
Unveiling the Promoting Mechanism of H2 Activation on CuFeOx Catalyst for Low-Temperature CO Oxidation

Abstract
:1. Introduction
2. Results and Discussion
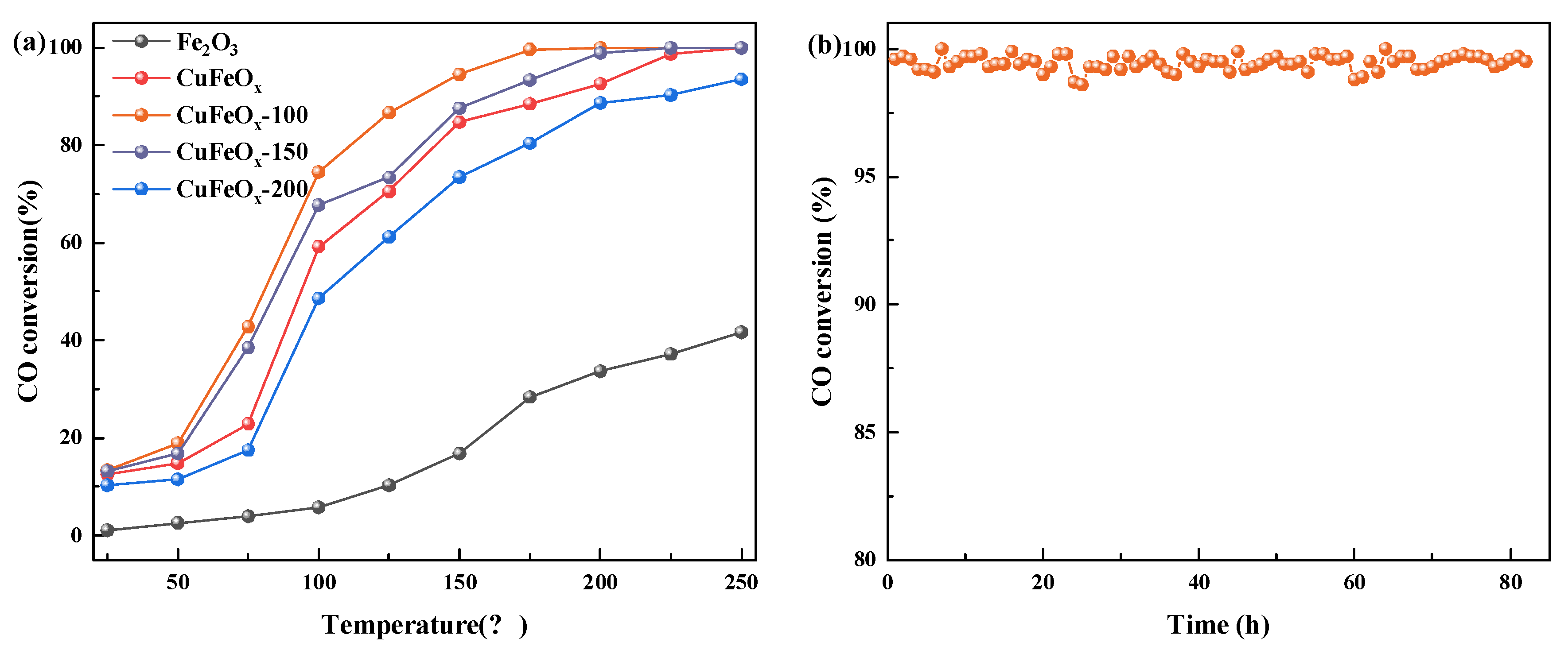
2.1. Catalytic Performance
2.2. Structural and Textural Properties
2.3. Surface Chemical States
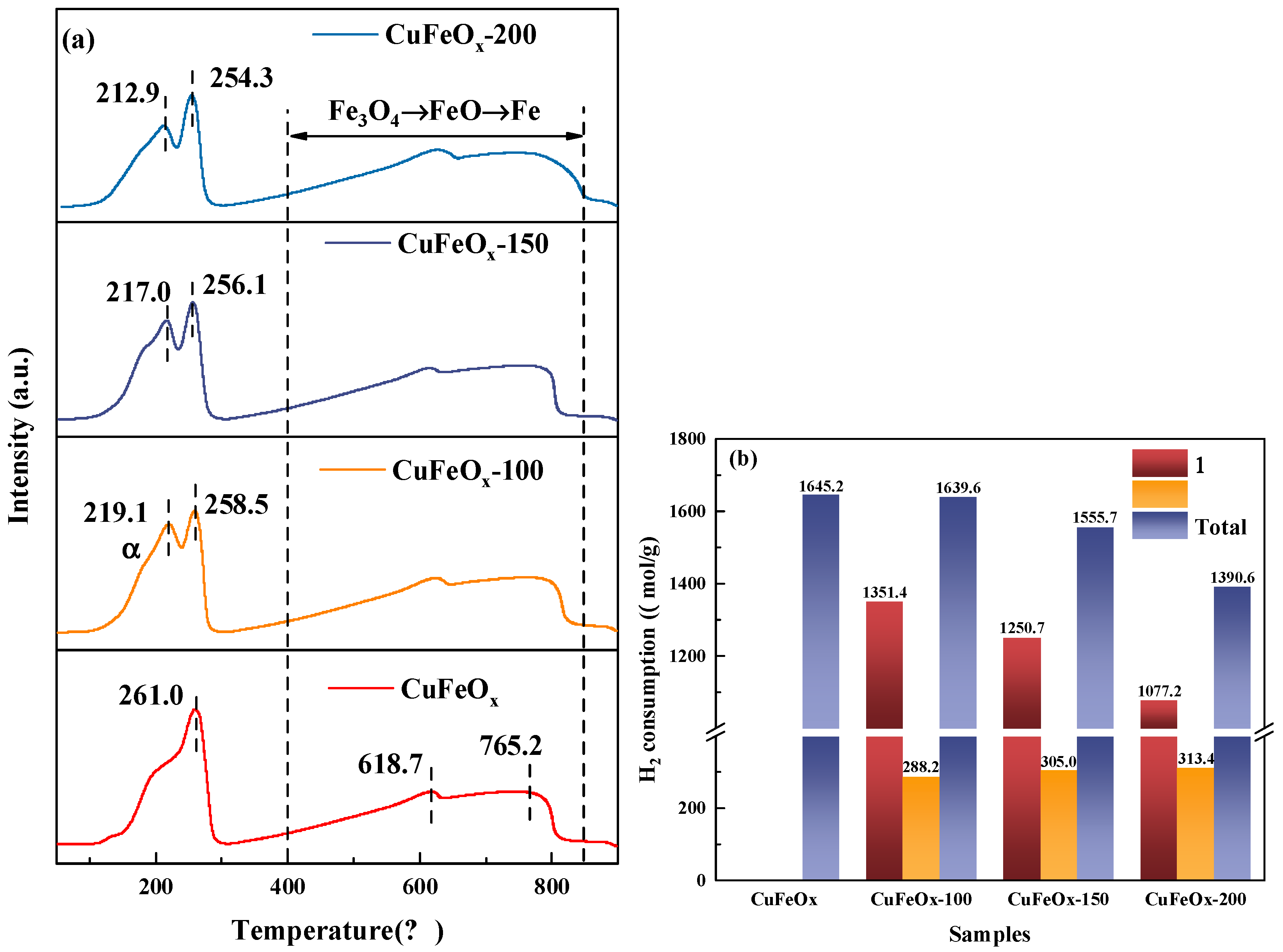
2.4. Redox Properties
2.5. Temperature-Programmed Studies
2.6. Reaction Intermediates Analysis
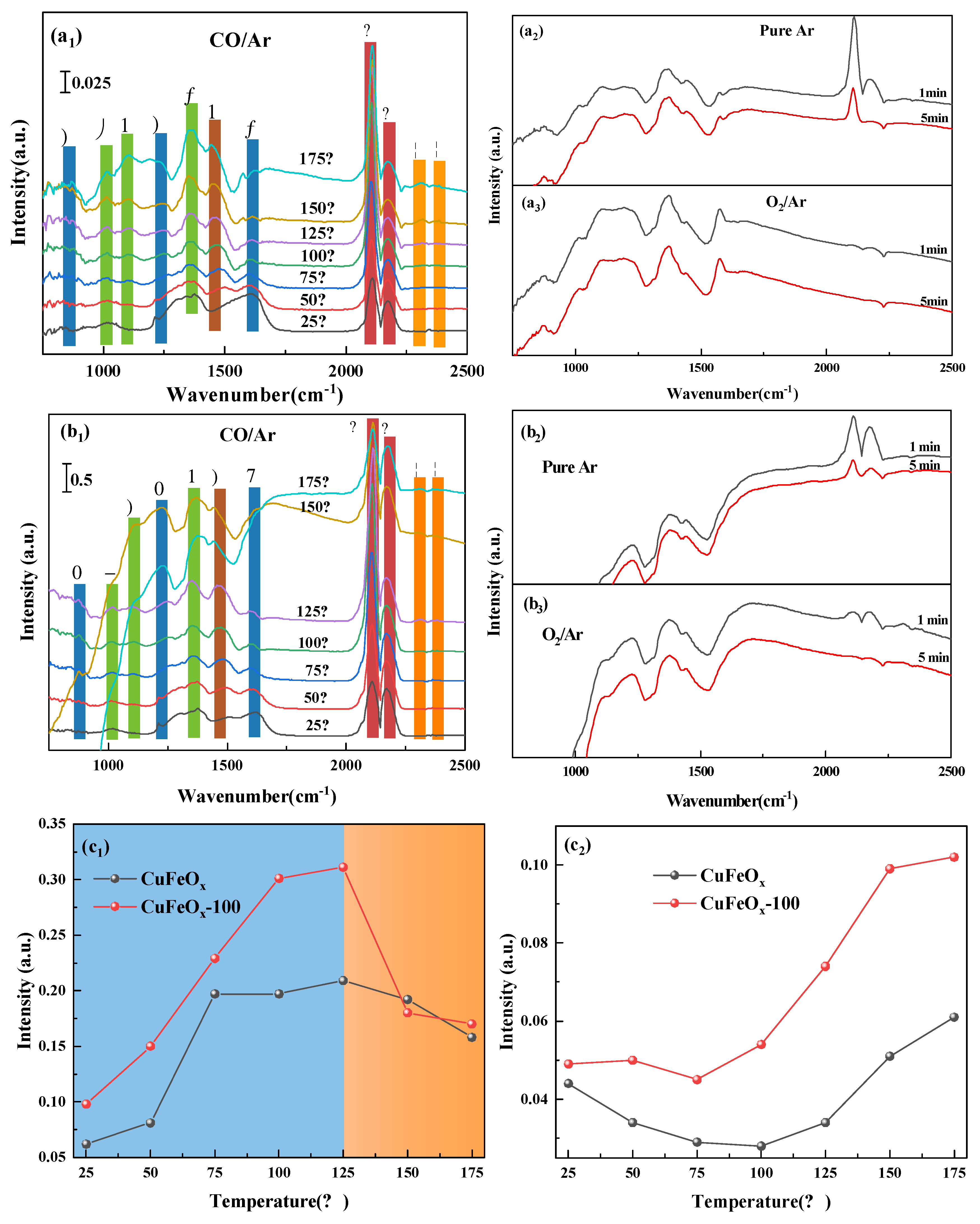
2.6.1. In Situ DRIFT Spectra of CO Adsorption, Ar and O2 Purging
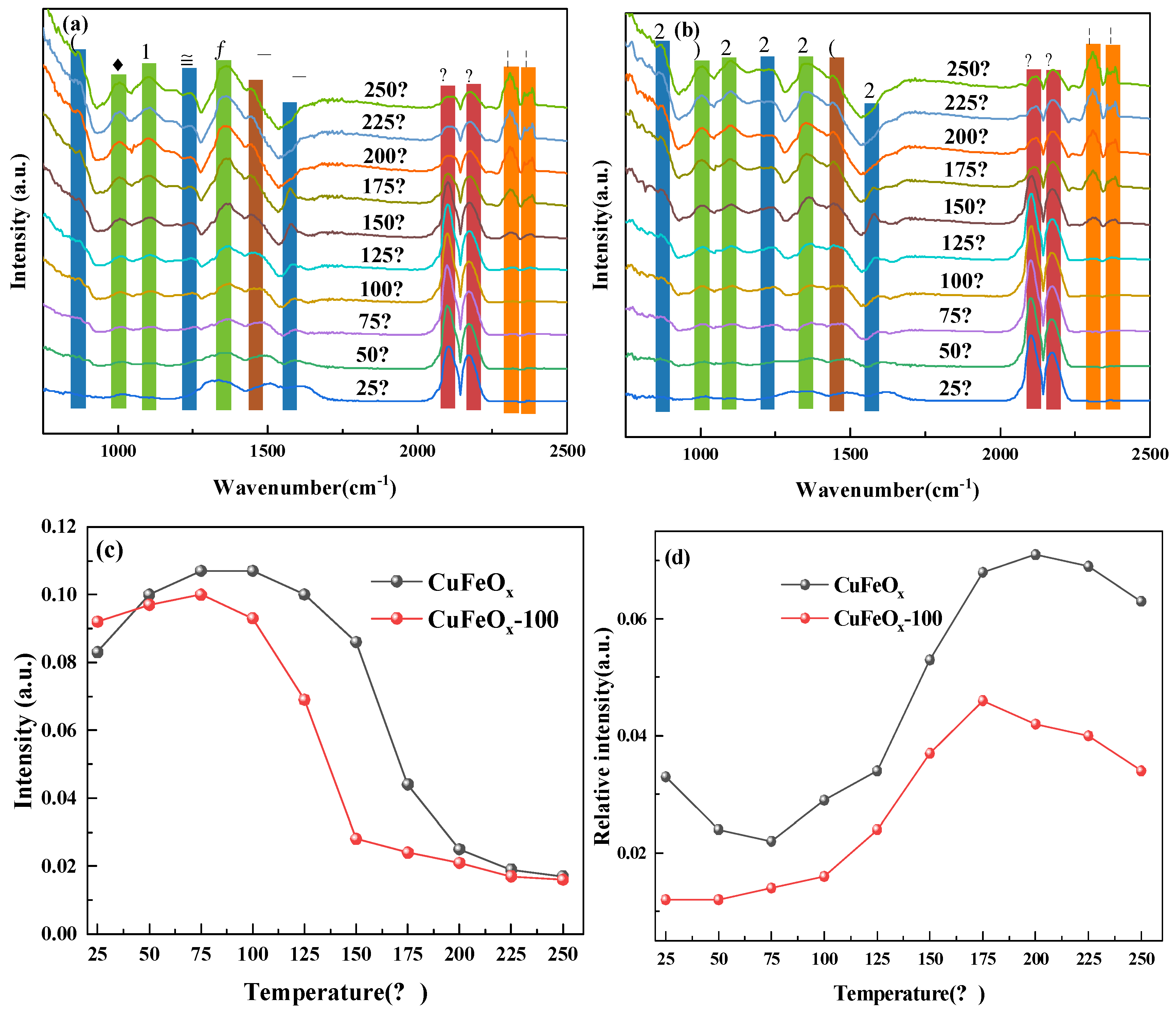
2.6.2. CO+O2 Coadsorption
3. Discussion





4. Materials and Methods
4.1. Preparation of Catalyst
4.2. Catalytic Performance Test
4.3. Catalyst Characterization
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shen, Z.H.; Xing, X.D.; Wang, S.X.; Zheng, Z.Y.; Lv, M. Low temperature CO oxidation from sintering flue gas on CuO-CeO2/AC-Fe catalyst. Catal. Today 2023, 423, 113988. [Google Scholar] [CrossRef]
- Wang, J.; Xing, Y.; Su, W.; Li, K.; Zhang, W. Bifunctional Mn-Cu-CeOx/γ-Al2O3 catalysts for low-temperature simultaneous removal of NOx and CO. Fuel 2022, 321, 124050. [Google Scholar] [CrossRef]
- Xu, J.C.; Wang, S.; Zhang, C.Y.; Zhang, J.; Xu, L.H.; Shi, Y.; Xu, G.Q.; Yao, S.L. Exploring the role of oxygen species on /3-and γ-MnO2 catalyst under in-plasma catalysis configuration at different temperature in CO oxidation using operand TPR-DRIFTS-MS. J. Alloys Compd. 2024, 978, 173462. [Google Scholar] [CrossRef]
- Waikar, J.; More, P. Oxygen deficient Ce doped CO supported on alumina catalyst for low-temperature CO oxidation in presence of H2O and SO2. Fuel 2023, 331, 125880. [Google Scholar] [CrossRef]
- Cai, J.Y.; Yu, Z.H.; Fan, X.; Li, J. Effect of TiO2 calcination pretreatment on the performance of Pt/TiO2 catalyst for CO Oxidation. Molecules 2022, 27, 3875. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.H.; Xing, X.D.; Wang, S.X.; Ren, S.; Lv, M.; Zheng, Z.Y.; Jiang, X. Removal of CO in flue gas by catalytic oxidation: A review. Z. Phys. Chem. 2024, 238. [Google Scholar] [CrossRef]
- Feng, C.L.; Liu, X.L.; Zhu, T.Y.; Tian, M.K. Catalytic oxidation of CO on noble metal-based catalysts. Environ. Sci. Pollut. Res. 2021, 28, 24847–24871. [Google Scholar] [CrossRef] [PubMed]
- Khalafallah, D.; Miao, J.; Zhi, M.; Hong, Z. Structuring graphene quantum dots anchored CuO for high-performance hybrid supercapacitors. J. Taiwan Inst. Chem. Eng. 2021, 122, 168–175. [Google Scholar] [CrossRef]
- Xu, L.L.; Yang, X.; Shi, Y.Y.; Chen, M.D.; Xue, Y.Y.; Wu, C.E.; Qiu, J.; Cheng, G.; Wang, N.; Xu, J.X.; et al. CO oxidation over the Cu2O/CuO hollow sphere heterojunction catalysts with enhanced low-temperature activities. Int. J. Hydrogen Energy 2023, 48, 24845–24859. [Google Scholar] [CrossRef]
- Cui, Y.; Song, H.K.; Shi, Y.Y.; Ge, P.X.; Chen, M.D.; Xu, L.L. Enhancing the low-temperature CO oxidation over CuO-based α-MnO2 nanowire catalysts. Nanomaterials 2022, 12, 2083. [Google Scholar] [CrossRef]
- Gong, S.W.; Zhu, G.Q.; Xie, X.Q.; Rao, F.; Zhu, L.J.; An, Y.R.; Shi, X.J.; Huang, Y.; Liu, P.; Hojamberdiev, M. Engineering the CuO/MnO2 interface with multivalent conversion for low-temperature and efficient CO oxidation. Appl. Surf. Sci. 2024, 652, 159326. [Google Scholar] [CrossRef]
- Bulavchenko, O.A.; Pochtar, A.A.; Gerasimov, E.Y.; Fedorov, A.V.; Chesalov, Y.A.; Saraev, A.A.; Yakovlev, V.A.; Kaichev, V.V. Chemical and texture promoters in Cu-Fe-Al oxide nanocomposite catalysts for combustion of solid fuel gasification products. Appl. Catal. A 2020, 590, 117364. [Google Scholar] [CrossRef]
- Wang, L.P.; Li, Q.; Liu, X.; Li, C.; Zhao, Z.Z.; Diao, S.T.; Cao, D.F.; Xiang, D.C.; Wu, C.N.; Liu, K. Improved CO-PROX selectivity of CuO/CeO2 catalysts by decorating with lanthanum via surface Cuξ+ redox site. Appl. Surf. Sci. 2024, 649, 159087. [Google Scholar] [CrossRef]
- Kerkar, R.D.; Salker, A.V. Highly active nano-composite of cobalt-copper-manganese oxides for room temperature CO oxidation. Appl. Nanosci. 2021, 11, 2861–2867. [Google Scholar] [CrossRef]
- Zhao, F.; Shi, Y.Y.; Xu, L.L.; Chen, M.D.; Xue, Y.Y.; Wu, C.E.; Qiu, J.; Cheng, G.; Xu, J.X.; Hu, X. Designing highly efficient Cu2O-CuO heterojunction CO oxidation catalysts: The roles of the support type and Cu2O-CuO interface effect. Nanomaterials 2022, 12, 3020. [Google Scholar] [CrossRef]
- Rezaei, P.; Rezaei, M.; Meshkani, F. Low temperature CO oxidation over mesoporous iron and copper mixed oxides nanopowders synthesized by a simple one-pot solid-state method. Process Saf. Environ. Prot. 2018, 119, 379–388. [Google Scholar] [CrossRef]
- Amini, E.; Rezaei, M. Preparation of mesoporous Fe-Cu mixed metal oxide nanopowder as active and stable catalyst for low-temperature CO oxidation. Chin. J. Catal. 2015, 36, 1711–1718. [Google Scholar] [CrossRef]
- Yeste, M.P.; Vidal, H.; García-Cabeza, A.L.; Hernández-Garrido, J.C.; Guerra, F.M.; Cifredo, G.A.; González-Leal, J.M.; Gatica, J.M. Low temperature prepared copper-iron mixed oxides for the selective CO oxidation in the presence of hydrogen. Appl. Catal. A 2018, 552, 58–69. [Google Scholar] [CrossRef]
- Shen, Z.H.; Xing, X.D.; She, Y.; Wang, S.X.; Lv, M.; Li, J.X.; Li, H.Z. Revealing of K and SO2 poisoning mechanism on CuCeOx catalyst for low-temperature CO oxidation. Chem. Eng. J. 2024, 485, 149373. [Google Scholar] [CrossRef]
- Shen, Z.H.; Xing, X.D.; Wang, S.X.; Lv, M.; Zheng, Z.Y.; Li, J.X.; Li, H.Z. High activity of CuO/#-Fe2O3 for low temperature CO oxidation: Effect of support crystal types in catalyst design. J. Energy Inst. 2023, 110, 101339. [Google Scholar]
- Wang, L.; Pu, C.; Xu, L.; Cai, Y.; Guo, Y.; Guo, Y.; Lu, G. Effect of supports over Pd/Fe2O3 on CO oxidation at low temperature. Fuel Process. Technol. 2017, 160, 152–157. [Google Scholar] [CrossRef]
- Wang, W.W.; Du, P.P.; Zou, S.H.; He, H.Y.; Wang, R.X.; Jin, Z.; Shi, S.; Huang, Y.Y.; Si, R.; Song, Q.S.; et al. Highly dispersed copper oxide clusters as active species in copper-ceria catalyst for preferential oxidation of carbon monoxide. ACS Catal. 2015, 5, 2088–2099. [Google Scholar] [CrossRef]
- Niu, L.; Liu, X.; Wen, X.; Yang, Y.; Xu, J.; Li, Y. Effect of potassium promoter on phase transformation during H2 pretreatment of a Fe2O3 Fischer Tropsch synthesis catalyst precursor. Catal. Today 2020, 343, 101–111. [Google Scholar] [CrossRef]
- Cam, T.S.; Omarov, S.O.; Chebanenko, M.I.; Sklyarova, A.S.; Nevedomskiy, V.N.; Popkov, V.I. One step closer to the low-temperature CO oxidation over non-noble CuO/CeO2 nanocatalyst: The effect of CuO loading. J. Environ. Chem. Eng. 2021, 9, 105373. [Google Scholar] [CrossRef]
- Khalafallah, D.; Zhi, M.; Hong, Z. Rational engineering of hierarchical mesoporous CuxFeySe battery-type electrodes for asymmetric hybrid supercapacitors. Ceram. Int. 2021, 47, 29081–29090. [Google Scholar] [CrossRef]
- Zhang, Z.H.; Fan, L.P.; Liao, W.Q.; Zhao, F.Y.; Tang, C.; Zhang, J.; Feng, M.; Lu, J.Q. Structure sensitivity of CuO in CO oxidation over CeO2-CuO/Cu2O catalysts. J. Catal. 2022, 405, 333–345. [Google Scholar] [CrossRef]
- Tang, X.Y.; Tariq, N.U.; Wang, J.C.; Cheng, Y.L.; Wang, Y.L.; Yuan, Z.G.; Liu, B.D. CuO/TiO2/Ti monolithic catalysts for low-temperature CO oxidation. ACS Appl. Nano Mater. 2023, 7, 809–817. [Google Scholar] [CrossRef]
- Gong, X.F.; Xu, J.C.; Zhang, T.T.; Sun, Y.; Fang, S.Y.; Li, N.; Zhu, J.L.; Wu, Z.L.; Li, J.; Gao, E.H.; et al. DRIFTS-MS investigation of low-temperature CO oxidation on Cu-doped manganese oxide prepared using nitrate aerosol decomposition. Molecules 2023, 28, 3511. [Google Scholar] [CrossRef]
- Xu, J.C.; Wu, Z.L.; Gao, E.R.; Zhu, J.L.; Yao, S.L.; Li, J. Revealing the role of oxygen vacancies on ?-MnO2 of different morphologies in CO oxidation using operando DRIFTS-MS. Appl. Surf. Sci. 2023, 618, 156643. [Google Scholar] [CrossRef]
- Shan, R.T.; Sheng, Z.T.; Hu, S.; Xiao, H.F.; Zhang, Y.H.; Zhang, J.H.; Wang, L.; Zhang, C.B.; Li, J.L. Enhancing oxidation reaction over Pt-MnO2 catalyst by activation of surface oxygen. J. Environ. Sci. 2023, 134, 117–125. [Google Scholar] [CrossRef]
- Teng, W.X.; Li, J.L.; Dai, X.Y.; Chen, Y.F.; Wu, H.M.; Liu, W.Z.; Ren, S.; Yang, J.; Liu, Q.C. Promoting the NH3-SCR performance of Fe2O3-TiO2 catalyst through regulation of the exposed crystal facets of Fe2O3. Appl. Surf. Sci. 2024, 647, 158938. [Google Scholar] [CrossRef]
- Wang, G.W.; Zhang, Y.K.; Lu, J.M.; Liu, Y.J.; Yang, M.; Peng, G.J.; Jia, L.J.; Wang, H.M.; Xia, F.T.; Zhang, Q.L. The improving effect of SO42- on environmental-friendly SO42-/a-Fe2O3 catalyst for NH3-SCR. J. Energy Inst. 2023, 110, 101347. [Google Scholar] [CrossRef]
- Li, X.Y.; Chen, J.; Lu, C.M.; Luo, G.Q.; Yao, H. Performance of Mo modified γ-Fe2O3 catalyst for selective catalytic reduction of NOx with ammonia: Presence of arsenic in flue gas. Fuel 2021, 294, 120552. [Google Scholar] [CrossRef]
- Zhang, Y.L.; Li, J.; Cai, J.Y.; Li, S.Y.; Fan, X.; Song, L.Y.; Guo, R.X.; Liu, J.S. Enhancement of low-temperature activity of γ-Fe2O3-modified V2O5-MoO3/TiO2 catalysts for selective catalytic reduction of NOx with NH3. J. Environ. Chem. Eng. 2024, 12, 112589. [Google Scholar] [CrossRef]
- Fu, Q.; Li, W.X.; Yao, Y.; Liu, H.; Su, H.Y.; Ma, D.; Gu, X.K.; Chen, L.; Wang, Z.; Zhang, H.J.S. Interface-confined ferrous centers for catalytic oxidation. Science 2010, 328, 1141–1144. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Qiao, B.; Liu, J.; Huang, Y.; Wang, A.; Li, L.; Zhang, W.; Allard, L.F.; Wang, X.; Zhang, T. Design of a highly active Ir/Fe(OH)x catalyst: Versatile application of Pt-group metals for the preferential oxidation of carbon monoxide. Angew. Chem., Int. Ed. 2012, 51, 2920–2924. [Google Scholar] [CrossRef] [PubMed]
- Gui, R.; Xiao, J.; Gao, Y.; Li, Y.; Zhu, T.; Wang, Q. Simultaneously achieving selective catalytic reduction of NOx with NH3 and catalytic oxida tion of CO with O2 over one finely optimized bifunctional catalyst Mn2Cu1Al1Ox at low temperatures. Appl. Catal., B 2022, 306, 121104. [Google Scholar] [CrossRef]
- Qiu, Z.H.; Guo, X.L.; Mao, J.X.; Zhou, R.X. Insights into the structure-performance relationship of CuOx-CeO2 catalysts for preferential oxidation of CO: Investigation on thermally induced copper migration process. Appl. Surf. Sci. 2022, 600, 154100. [Google Scholar] [CrossRef]
- Hou, X.L.; Zhao, Q.Y.; Liu, X.L.; Chong, M.B.; Cheng, D.G.; Chen, F.Q.; Zhan, X.L. CeO2 crystal plane engineering to modulate CuO-CeO2 interactions and modify the CO-PROX reaction pathway. Chem. Eng. J. 2024, 480, 147982. [Google Scholar] [CrossRef]
- Zeng, Y.; Rong, W.; Zhang, S.; Zhong, Q.; Zhong, Z. Ti species regulated CuO/CeO2 catalyst for efficient simultaneously NH3-SCR denitration and CO oxidation under oxygen-rich conditions. Fuel 2024, 358, 130277. [Google Scholar] [CrossRef]
- Wang, L.; Peng, H.; Shi, S.L.; Hu, Z.; Zhang, B.Z.; Ding, S.M.; Wang, S.H.; Chen, C. Metal-organic framework derived hollow CuO/CeO2 nano-sphere: To expose more highly dispersed Cu-O-Ce interface for enhancing preferential CO oxidation. Appl. Surf. Sci. 2022, 573, 151611. [Google Scholar] [CrossRef]
- Lu, J.C.; Wang, J.; Zou, Q.; He, D.D.; Zhang, L.M.; Xu, Z.Z.; He, S.F.; Luo, Y.M. Unravelling the nature of the active species as well as the doping effect over Cu/Ce-based catalyst for carbon monoxide preferential oxidation. ACS Catal. 2019, 9, 2177–2195. [Google Scholar] [CrossRef]
- Liu, Q.; Mi, J.; Chen, X.; Wang, S.; Chen, J.; Li, J. Effects of phosphorus modification on the catalytic properties and performance of CuCeZr mixed metal catalyst for simultaneous removal of CO and NOx. Chem. Eng. J. 2021, 423, 130228. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Specific Surface Area (m2/g) | Average Pore Diameter (nm) | Total Pore Volume (cm3/g) |
|---|---|---|---|
| Fe2O3 | 9.85 | 14.96 | 0.037 |
| CuFeOx | 11.06 | 14.98 | 0.041 |
| CuFeOx-100 | 12.60 | 13.36 | 0.042 |
| Sample | Cu 2p (%) | Fe 2p (%) | O 1s (%) | |||
|---|---|---|---|---|---|---|
| Cu+ | Cu2+ | Fe2+ | Fe3+ | Oα | Oβ | |
| CuFeOx | 17.5 | 82.5 | / | 100 | 30.1 | 69.9 |
| CuFeOx-100 | 10.2 | 89.8 | 17.9 | 82.1 | 39.6 | 60.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, Z.; Xing, X.; She, Y.; Meng, H.; Niu, W.; Ren, S. Unveiling the Promoting Mechanism of H2 Activation on CuFeOx Catalyst for Low-Temperature CO Oxidation. Molecules 2024, 29, 3347. https://doi.org/10.3390/molecules29143347
Shen Z, Xing X, She Y, Meng H, Niu W, Ren S. Unveiling the Promoting Mechanism of H2 Activation on CuFeOx Catalyst for Low-Temperature CO Oxidation. Molecules. 2024; 29(14):3347. https://doi.org/10.3390/molecules29143347
Chicago/Turabian StyleShen, Zhenghua, Xiangdong Xing, Yuan She, Hao Meng, Wenkang Niu, and Shan Ren. 2024. "Unveiling the Promoting Mechanism of H2 Activation on CuFeOx Catalyst for Low-Temperature CO Oxidation" Molecules 29, no. 14: 3347. https://doi.org/10.3390/molecules29143347