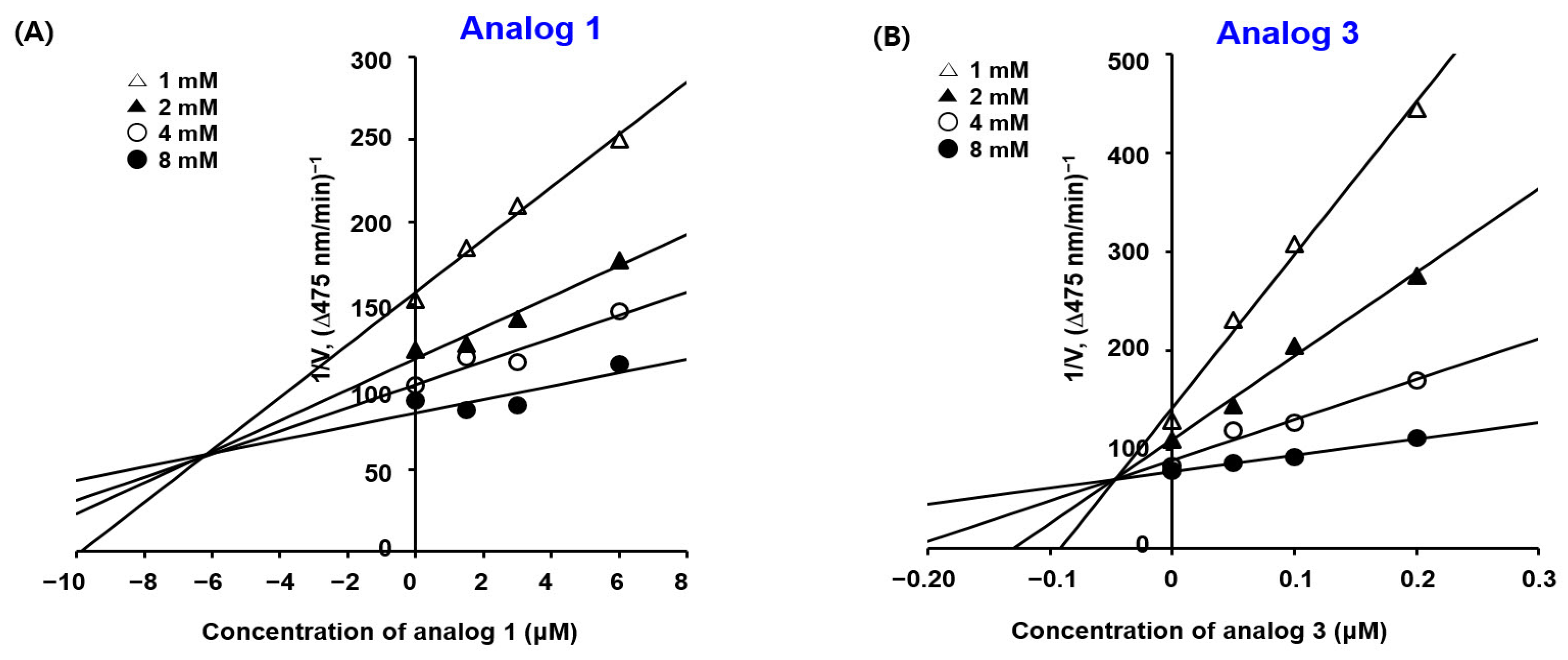
2.3. Kinetic Studies of BMTTZD Analogs 1–3 and 5 against Mushroom Tyrosinase
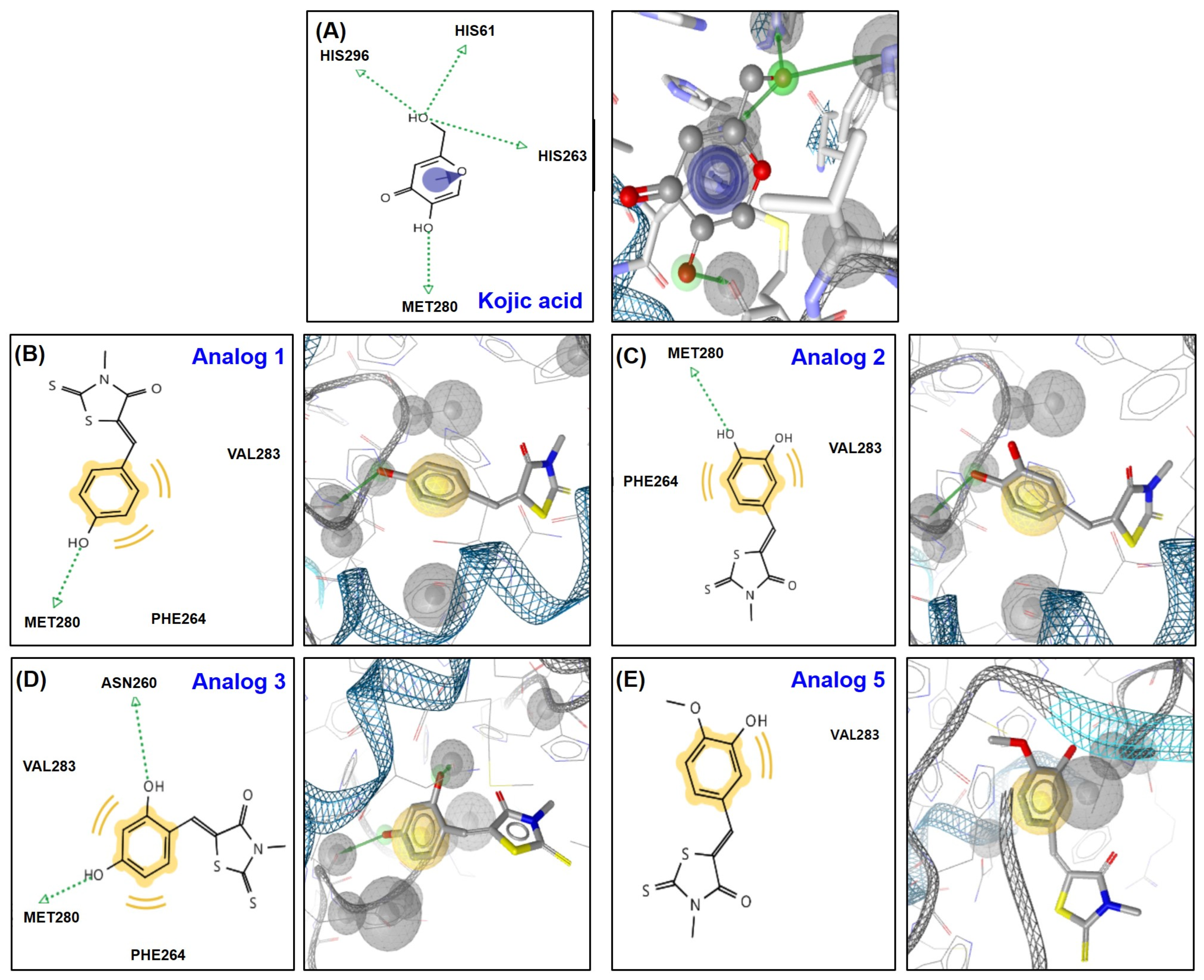
2.4. In Silico Docking Simulation Studies of BMTTZD Analogs 1–3 and 5 and Mushroom Tyrosinase at the Active and Allosteric Sites
2.5. Cytotoxic Effects of Analogs 1–3 and 5 in B16F10 Cells
2.6. Cellular Tyrosinase Inhibition Effects of Analogs in B16F10 Cells
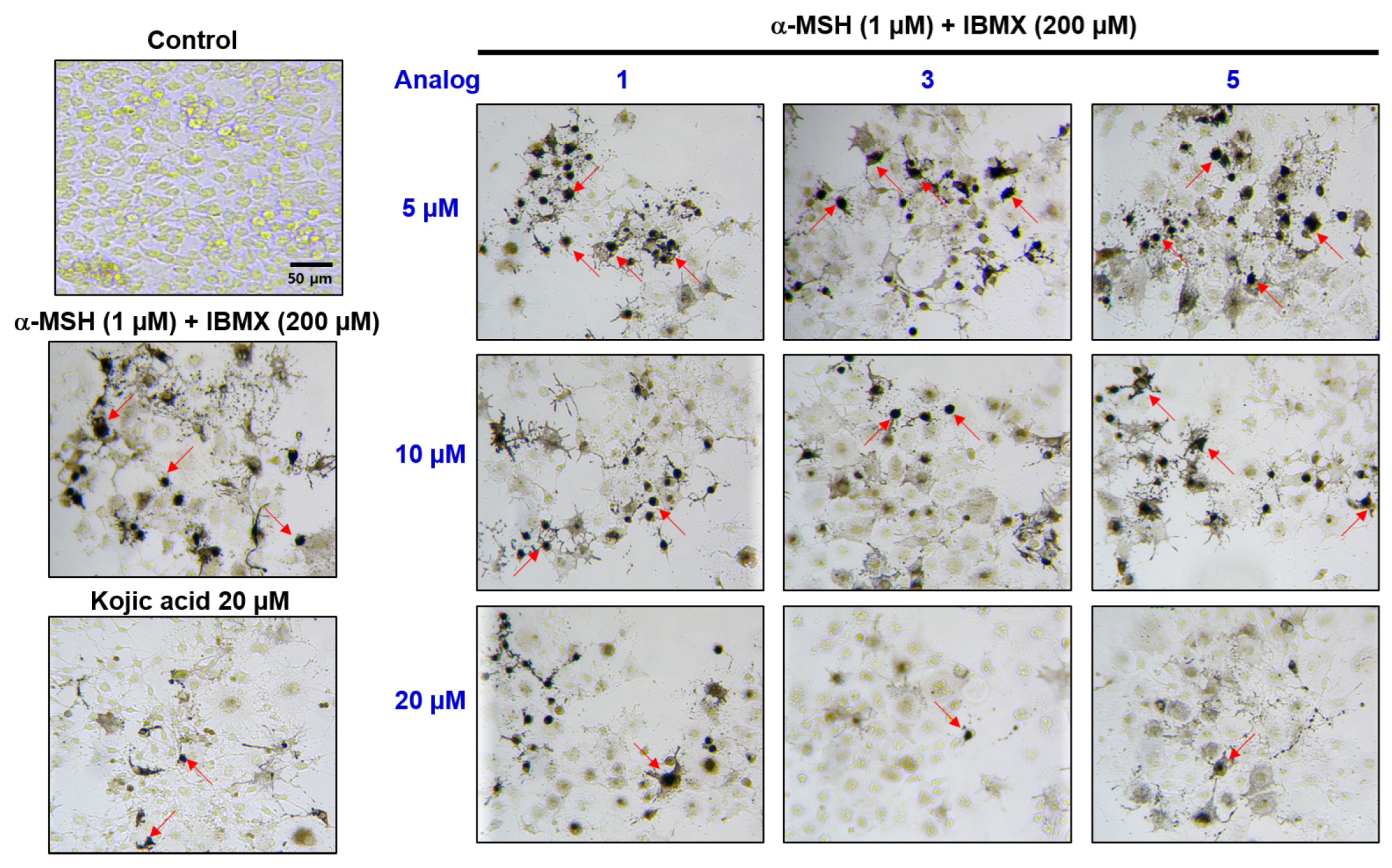
2.7. Effect of Analogs 1, 3, and 5 on B16F10 Intracellular Melanin Levels
2.8. Evaluation of In Situ Intracellular Tyrosinase Activities of Analogs in B16F10 Cells
2.9. Antioxidant Efficacy of BMTTZD Analogs 1–8
3. Materials and Methods
3.1. Chemistry
3.1.1. General Methods
3.1.2. Procedure for the Synthesis of BMTTZD Analogs 1–8
- (Z)-5-(4-Hydroxybenzylidene)-3-methyl-2-thioxothiazolidin-4-one (analog 1)
- 4 h; 92%; 1H NMR (DMSO-d6, 500 MHz) δ 10.46 (brs, 1H), 7.68 (s, 1H), 7.47 (d, J = 8.5 Hz, 2H), 6.91 (d, J = 8.5 Hz, 2H), 3.36 (s, 3H); 13C NMR (DMSO-d6, 125 MHz) δ 193.8, 167.5, 161.0, 134.0, 133.7, 124.4, 118.4, 117.0, 31.5; HRMS (EDA) m/z C11H10NO2S2 (M + H)+ calcd 252.0147, obsd 252.0148.
- (Z)-5-(3,4-Dihydroxybenzylidene)-3-methyl-2-thioxothiazolidin-4-one (analog 2)
- 9 h; 92%; 1H NMR (DMSO-d6, 500 MHz) δ 9.75 (brs, 2H), 7.58 (s, 1H), 7.00 (s, 1H), 6.99 (d, J = 8.0 Hz, 1H), 6.86 (d, J = 8.0 Hz, 1H), 3.34 (s, 3H); 13C NMR (DMSO-d6, 125 MHz) δ 193.8, 167.5, 149.9, 146.5, 134.4, 125.7, 124.9, 118.2, 117.2, 116.9, 31.5.
- (Z)-5-(2,4-Dihydroxybenzylidene)-3-methyl-2-thioxothiazolidin-4-one (analog 3)
- 9 h; 86%; 1H NMR (DMSO-d6, 500 MHz) δ 10.68 (s, 1H), 10.35 (s, 1H), 7.94 (s, 1H), 7.18 (d, J = 8.5 Hz, 1H), 6.44–6.39 (m, 2H), 3.36 (s, 3H); 13C NMR (DMSO-d6, 125 MHz) δ 194.0, 167.7 (dq, J = 5.8, 2.0 Hz), 163.1, 160.4, 131.9, 129.7, 116.2, 112.4, 109.4, 102.9, 31.5; HRMS (EDA) m/z C11H10NO3S2 (M + H)+ calcd 268.0097, obsd 268.0096.
- (Z)-5-(4-Hydroxy-3-methoxybenzylidene)-3-methyl-2-thioxothiazolidin-4-one (analog 4)
- 5 h; 89%; 1H NMR (DMSO-d6, 500 MHz) δ 10.14 (brs, 1H, OH), 7.74 (s, 1H, vinylic H), 7.20 (s, 1H), 7.13 (d, J = 7.5 Hz, 1H), 6.95 (d, J = 7.5 Hz, 1H), 3.84 (s, 3H), 3.39 (s, 3H); 13C NMR (DMSO-d6, 100 MHz) δ 193.8, 167.5, 151.0, 148.9, 134.4, 125.9, 125.2, 118.9, 117.1, 115.5, 56.5, 31.6.
- (Z)-5-(3-Hydroxy-4-methoxybenzylidene)-3-methyl-2-thioxothiazolidin-4-one (analog 5)
- 14 h; 90%; 1H NMR (DMSO-d6, 500 MHz) δ 9.53 (s, 1H), 7.58 (s, 1H), 7.07 (dd, J = 8.5, 2.0 Hz, 1H), 7.02 (d, J = 8.5 Hz, 1H), 6.99 (d, J = 2.0 Hz, 1H), 3.82 (s, 3H), 3.33 (s, 3H); 13C NMR (DMSO-d6, 125 MHz) δ 193.8, 167.4, 151.1, 147.5, 133.9, 126.1, 125.1, 119.4, 116.6, 112.9, 56.2, 31.5; HRMS (EDA) m/z C12H12NO3S2 (M + H)+ calcd 282.0253, obsd 282.0257.
- (Z)-5-(4-Hydroxy-3,5-dimethoxybenzylidene)-3-methyl-2-thioxothiazolidin-4-one (analog 6)
- 6 h; 91%; 1H NMR (DMSO-d6, 500 MHz) δ 9.51 (brs, 1H), 7.66 (s, 1H), 6.86 (s, 2H), 3.81 (s, 6H), 3.33 (s, 3H); 13C NMR (DMSO-d6, 125 MHz) δ 193.5, 167.4, 148.7, 139.9, 134.6, 123.7, 118.8, 109.1, 56.5, 31.5.
- (Z)-5-(3-Bromo-4-hydroxybenzylidene)-3-methyl-2-thioxothiazolidin-4-one (analog 7)
- 5 h; 85%; 1H NMR (DMSO-d6, 500 MHz) δ 11.28 (brs, 1H), 7.81 (d, J = 2.0 Hz, 1H), 7.68 (s, 1H), 7.44 (dd, J = 8.5, 2.0 Hz, 1H), 7.09 (d, J = 8.5 Hz, 1H), 3.36 (s, 3H); 13C NMR (DMSO-d6, 100 MHz) δ 193.6, 167.4, 157.4, 136.8, 132.4, 131.7, 126.1, 120.2, 117.6, 110.9, 31.7.
- (Z)-5-(3,5-Dibromo-4-hydroxybenzylidene)-3-methyl-2-thioxothiazolidin-4-one (analog 8)
- 3 h; 89%; 1H NMR (DMSO-d6, 500 MHz) δ 10.91 (brs, 1H), 7.75 (s, 2H), 7.66 (s, 1H), 3.36 (s, 3H); 13C NMR (DMSO-d6, 100 MHz) δ 193.3, 167.3, 153.8, 134.9, 130.7, 127.8, 122.3, 113.0, 31.7.
3.2. Mushroom Tyrosinase Inhibition Assay
3.3. The Kinetic Study of Mushroom Tyrosinase Inhibition of BMTTZD Analogs
3.4. In Silico Docking Simulation of Mushroom Tyrosinase and BMTTZD Analogs
3.5. B16F10 Cell Culture
3.6. Cell Cytotoxicity Test
3.7. Cellular Tyrosinase Activity Assay
3.8. Melanin Content Assay in B16F10 Cells
3.9. DPPH Radical Scavenging Activity Assay
3.10. ABTS+ Radical Scavenging Activity Assay
3.11. ROS Scavenging Activity Assay
3.12. In Vitro and In Situ Cellular Tyrosinase Activity Assay Using l-Dopa Staining
3.13. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Derardja, A.E.; Pretzler, M.; Kampatsikas, I.; Barkat, M.; Rompel, A. Purification and Characterization of Latent Polyphenol Oxidase from Apricot (Prunus armeniaca L.). J. Agric. Food Chem. 2017, 65, 8203–8212. [Google Scholar] [CrossRef] [PubMed]
- Ramsden, C.A.; Riley, P.A. Tyrosinase: The four oxidation states of the active site and their relevance to enzymatic activation, oxidation and inactivation. Bioorg. Med. Chem. 2014, 22, 2388–2395. [Google Scholar] [CrossRef] [PubMed]
- Solano, F. Melanins: Skin Pigments and Much More—Types, Structural Models, Biological Functions, and Formation Routes. N. J. Sci. 2014, 2014, 498276. [Google Scholar] [CrossRef]
- Friedman, M. Food Browning and Its Prevention: An Overview. J. Agric. Food Chem. 1996, 44, 631–653. [Google Scholar] [CrossRef]
- Chen, L.-H.; Hu, Y.-H.; Song, W.; Song, K.-K.; Liu, X.; Jia, Y.-L.; Zhuang, J.-X.; Chen, Q.-X. Synthesis and Antityrosinase Mechanism of Benzaldehyde Thiosemicarbazones: Novel Tyrosinase Inhibitors. J. Agric. Food Chem. 2012, 60, 1542–1547. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Wang, G.; Zeng, Q.-H.; Li, Y.; Liu, H.; Wang, J.J.; Zhao, Y. A systematic review of synthetic tyrosinase inhibitors and their structure-activity relationship. Crit. Rev. Food Sci. Nutr. 2022, 62, 4053–4094. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.S. An updated review of tyrosinase inhibitors. Int. J. Mol. Sci. 2009, 10, 2440–2475. [Google Scholar] [CrossRef] [PubMed]
- Pillaiyar, T.; Manickam, M.; Namasivayam, V. Skin whitening agents: Medicinal chemistry perspective of tyrosinase inhibitors. J. Enzym. Inhib. Med. Chem. 2017, 32, 403–425. [Google Scholar] [CrossRef]
- Yuan, Y.; Jin, W.; Nazir, Y.; Fercher, C.; Blaskovich, M.A.T.; Cooper, M.A.; Barnard, R.T.; Ziora, Z.M. Tyrosinase inhibitors as potential antibacterial agents. Eur. J. Med. Chem. 2020, 187, 111892. [Google Scholar] [CrossRef]
- Körner, A.; Pawelek, J. Mammalian tyrosinase catalyzes three reactions in the biosynthesis of melanin. Science 1982, 217, 1163–1165. [Google Scholar] [CrossRef]
- Hearing, V.J.; Tsukamoto, K. Enzymatic control of pigmentation in mammals. FASEB J. 1991, 5, 2902–2909. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-J.; Uyama, H. Tyrosinase inhibitors from natural and synthetic sources: Structure, inhibition mechanism and perspective for the future. Cell. Mol. Life Sci. 2005, 62, 1707–1723. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, N.; Onodera, H.; Mitsumori, K.; Tamura, T.; Maruyama, S.; Ito, A. Changes in thyroid function during development of thyroid hyperplasia induced by kojic acid in F344 rats. Carcinogenesis 1999, 20, 1567–1572. [Google Scholar] [CrossRef]
- McGregor, D. Hydroquinone: An evaluation of the human risks from its carcinogenic and mutagenic properties. Crit. Rev. Toxicol. 2007, 37, 887–914. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.S.; Choi, J.H.; Cho, W.K.; Park, J.C.; Choi, J.S. A sphingolipid and tyrosinase inhibitors from the fruiting body of Phellinus linteus. Arch. Pharm. Res. 2004, 27, 742–750. [Google Scholar] [CrossRef] [PubMed]
- Bekier, A.; Węglińska, L.; Paneth, A.; Paneth, P.; Dzitko, K. 4-Arylthiosemicarbazide derivatives as a new class of tyrosinase inhibitors and anti-Toxoplasma gondii agents. J. Enzyme Inhib. Med. Chem. 2021, 36, 1145–1164. [Google Scholar] [CrossRef]
- Kim, Y.M.; Yun, J.; Lee, C.K.; Lee, H.; Min, K.R.; Kim, Y. Oxyresveratrol and hydroxystilbene compounds. Inhibitory effect on tyrosinase and mechanism of action. J. Biol. Chem. 2002, 277, 16340–16344. [Google Scholar] [CrossRef]
- Xie, W.; Zhang, J.; Ma, X.; Yang, W.; Zhou, Y.; Tang, X.; Zou, Y.; Li, H.; He, J.; Xie, S.; et al. Synthesis and biological evaluation of kojic acid derivatives containing 1,2,4-triazole as potent tyrosinase inhibitors. Chem. Biol. Drug Des. 2015, 86, 1087–1092. [Google Scholar] [CrossRef]
- Chen, Q.X.; Ke, L.N.; Song, K.K.; Huang, H.; Liu, X.D. Inhibitory effects of hexylresorcinol and dodecylresorcinol on mushroom (Agaricus bisporus) tyrosinase. Protein J. 2004, 23, 135–141. [Google Scholar] [CrossRef]
- Ullah, S.; Kang, D.; Lee, S.; Ikram, M.; Park, C.; Park, Y.; Yoon, S.; Chun, P.; Moon, H.R. Synthesis of cinnamic amide derivatives and their anti-melanogenic effect in α-MSH-stimulated B16F10 melanoma cells. Eur. J. Med. Chem. 2019, 161, 78–92. [Google Scholar] [CrossRef]
- Ryu, Y.; Ha, T.; Curtis-Long, M.; Ryu, H.; Gal, S.; Park, K. Inhibitory effects on mushroom tyrosinase by flavones from the stem barks of Morus lhou (S.) Koidz. J. Enzyme Inhib. Med. Chem. 2008, 23, 922–930. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Jeong, Y.; Jin Jung, H.; Ullah, S.; Ko, J.; Young Kim, G.; Yoon, D.; Hong, S.; Kang, D.; Park, Y.; et al. Anti-tyrosinase flavone derivatives and their anti-melanogenic activities: Importance of the β-phenyl-α,β-unsaturated carbonyl scaffold. Bioorg. Chem. 2023, 135, 106504. [Google Scholar] [CrossRef] [PubMed]
- Athipornchai, A.; Niyomtham, N.; Pabuprapap, W.; Ajavakom, V.; Duca, M.; Azoulay, S.; Suksamrarn, A. Potent Tyrosinase Inhibitory Activity of Curcuminoid Analogues and Inhibition Kinetics Studies. Cosmetics 2021, 8, 35. [Google Scholar] [CrossRef]
- Khan, M.T.; Choudhary, M.I.; Khan, K.M.; Rani, M.; Attaur, R. Structure-activity relationships of tyrosinase inhibitory combinatorial library of 2,5-disubstituted-1,3,4-oxadiazole analogues. Bioorg. Med. Chem. 2005, 13, 3385–3395. [Google Scholar] [CrossRef] [PubMed]
- Mermer, A.; Demirci, S. Recent advances in triazoles as tyrosinase inhibitors. Eur. J. Med. Chem. 2023, 259, 115655. [Google Scholar] [CrossRef] [PubMed]
- Criton, M.; Le Mellay-Hamon, V. Analogues of N-hydroxy-N′-phenylthiourea and N-hydroxy-N’-phenylurea as inhibitors of tyrosinase and melanin formation. Bioorg. Med. Chem. Lett. 2008, 18, 3607–3610. [Google Scholar] [CrossRef] [PubMed]
- Jun, N.; Hong, G.; Jun, K. Synthesis and evaluation of 2’,4’,6’-trihydroxychalcones as a new class of tyrosinase inhibitors. Bioorg. Med. Chem. 2007, 15, 2396–2402. [Google Scholar] [CrossRef]
- Strothkamp, K.G.; Jolley, R.L.; Mason, H.S. Quaternary structure of mushroom tyrosinase. Biochem. Biophys. Res. Commun. 1976, 70, 519–524. [Google Scholar] [CrossRef] [PubMed]
- Mendes, E.; Perry, M.d.J.; Francisco, A.P. Design and discovery of mushroom tyrosinase inhibitors and their therapeutic applications. Expert Opin. Drug Discov. 2014, 9, 533–554. [Google Scholar] [CrossRef]
- Li, J.; Feng, L.; Liu, L.; Wang, F.; Ouyang, L.; Zhang, L.; Hu, X.; Wang, G. Recent advances in the design and discovery of synthetic tyrosinase inhibitors. Eur. J. Med. Chem. 2021, 224, 113744. [Google Scholar] [CrossRef]
- Martinez, A.; Alonso, M.; Castro, A.; Dorronsoro, I.; Gelpí, J.L.; Luque, F.J.; Pérez, C.; Moreno, F.J. SAR and 3D-QSAR studies on thiadiazolidinone derivatives: Exploration of structural requirements for glycogen synthase kinase 3 inhibitors. J. Med. Chem. 2005, 48, 7103–7112. [Google Scholar] [CrossRef] [PubMed]
- Muro, C.; Yasuda, M.; Sakagami, Y.; Yamada, T.; Tsujibo, H.; NUMATA, A.; Inamori, Y. Inhibitory activities of rhodanine derivatives on plant growth. Biosci. Biotech. Biochem. 1996, 60, 1368–1371. [Google Scholar] [CrossRef]
- Tong, H.; Skidmore, D.; Maibach, H.; Skinner, W. Topical mosquito repellents VIII: Substituted 2-thio-4-thiazolidineones and 2, 4-thiazolidinediones. Mosq. News 1975, 35, 76–82. [Google Scholar]
- Akunuri, R.; Unnissa, T.; Kaul, G.; Akhir, A.; Saxena, D.; Wajidali, M.; Veerareddy, V.; Yaddanapudi, V.M.; Chopra, S.; Nanduri, S. Synthesis and antibacterial evaluation of rhodanine and its related heterocyclic compounds against S. aureus and A. baumannii. Chem. Biodivers. 2022, 19, e202200213. [Google Scholar] [CrossRef] [PubMed]
- Russell, A.J.; Westwood, I.M.; Crawford, M.H.; Robinson, J.; Kawamura, A.; Redfield, C.; Laurieri, N.; Lowe, E.D.; Davies, S.G.; Sim, E. Selective small molecule inhibitors of the potential breast cancer marker, human arylamine N-acetyltransferase 1, and its murine homologue, mouse arylamine N-acetyltransferase 2. Bioorg. Med. Chem. 2009, 17, 905–918. [Google Scholar] [CrossRef] [PubMed]
- Eggers, H.J.; Koch, M.A.; Furst, A.; Daves, G.D.; Wilczynski, J.J.; Folkers, K. Rhodanine: A Selective Inhibitor of the Multiplication of Echovirus 12. Science 1970, 167, 294–297. [Google Scholar] [CrossRef]
- Jung, H.J.; Choi, D.C.; Noh, S.G.; Choi, H.; Choi, I.; Ryu, I.Y.; Chung, H.Y.; Moon, H.R. New Benzimidazothiazolone Derivatives as Tyrosinase Inhibitors with Potential Anti-Melanogenesis and Reactive Oxygen Species Scavenging Activities. Antioxidants 2021, 10, 1078. [Google Scholar] [CrossRef] [PubMed]
- Jung Park, Y.; Jin Jung, H.; Jin Kim, H.; Soo Park, H.; Lee, J.; Yoon, D.; Kyung Kang, M.; Young Kim, G.; Ullah, S.; Kang, D.; et al. Thiazol-4(5H)-one analogs as potent tyrosinase inhibitors: Synthesis, tyrosinase inhibition, antimelanogenic effect, antioxidant activity, and in silico docking simulation. Bioorg. Med. Chem. 2024, 98, 117578. [Google Scholar] [CrossRef] [PubMed]
- Grigalius, I.; Petrikaite, V. Relationship between Antioxidant and Anticancer Activity of Trihydroxyflavones. Molecules 2017, 22, 2169. [Google Scholar] [CrossRef]
- Vögeli, U.; von Philipsborn, W.; Nagarajan, K.; Nair, M.D. Structures of Addition Products of Acetylenedicarboxylic Acid Esters with Various Dinucleophiles. An application of C, H-spin-coupling constants. Helv. Chim. Acta 1978, 61, 607–617. [Google Scholar] [CrossRef]
- Panich, U.; Tangsupa-a-nan, V.; Onkoksoong, T.; Kongtaphan, K.; Kasetsinsombat, K.; Akarasereenont, P.; Wongkajornsilp, A. Inhibition of UVA-mediated melanogenesis by ascorbic acid through modulation of antioxidant defense and nitric oxide system. Arch. Pharm. Res. 2011, 34, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-C.; Wang, H.-F.; Yih, K.-H.; Chang, L.-Z.; Chang, T.-M. The Dual Antimelanogenic and Antioxidant Activities of the Essential Oil Extracted from the Leaves of Acorus macrospadiceus (Yamamoto) F. N. Wei et Y. K. Li. Evid. Based Complement. Alternat. Med. 2012, 2012, 781280. [Google Scholar] [CrossRef] [PubMed]
- Newton, R.A.; Cook, A.L.; Roberts, D.W.; Leonard, J.H.; Sturm, R.A. Post-transcriptional regulation of melanin biosynthetic enzymes by cAMP and resveratrol in human melanocytes. J. Investig. Dermatol. 2007, 127, 2216–2227. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||
|---|---|---|---|---|---|---|
| Analog | R1 | R2 | R3 | R4 | IC50 (Mean ± SEM, µM) | |
| l-Dopa | l-Tyrosine | |||||
| 1 | H | H | OH | H | 17.62 ± 0.62 | 3.82 ± 0.22 |
| 2 | H | OH | OH | H | 6.18 ± 0.17 | 3.77 ± 0.47 |
| 3 | OH | H | OH | H | 1.12 ± 0.06 | 0.08 ± 0.02 |
| 4 | H | CH3O | OH | H | >200 | 54.81 ± 16.07 |
| 5 | H | OH | CH3O | H | 13.75 ± 2.64 | 3.60 ± 0.52 |
| 6 | H | CH3O | OH | CH3O | >200 | 57.40 ± 10.94 |
| 7 | H | Br | OH | H | >200 | 33.23 ± 8.42 |
| 8 | H | Br | OH | Br | >200 | >200 |
| a Kojic acid | 24.09 ± 0.32 | 17.68 ± 2.23 | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.J.; Jung, H.J.; Kim, Y.E.; Jeong, D.; Park, H.S.; Park, H.S.; Kang, D.; Park, Y.; Chun, P.; Chung, H.Y.; et al. Investigation of the Efficacy of Benzylidene-3-methyl-2-thioxothiazolidin-4-one Analogs with Antioxidant Activities on the Inhibition of Mushroom and Mammal Tyrosinases. Molecules 2024, 29, 2887. https://doi.org/10.3390/molecules29122887
Kim HJ, Jung HJ, Kim YE, Jeong D, Park HS, Park HS, Kang D, Park Y, Chun P, Chung HY, et al. Investigation of the Efficacy of Benzylidene-3-methyl-2-thioxothiazolidin-4-one Analogs with Antioxidant Activities on the Inhibition of Mushroom and Mammal Tyrosinases. Molecules. 2024; 29(12):2887. https://doi.org/10.3390/molecules29122887
Chicago/Turabian StyleKim, Hye Jin, Hee Jin Jung, Young Eun Kim, Daeun Jeong, Hyeon Seo Park, Hye Soo Park, Dongwan Kang, Yujin Park, Pusoon Chun, Hae Young Chung, and et al. 2024. "Investigation of the Efficacy of Benzylidene-3-methyl-2-thioxothiazolidin-4-one Analogs with Antioxidant Activities on the Inhibition of Mushroom and Mammal Tyrosinases" Molecules 29, no. 12: 2887. https://doi.org/10.3390/molecules29122887
APA StyleKim, H. J., Jung, H. J., Kim, Y. E., Jeong, D., Park, H. S., Park, H. S., Kang, D., Park, Y., Chun, P., Chung, H. Y., & Moon, H. R. (2024). Investigation of the Efficacy of Benzylidene-3-methyl-2-thioxothiazolidin-4-one Analogs with Antioxidant Activities on the Inhibition of Mushroom and Mammal Tyrosinases. Molecules, 29(12), 2887. https://doi.org/10.3390/molecules29122887