
Density Functional Theory Study of Triple Transition Metal Cluster Anchored on the C2N Monolayer for Nitrogen Reduction Reactions



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
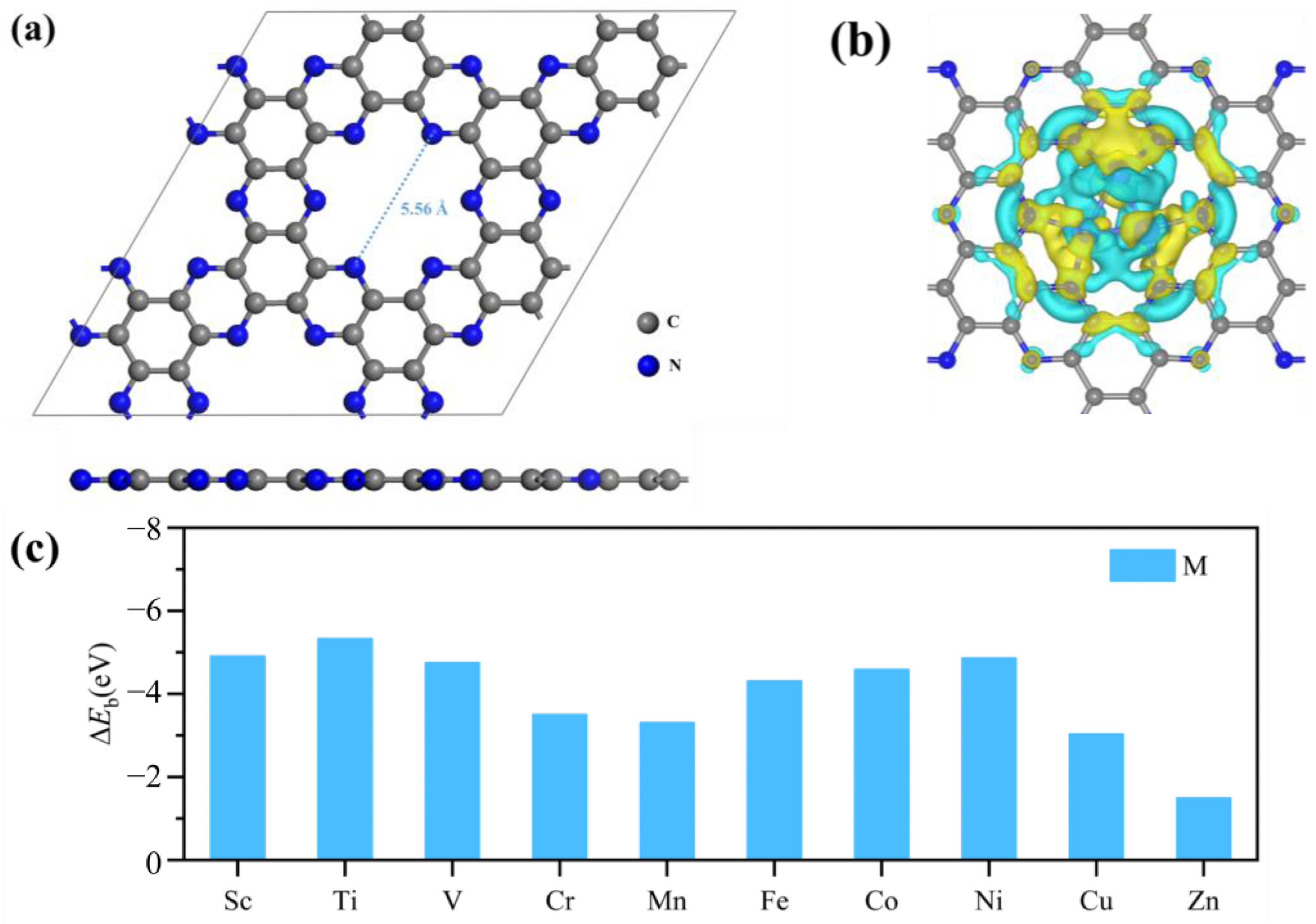
2.1. Structure and Stability of M3-C2N
2.2. N2 Adsorption on M3-C2N
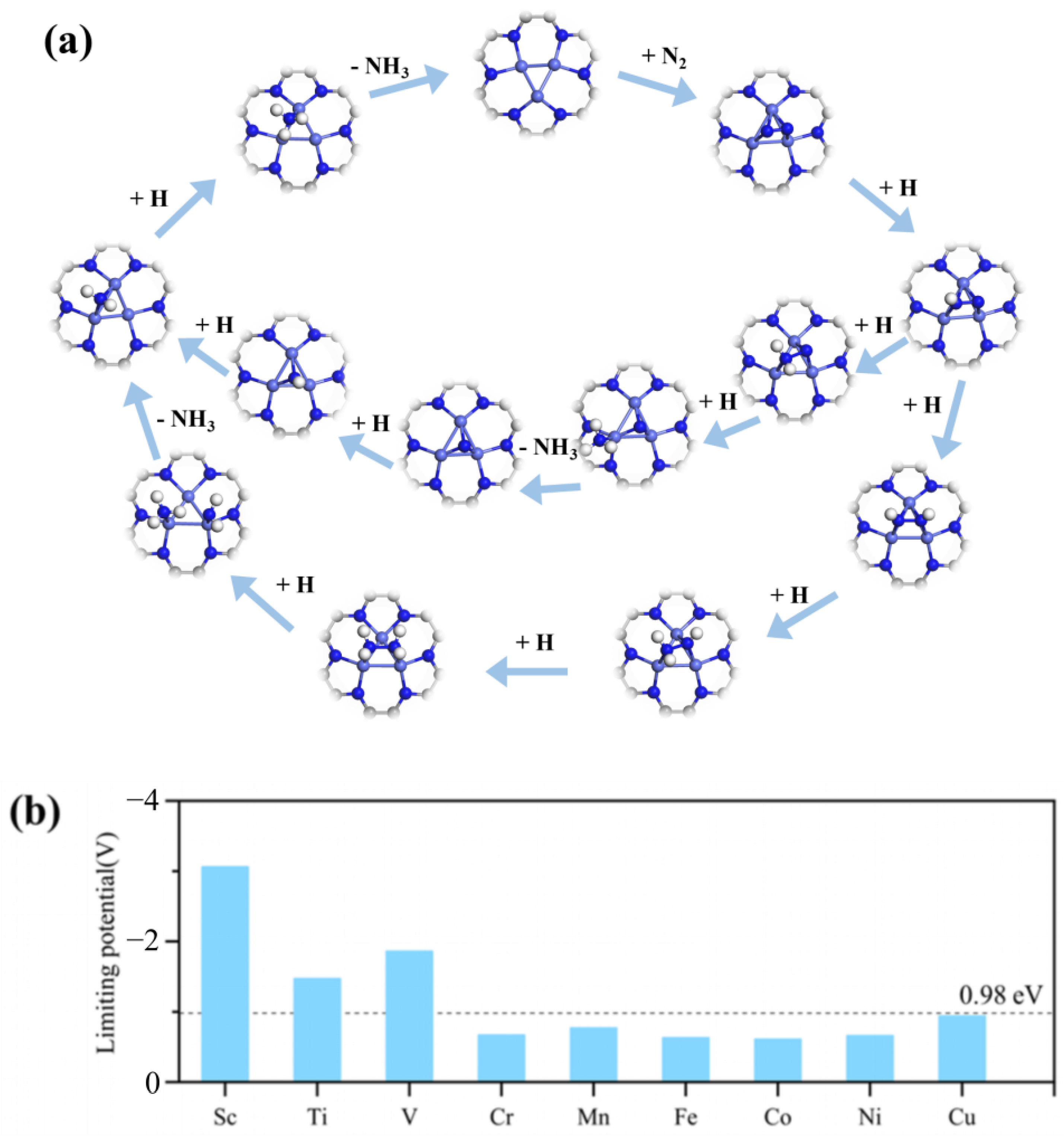
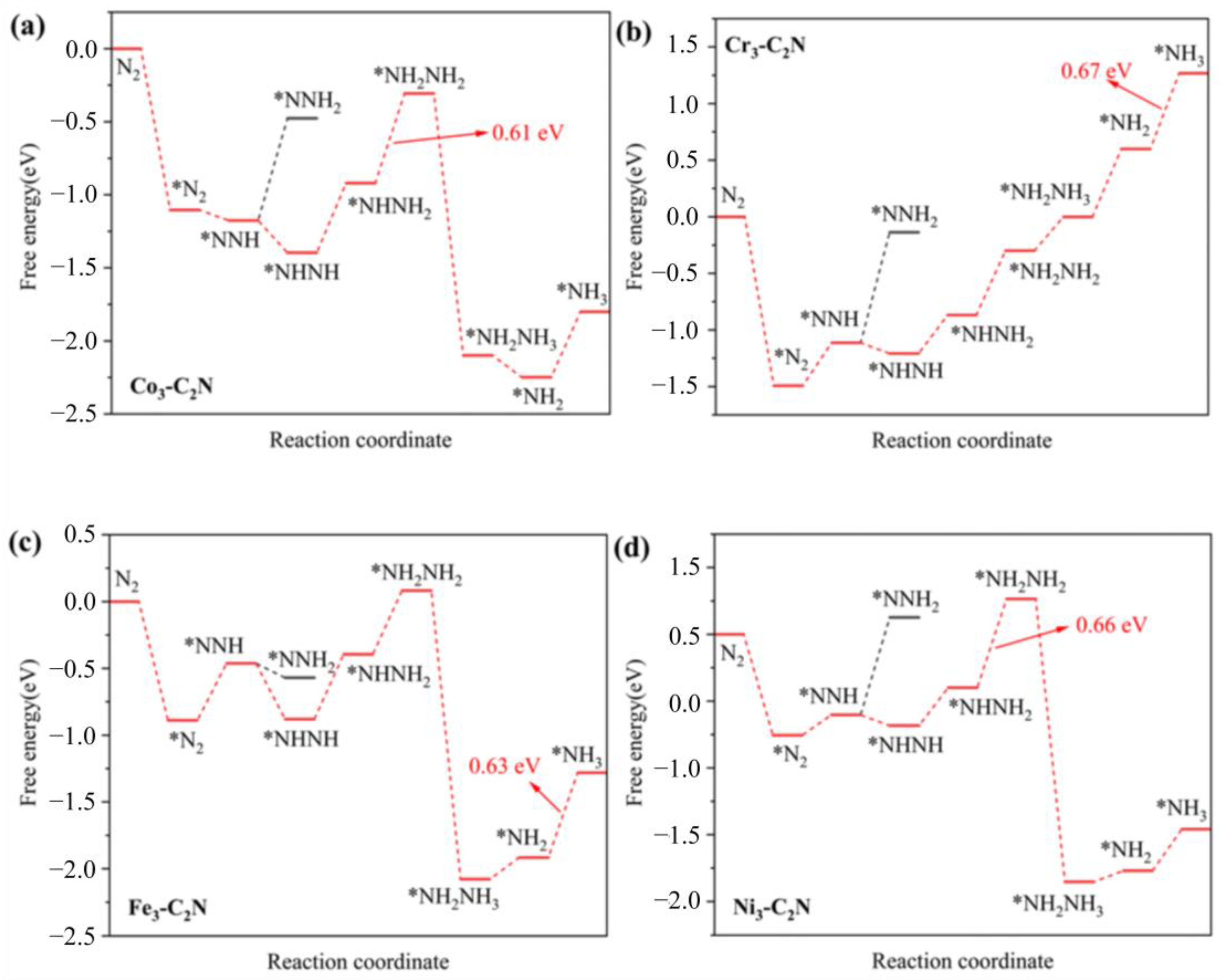
2.3. NRR Mechanism and Activity on M3-C2N
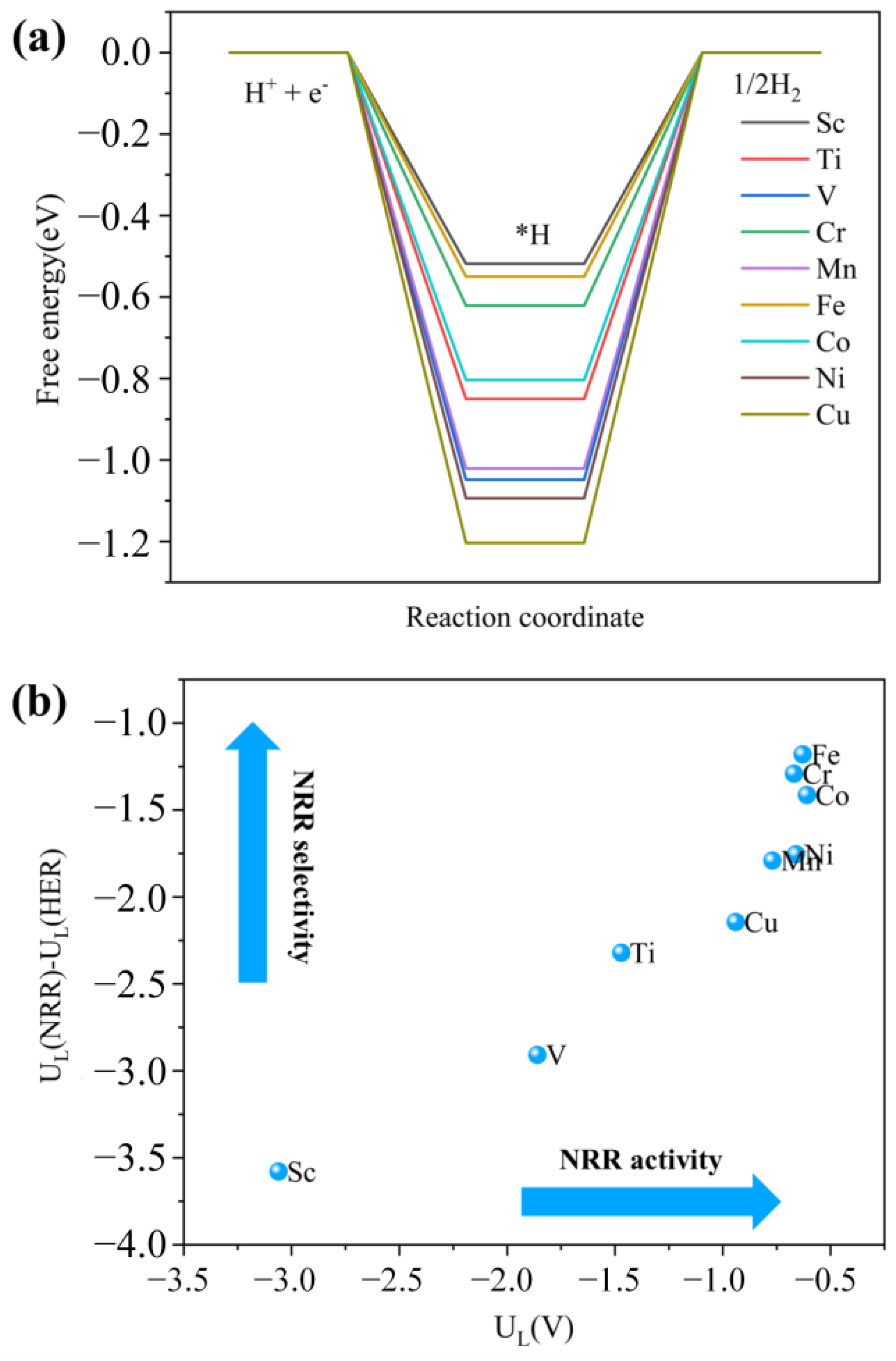
2.4. Selectivity Evaluation for NRR on M3-C2N
3. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Valera-Medina, A.; Xiao, H.; Owen-Jones, M.; David, W.I.F.; Bowen, P.J. Ammonia for power. Prog. Energy Combust. Sci. 2018, 69, 63–102. [Google Scholar] [CrossRef]
- Kobayashi, H.; Hayakawa, A.; Somarathne, K.D.; Kunkuma, A.; Okafor, E.C. Science and technology of ammonia combustion. Proc. Combust. Inst. 2019, 37, 109–133. [Google Scholar] [CrossRef]
- Sadeek, S.; Lee Chan, T.; Ramdath, R.; Rajkumar, A.; Guo, M.; Ward, K. The influence of raw material availability and utility power consumption on the sustainability of the ammonia process. Chem. Eng. Res. Des. 2020, 158, 177–192. [Google Scholar] [CrossRef]
- Smith, C.; Hill, A.K.; Torrente-Murciano, L. Current and future role of Haber–Bosch ammonia in a carbon-free energy landscape. Energy Environ. Sci. 2020, 13, 331–344. [Google Scholar] [CrossRef]
- Humphreys, J.; Lan, R.; Tao, S. Development and Recent Progress on Ammonia Synthesis Catalysts for Haber–Bosch Process. Adv. Energy Sustain. Res. 2021, 2, 2000043. [Google Scholar] [CrossRef]
- Lee, C.; Yan, Q. Electrochemical reduction of nitrogen to ammonia: Progress, challenges and future outlook. Curr. Opin. Electrochem. 2021, 29, 100808. [Google Scholar] [CrossRef]
- Huang, Z.; Rafiq, M.; Woldu, A.R.; Tong, Q.-X.; Astruc, D.; Hu, L. Recent progress in electrocatalytic nitrogen reduction to ammonia (NRR). Coord. Chem. Rev. 2023, 478, 214981. [Google Scholar] [CrossRef]
- Wang, J.; Chen, S.; Li, Z.; Li, G.; Liu, X. Recent Advances in Electrochemical Synthesis of Ammonia through Nitrogen Reduction under Ambient Conditions. ChemElectroChem 2020, 7, 1067–1079. [Google Scholar] [CrossRef]
- Zheng, J.; Jiang, L.; Lyu, Y.; Jiang, S.P.; Wang, S. Green Synthesis of Nitrogen-to-Ammonia Fixation: Past, Present, and Future. Energy Environ. Mater. 2022, 5, 452–457. [Google Scholar] [CrossRef]
- Luo, Y.; Li, M.; Dai, Y.; Zhang, X.; Zhao, R.; Jiang, F.; Ling, C.; Huang, Y. Screening of effective NRR electrocatalysts among the Si-based MSi2N4 (M = Ti, Zr, Hf, V, Nb, Ta, Cr, Mo, and W) monolayers. J. Mater. Chem. A 2021, 9, 15217–15225. [Google Scholar] [CrossRef]
- Ferrara, M.; Bevilacqua, M.; Tavagnacco, C.; Vizza, F.; Fornasiero, P. Fast Screening Method for Nitrogen Reduction Reaction (NRR) Electrocatalytic Activity with Rotating Ring-Disc Electrode (RRDE) Analysis in Alkaline Environment. ChemCatChem 2020, 12, 6205–6213. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, L.; Hu, Z.; Zheng, Y.; Tang, C.; Chen, P.; Wang, R.; Qiu, K.; Mao, J.; Ling, T.; et al. The Crucial Role of Charge Accumulation and Spin Polarization in Activating Carbon-Based Catalysts for Electrocatalytic Nitrogen Reduction. Angew. Chem. Int. Ed. 2020, 59, 4525–4531. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Ma, Y.; Liu, Y.; Wang, K.; Wang, Y.; Liu, M.; Qiu, X.; Li, W.; Li, J. Defect-Induced Ce-Doped Bi2WO6 for Efficient Electrocatalytic N2 Reduction. ACS Appl. Mater. Interfaces 2021, 13, 19864–19872. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Zhao, L.; Huang, G.; Liu, Z.; Yu, S.; Wang, K.; Yuan, S.; Sun, Q.; Li, X.; Li, N. Electrochemical Fixation of Nitrogen by Promoting N2 Adsorption and N–N Triple Bond Cleavage on the CoS2/MoS2 Nanocomposite. ACS Appl. Mater. Interfaces 2021, 13, 21474–21481. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Li, Q.; Ling, C.; Zhang, Y.; Ouyang, Y.; Bai, X.; Wang, J. Metal-free electrocatalyst for reducing nitrogen to ammonia using a Lewis acid pair. J. Mater. Chem. A 2019, 7, 4865–4871. [Google Scholar] [CrossRef]
- Wang, H.; Chen, Y.; Fan, R.; Chen, J.; Wang, Z.; Mao, S.; Wang, Y. Selective Electrochemical Reduction of Nitrogen to Ammonia by Adjusting the Three-Phase Interface. Research 2019, 2019, 1401209. [Google Scholar] [CrossRef] [PubMed]
- Shinde, S.S.; Lee, C.H.; Yu, J.-Y.; Kim, D.-H.; Lee, S.U.; Lee, J.-H. Hierarchically Designed 3D Holey C2N Aerogels as Bifunctional Oxygen Electrodes for Flexible and Rechargeable Zn-Air Batteries. ACS Nano 2018, 12, 596–608. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; López-Salas, N.; Liu, C.; Liu, T.; Antonietti, M. C2N: A Class of Covalent Frameworks with Unique Properties. Adv. Sci. 2020, 7, 2001767. [Google Scholar] [CrossRef] [PubMed]
- Yong, Y.; Cui, H.; Zhou, Q.; Su, X.; Kuang, Y.; Li, X. C2N monolayer as NH3 and NO sensors: A DFT study. Appl. Surf. Sci. 2019, 487, 488–495. [Google Scholar] [CrossRef]
- Zhao, D.; Tang, X.; Xing, W.; Zhang, Y.; Gao, X.; Zhang, M.; Xie, Z.; Yan, X.; Ju, L. Designing Organic Spin-Gapless Semiconductors via Molecular Adsorption on C4N3 Monolayer. Molecules 2024, 29, 3138. [Google Scholar] [CrossRef]
- Gao, Z.; Meng, Y.; Shen, H.; Xie, B.; Ni, Z.; Xia, S. Theoretical study on low temperature reverse water gas shift (RWGS) mechanism on monatomic transition metal M doped C2N catalyst (M=Cu, Co, Fe). Mol. Catal. 2021, 516, 111992. [Google Scholar] [CrossRef]
- Peng, M.; Dong, C.; Gao, R.; Xiao, D.; Liu, H.; Ma, D. Fully Exposed Cluster Catalyst (FECC): Toward Rich Surface Sites and Full Atom Utilization Efficiency. ACS Cent. Sci. 2021, 7, 262–273. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Xu, R.; Li, Y.; Cai, L.; Yang, Y.; Zheng, Y.; Zuo, C.; Huang, H.; Wen, Z.; Wang, Q. Facile preparation of single-atom Ru catalysts via a two-dimensional interface directed synthesis technique for the NRR. Chem. Commun. 2023, 59, 5403–5406. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Liang, X.; Yang, Q.; Ren, X.; Gao, M.; Yang, Y.; Ma, T. Metal-Organic-Framework-Derived Cobalt-Doped Carbon Material for Electrochemical Ammonia Synthesis under Ambient Conditions. ChemElectroChem 2020, 7, 4900–4905. [Google Scholar] [CrossRef]
- Xiong, W.; Cheng, X.; Wang, T.; Luo, Y.; Feng, J.; Lu, S.; Asiri, A.M.; Li, W.; Jiang, Z.; Sun, X. Co3(hexahydroxytriphenylene)2: A conductive metal—Organic framework for ambient electrocatalytic N2 reduction to NH3. Nano Res. 2020, 13, 1008–1012. [Google Scholar] [CrossRef]
- Wang, Y.; Cui, X.; Zhao, J.; Jia, G.; Gu, L.; Zhang, Q.; Meng, L.; Shi, Z.; Zheng, L.; Wang, C.; et al. Rational Design of Fe–N/C Hybrid for Enhanced Nitrogen Reduction Electrocatalysis under Ambient Conditions in Aqueous Solution. ACS Catal. 2019, 9, 336–344. [Google Scholar] [CrossRef]
- Ma, Y.; Yang, T.; Zou, H.; Zang, W.; Kou, Z.; Mao, L.; Feng, Y.; Shen, L.; Pennycook, S.J.; Duan, L.; et al. Synergizing Mo Single Atoms and Mo2C Nanoparticles on CNTs Synchronizes Selectivity and Activity of Electrocatalytic N2 Reduction to Ammonia. Adv. Mater. 2020, 32, 2002177. [Google Scholar] [CrossRef]
- Ahmed, M.I.; Arachchige, L.J.; Su, Z.; Hibbert, D.B.; Sun, C.; Zhao, C. Nitrogenase-Inspired Atomically Dispersed Fe–S–C Linkages for Improved Electrochemical Reduction of Dinitrogen to Ammonia. ACS Catal. 2022, 12, 1443–1451. [Google Scholar] [CrossRef]
- Wang, X.; Wu, D.; Liu, S.; Zhang, J.; Fu, X.-Z.; Luo, J.-L. Folic Acid Self-Assembly Enabling Manganese Single-Atom Electrocatalyst for Selective Nitrogen Reduction to Ammonia. Nano-Micro Lett. 2021, 13, 125. [Google Scholar] [CrossRef]
- Zhang, Z.; Huang, X.; Xu, H. Anchoring an Fe Dimer on Nitrogen-Doped Graphene toward Highly Efficient Electrocatalytic Ammonia Synthesis. ACS Appl. Mater. Interfaces 2021, 13, 43632–43640. [Google Scholar] [CrossRef]
- Ma, D.; Wang, Y.; Liu, L.; Jia, Y. Electrocatalytic nitrogen reduction on the transition-metal dimer anchored N-doped graphene: Performance prediction and synergetic effect. Phys. Chem. Chem. Phys. 2021, 23, 4018–4029. [Google Scholar] [CrossRef]
- Chen, Z.W.; Yan, J.-M.; Jiang, Q. Single or Double: Which Is the Altar of Atomic Catalysts for Nitrogen Reduction Reaction? Small Methods 2019, 3, 1800291. [Google Scholar] [CrossRef]
- Chen, Z.W.; Chen, L.X.; Yang, C.C.; Jiang, Q. Atomic (single, double, and triple atoms) catalysis: Frontiers, opportunities, and challenges. J. Mater. Chem. A 2019, 7, 3492–3515. [Google Scholar] [CrossRef]
- Liu, J.-C.; Ma, X.-L.; Li, Y.; Wang, Y.-G.; Xiao, H.; Li, J. Heterogeneous Fe3 single-cluster catalyst for ammonia synthesis via an associative mechanism. Nat. Commun. 2018, 9, 1610. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhong, W.; Cui, P.; Li, J.; Jiang, J. Design of Efficient Catalysts with Double Transition Metal Atoms on C2N Layer. J. Phys. Chem. Lett. 2016, 7, 1750–1755. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, J.; Lee, E.K.; Jung, M.; Shin, D.; Jeon, I.-Y.; Jung, S.-M.; Choi, H.-J.; Seo, J.-M.; Bae, S.-Y.; Sohn, S.-D.; et al. Nitrogenated holey two-dimensional structures. Nat. Commun. 2015, 6, 6486. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Hafner, J. Ab initio Hellmann-Feynman molecular dynamics for liquid metals. J. Non-Cryst. Solids 1993, 156–158, 956–960. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Allouche, A.-R. Gabedit—A graphical user interface for computational chemistry softwares. J. Comput. Chem. 2011, 32, 174–182. [Google Scholar] [CrossRef]
- Mathew, K.; Sundararaman, R.; Letchworth-Weaver, K.; Arias, T.A.; Hennig, R.G. Implicit solvation model for density-functional study of nanocrystal surfaces and reaction pathways. J. Chem. Phys. 2014, 140, 084106. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Gan, S.; Mao, Y.; Xiao, J.; Xu, C.; Zhou, T. Transition metals anchored on nitrogen-doped graphdiyne for an efficient oxygen reduction reaction: A DFT study. Phys. Chem. Chem. Phys. 2024, 26, 2449–2456. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, S.; Zhang, D.; Wang, G.; Zhou, T.; Wang, N. Density Functional Theory Study of Triple Transition Metal Cluster Anchored on the C2N Monolayer for Nitrogen Reduction Reactions. Molecules 2024, 29, 3314. https://doi.org/10.3390/molecules29143314
Xiao S, Zhang D, Wang G, Zhou T, Wang N. Density Functional Theory Study of Triple Transition Metal Cluster Anchored on the C2N Monolayer for Nitrogen Reduction Reactions. Molecules. 2024; 29(14):3314. https://doi.org/10.3390/molecules29143314
Chicago/Turabian StyleXiao, Shifa, Daoqing Zhang, Guangzhao Wang, Tianhang Zhou, and Ning Wang. 2024. "Density Functional Theory Study of Triple Transition Metal Cluster Anchored on the C2N Monolayer for Nitrogen Reduction Reactions" Molecules 29, no. 14: 3314. https://doi.org/10.3390/molecules29143314
APA StyleXiao, S., Zhang, D., Wang, G., Zhou, T., & Wang, N. (2024). Density Functional Theory Study of Triple Transition Metal Cluster Anchored on the C2N Monolayer for Nitrogen Reduction Reactions. Molecules, 29(14), 3314. https://doi.org/10.3390/molecules29143314