Primed for Interactions: Investigating the Primed Substrate Channel of the Proteasome for Improved Molecular Engagement


Abstract
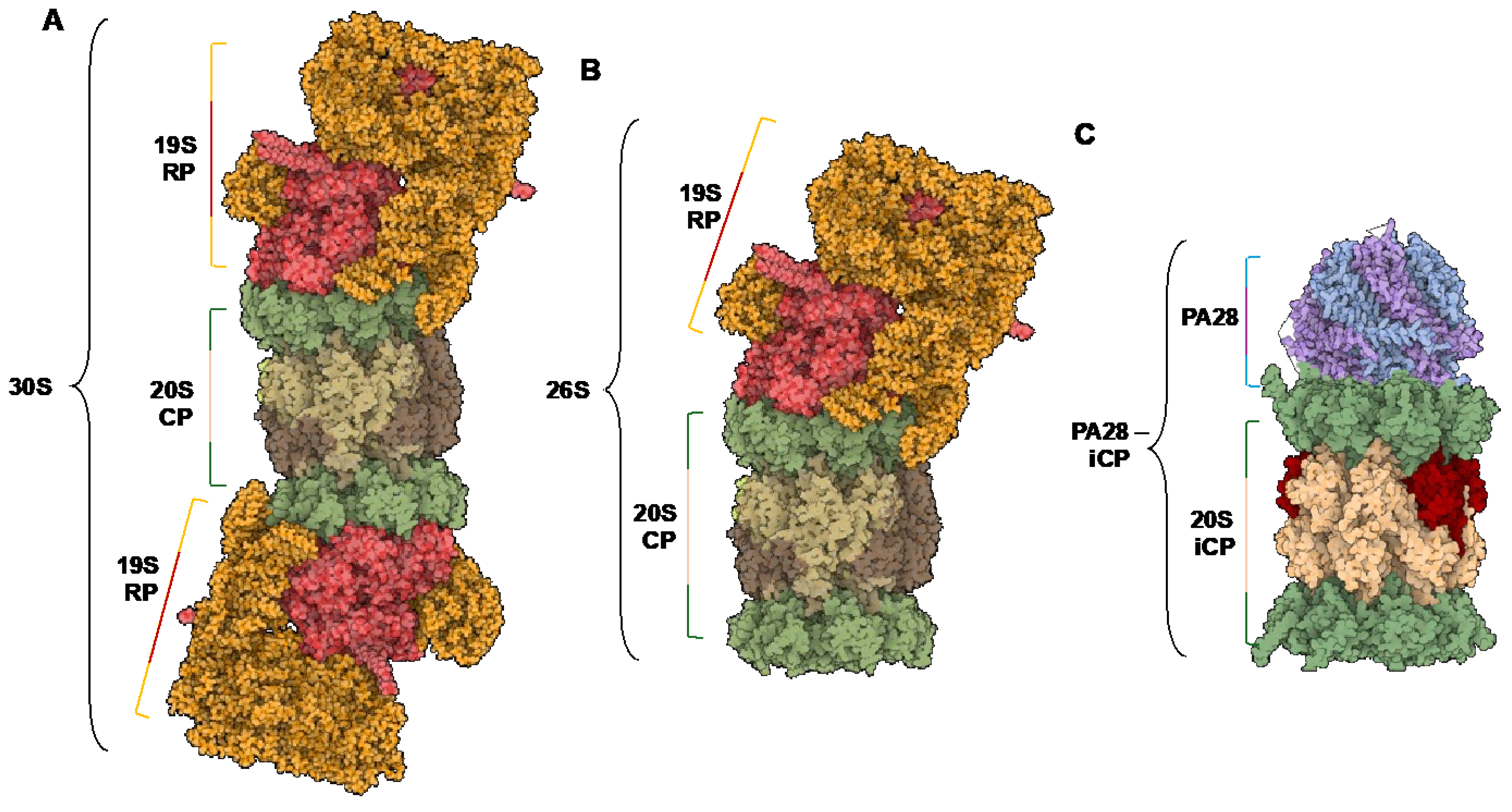
:1. Introduction to the Proteasome’s Structure
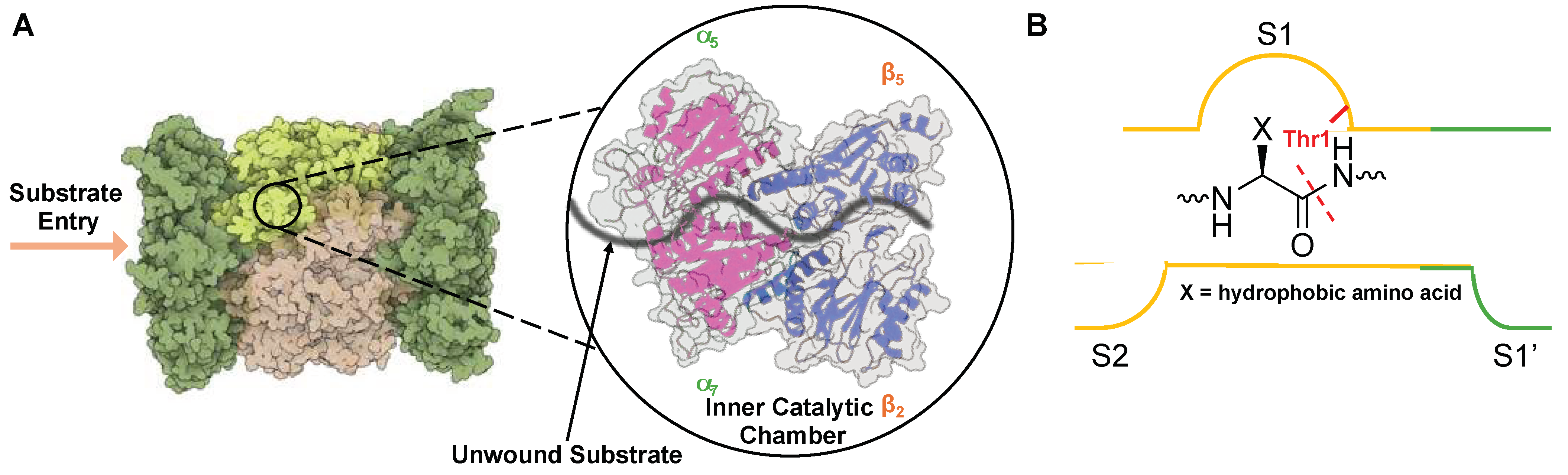
1.1. 20S Core Particle
1.2. Ubiquitin Proteasome System Degradation
1.3. Endogenous Activators
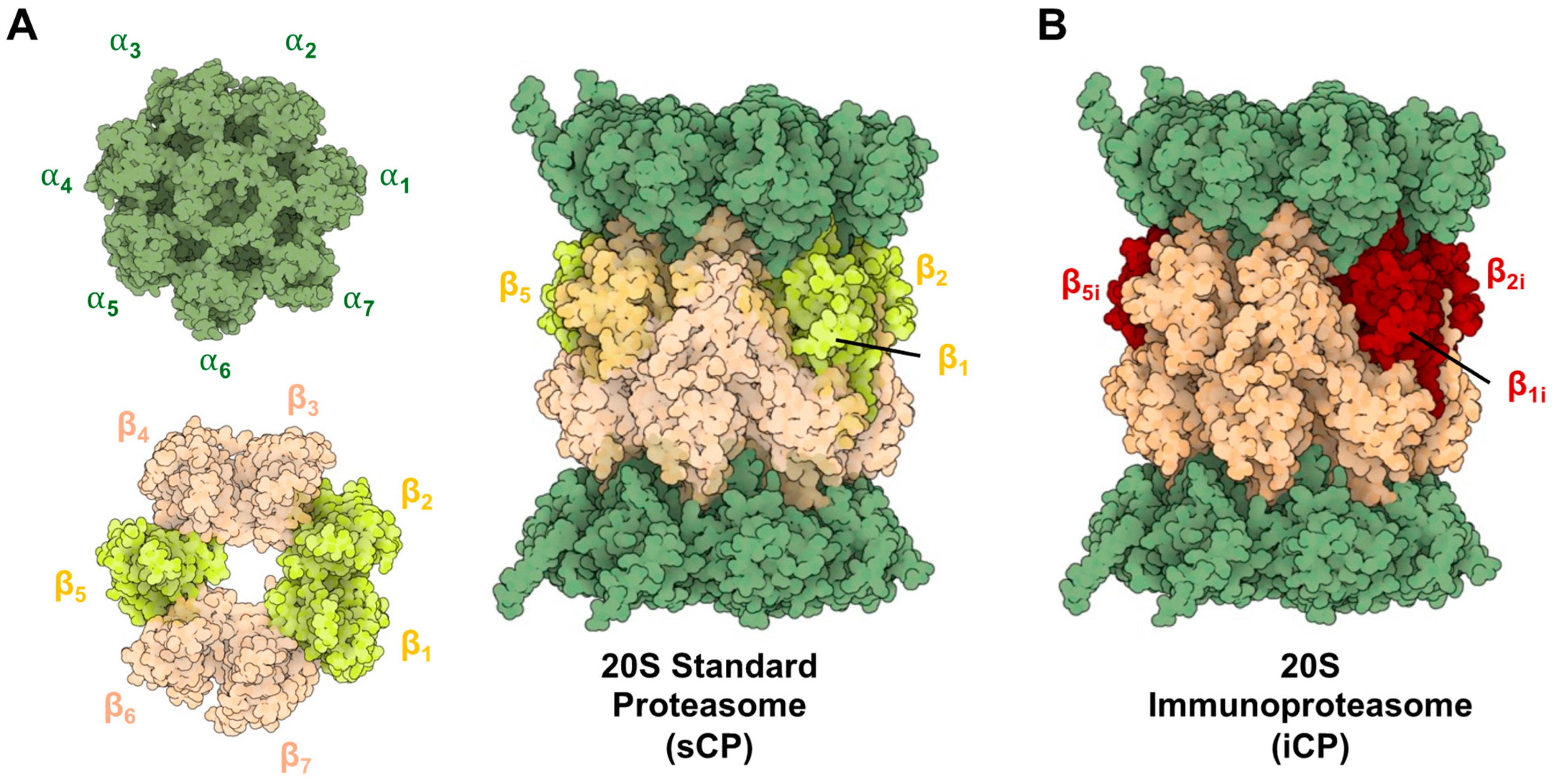
2. Structural Differences in Core Particles
2.1. Structural Overview
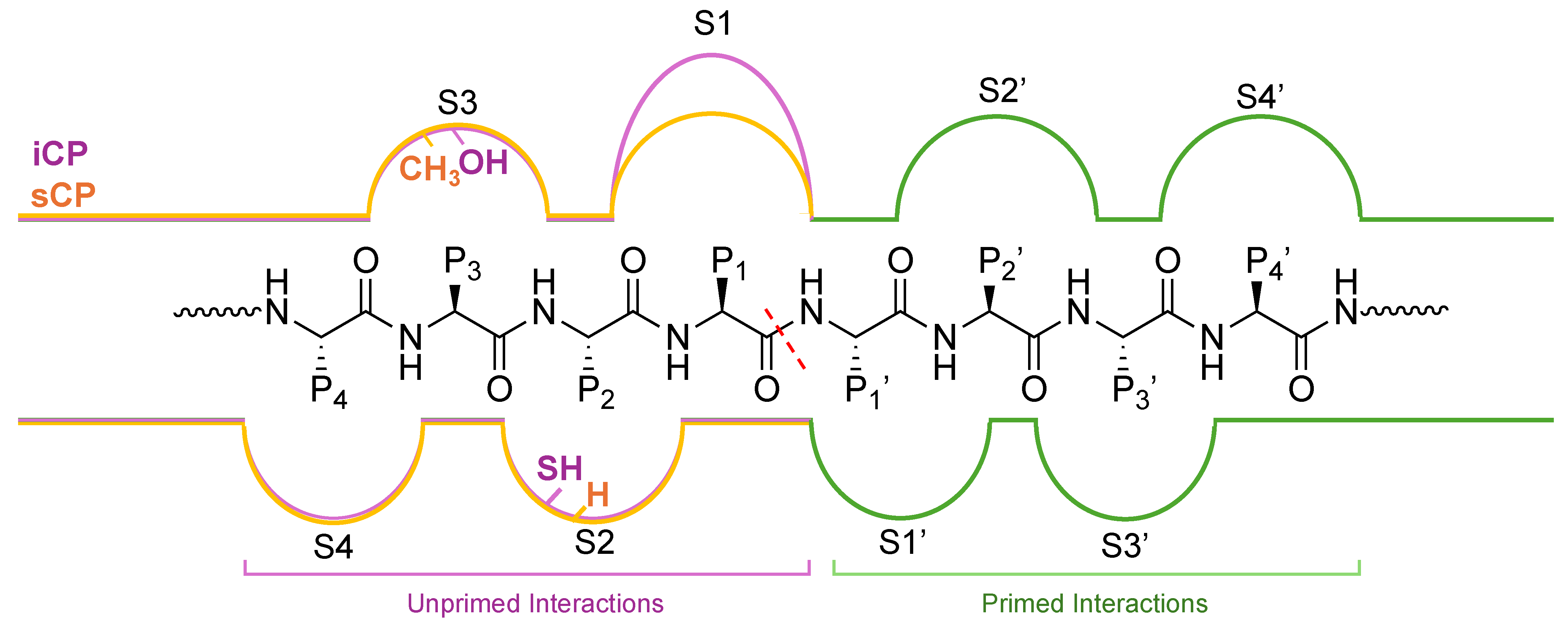
2.2. Substrate Channel Preferences
3. Rational Design of Small Molecule Proteasome Interactors
3.1. Non-Primed Substrate Binders
3.1.1. Boronic Acids
3.1.2. Epoxides
3.2. Primed Substrate Binders
3.2.1. Natural Products
3.2.2. Peptide Based Interactors
3.3. Primed Substrate Chemical Probe Interactors
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Groll, M.; Ditzel, L.; Löwe, J.; Stock, D.; Bochtler, M.; Bartunik, H.D.; Huber, R. Structure of 20S Proteasome from Yeast at 2.4Å Resolution. Nature 1997, 386, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Coux, O.; Tanaka, K.; Goldberg, A.L. Structure and functions of the 20S and 26S proteasomes. Annu. Rev. Biochem. 1996, 65, 801–847. [Google Scholar] [CrossRef] [PubMed]
- Boes, B.; Hengel, H.; Ruppert, T.; Multhaup, G.; Koszinowski, U.H.; Kloetzel, P.M. Interferon Gamma Stimulation Modulates the Proteolytic Activity and Cleavage Site Preference of 20S Mouse Proteasomes. J. Exp. Med. 1994, 179, 901–909. [Google Scholar] [CrossRef] [PubMed]
- Myers, N.; Olender, T.; Savidor, A.; Levin, Y.; Reuven, N.; Shaul, Y. The Disordered Landscape of the 20S Proteasome Substrates Reveals Tight Association with Phase Separated Granules. Proteomics 2018, 18, 1800076. [Google Scholar] [CrossRef] [PubMed]
- Chondrogianni, N.; Georgila, K.; Kourtis, N.; Tavernarakis, N.; Gonos, E.S. 20S Proteasome Activation Promotes Life Span Extension and Resistance to Proteotoxicity in Caenorhabditis Elegans. FASEB J. 2015, 29, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Arrigo, A.-P.; Tanaka, K.; Goldberg, A.L.; Welch, W.J. Identity of the 19S “prosome” Particle with the Large Multifunctional Protease Complex of Mammalian Cells (the Proteasome). Nature 1988, 331, 192–194. [Google Scholar] [CrossRef]
- Davies, K.J.A. Degradation of Oxidized Proteins by the 20S Proteasome. Biochimie 2001, 83, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Suskiewicz, M.J.; Sussman, J.L.; Silman, I.; Shaul, Y. Context-Dependent Resistance to Proteolysis of Intrinsically Disordered Proteins. Protein Sci. 2011, 20, 1285–1297. [Google Scholar] [CrossRef]
- Tonoki, A.; Kuranaga, E.; Tomioka, T.; Hamazaki, J.; Murata, S.; Tanaka, K.; Miura, M. Genetic Evidence Linking Age-Dependent Attenuation of the 26S Proteasome with the Aging Process. Mol. Cell. Biol. 2009, 29, 1095–1106. [Google Scholar] [CrossRef]
- Shringarpure, R.; Grune, T.; Davies, K.J.A. Protein Oxidation and 20S Proteasome-Dependent Proteolysis in Mammalian Cells. Cell. Mol. Life Sci. CMLS 2001, 58, 1442–1450. [Google Scholar] [CrossRef]
- Arellano-Garcia, M.E.; Misuno, K.; Tran, S.D.; Hu, S. Interferon-γ Induces Immunoproteasomes and the Presentation of MHC I-Associated Peptides on Human Salivary Gland Cells. PLoS ONE 2014, 9, e102878. [Google Scholar] [CrossRef] [PubMed]
- Aki, M.; Shimbara, N.; Takashina, M.; Akiyama, K.; Kagawa, S.; Tamura, T.; Tanahashi, N.; Yoshimura, T.; Tanaka, K.; Ichihara, A. Interferon-γ Induces Different Subunit Organizations and Functional Diversity of Proteasomes1. J. Biochem. 1994, 115, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Waters, J.B.; Früh, K.; Peterson, P.A. Proteasomes Are Regulated by Interferon Gamma: Implications for Antigen Processing. Proc. Natl. Acad. Sci. USA 1992, 89, 4928–4932. [Google Scholar] [CrossRef] [PubMed]
- Ferrington, D.A.; Gregerson, D.S. Immunoproteasomes: Structure, Function, and Antigen Presentation. Prog. Mol. Biol. Transl. Sci. 2012, 109, 75–112. [Google Scholar] [CrossRef]
- Rock, K.L.; Farfán-Arribas, D.J.; Shen, L. Proteases in MHC Class I Presentation and Cross-Presentation. J. Immunol. Baltim. 2010, 184, 9–15. [Google Scholar] [CrossRef]
- Driscoll, J.J., II; Rana, P.S.; Malek, E.; Ignatz-Hoover, J.J. An Immunoproteasome Activator That Increases MHC Class I Antigen Presentation to Enhance Anti-Tumor Immunity. Blood 2023, 142 (Suppl. S1), 3642. [Google Scholar] [CrossRef]
- McCarthy, M.K.; Weinberg, J.B. The Immunoproteasome and Viral Infection: A Complex Regulator of Inflammation. Front. Microbiol. 2015, 6, 21. [Google Scholar] [CrossRef]
- Immunoproteasome Assembly: Cooperative Incorporation of Interferon γ (IFN-γ)–inducible Subunits|Journal of Experimental Medicine|Rockefeller University Press. Available online: https://rupress.org/jem/article/187/1/97/25375/Immunoproteasome-Assembly-Cooperative (accessed on 24 April 2024).
- Greene, E.R.; Dong, K.C.; Martin, A. Understanding the 26S Proteasome Molecular Machine from a Structural and Conformational Dynamics Perspective. Curr. Opin. Struct. Biol. 2020, 61, 33–41. [Google Scholar] [CrossRef]
- Davis, C.; Spaller, B.L.; Matouschek, A. Mechanisms of Substrate Recognition by the 26S Proteasome. Curr. Opin. Struct. Biol. 2021, 67, 161–169. [Google Scholar] [CrossRef]
- Collins, G.A.; Goldberg, A.L. The Logic of the 26S Proteasome. Cell 2017, 169, 792–806. [Google Scholar] [CrossRef]
- Bard, J.A.M.; Goodall, E.A.; Greene, E.R.; Jonsson, E.; Dong, K.C.; Martin, A. Structure and Function of the 26S Proteasome. Annu. Rev. Biochem. 2018, 87, 697–724. [Google Scholar] [CrossRef] [PubMed]
- Ziv, I.; Matiuhin, Y.; Kirkpatrick, D.S.; Erpapazoglou, Z.; Leon, S.; Pantazopoulou, M.; Kim, W.; Gygi, S.P.; Haguenauer-Tsapis, R.; Reis, N.; et al. A Perturbed Ubiquitin Landscape Distinguishes Between Ubiquitin in Trafficking and in Proteolysis. Mol. Cell. Proteom. 2011, 10, M111.009753. [Google Scholar] [CrossRef] [PubMed]
- Clague, M.J.; Heride, C.; Urbé, S. The Demographics of the Ubiquitin System. Trends Cell Biol. 2015, 25, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Longworth, J.; Dittmar, G. Assessment of Ubiquitin Chain Topology by Targeted Mass Spectrometry. In Mass Spectrometry of Proteins: Methods and Protocols; Evans, C.A., Wright, P.C., Noirel, J., Eds.; Springer: New York, NY, USA, 2019; pp. 25–34. [Google Scholar] [CrossRef]
- Neklesa, T.K.; Winkler, J.D.; Crews, C.M. Targeted Protein Degradation by PROTACs. Pharmacol. Ther. 2017, 174, 138–144. [Google Scholar] [CrossRef]
- Lu, J.; Qian, Y.; Altieri, M.; Dong, H.; Wang, J.; Raina, K.; Hines, J.; Winkler, J.D.; Crew, A.P.; Coleman, K.; et al. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem. Biol. 2015, 22, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Ali, E.M.H.; Loy, C.A.; Trader, D.J. ByeTAC: Bypassing an E3 Ligase for Targeted Protein Degradation. bioRxiv 2024, bioRxiv:2024.01.20.576376. [Google Scholar] [CrossRef]
- Rosenzweig, R.; Osmulski, P.A.; Gaczynska, M.; Glickman, M.H. The Central Unit within the 19S Regulatory Particle of the Proteasome. Nat. Struct. Mol. Biol. 2008, 15, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Ehlinger, A.; Walters, K.J. Structural Insights into Proteasome Activation by the 19S Regulatory Particle. Biochemistry 2013, 52, 3618–3628. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Fonts, K.; Davis, C.; Tomita, T.; Elsasser, S.; Nager, A.R.; Shi, Y.; Finley, D.; Matouschek, A. The Proteasome 19S Cap and Its Ubiquitin Receptors Provide a Versatile Recognition Platform for Substrates. Nat. Commun. 2020, 11, 477. [Google Scholar] [CrossRef]
- Shi, Y.; Chen, X.; Elsasser, S.; Stocks, B.B.; Tian, G.; Lee, B.-H.; Shi, Y.; Zhang, N.; de Poot, S.A.H.; Tuebing, F.; et al. Rpn1 Provides Adjacent Receptor Sites for Substrate Binding and Deubiquitination by the Proteasome. Science 2016, 351, aad9421. [Google Scholar] [CrossRef]
- Van Nocker, S.; Sadis, S.; Rubin, D.M.; Glickman, M.; Fu, H.; Coux, O.; Wefes, I.; Finley, D.; Vierstra, R.D. The Multiubiquitin-Chain-Binding Protein Mcb1 Is a Component of the 26S Proteasome in Saccharomyces Cerevisiae and Plays a Nonessential, Substrate-Specific Role in Protein Turnover. Mol. Cell. Biol. 1996, 16, 6020–6028. [Google Scholar] [CrossRef] [PubMed]
- Husnjak, K.; Elsasser, S.; Zhang, N.; Chen, X.; Randles, L.; Shi, Y.; Hofmann, K.; Walters, K.J.; Finley, D.; Dikic, I. Proteasome Subunit Rpn13 Is a Novel Ubiquitin Receptor. Nature 2008, 453, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Mulder, M.P.C.; Witting, K.; Berlin, I.; Pruneda, J.N.; Wu, K.-P.; Chang, J.-G.; Merkx, R.; Bialas, J.; Groettrup, M.; Vertegaal, A.C.O.; et al. A Cascading Activity-Based Probe Sequentially Targets E1–E2–E3 Ubiquitin Enzymes. Nat. Chem. Biol. 2016, 12, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Scheffner, M.; Nuber, U.; Huibregtse, J.M. Protein Ubiquitination Involving an E1–E2–E3 Enzyme Ubiquitin Thioester Cascade. Nature 1995, 373, 81–83. [Google Scholar] [CrossRef] [PubMed]
- Kuehn, L.; Dahlmann, B. Structural and Functional Properties of Proteasome Activator PA28. Mol. Biol. Rep. 1997, 24, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, Y.; Xu, C.; Chen, K.; Zhao, Q.; Wang, S.; Yin, Y.; Peng, C.; Ding, Z.; Cong, Y. Cryo-EM of Mammalian PA28αβ-iCP Immunoproteasome Reveals a Distinct Mechanism of Proteasome Activation by PA28αβ. Nat. Commun. 2021, 12, 739. [Google Scholar] [CrossRef] [PubMed]
- Cascio, P. PA28αβ: The Enigmatic Magic Ring of the Proteasome? Biomolecules 2014, 4, 566–584. [Google Scholar] [CrossRef] [PubMed]
- Ahn, K.; Erlander, M.; Leturcq, D.; Peterson, P.A.; Früh, K.; Yang, Y. In Vivo Characterization of the Proteasome Regulator PA28. J. Biol. Chem. 1996, 271, 18237–18242. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, T.; Baumeister, W. Conformational Constraints in Protein Degradation by the 20S Proteasome. Nat. Struct. Biol. 1995, 2, 199–204. [Google Scholar] [CrossRef]
- Osmulski, P.A.; Gaczynska, M. Nanoenzymology of the 20S Proteasome: Proteasomal Actions Are Controlled by the Allosteric Transition. Biochemistry 2002, 41, 7047–7053. [Google Scholar] [CrossRef]
- Unno, M.; Mizushima, T.; Morimoto, Y.; Tomisugi, Y.; Tanaka, K.; Yasuoka, N.; Tsukihara, T. The Structure of the Mammalian 20S Proteasome at 2.75 Å Resolution. Structure 2002, 10, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K. The Proteasome: Overview of Structure and Functions. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2009, 85, 12–36. [Google Scholar] [CrossRef] [PubMed]
- Arendt, C.S.; Hochstrasser, M. Identification of the Yeast 20S Proteasome Catalytic Centers and Subunit Interactions Required for Active-Site Formation. Proc. Natl. Acad. Sci. USA 1997, 94, 7156–7161. [Google Scholar] [CrossRef] [PubMed]
- Groll, M.; Heinemeyer, W.; Jäger, S.; Ullrich, T.; Bochtler, M.; Wolf, D.H.; Huber, R. The Catalytic Sites of 20S Proteasomes and Their Role in Subunit Maturation: A Mutational and Crystallographic Study. Proc. Natl. Acad. Sci. USA 1999, 96, 10976–10983. [Google Scholar] [CrossRef] [PubMed]
- Huber, E.M.; Heinemeyer, W.; Li, X.; Arendt, C.S.; Hochstrasser, M.; Groll, M. A Unified Mechanism for Proteolysis and Autocatalytic Activation in the 20S Proteasome. Nat. Commun. 2016, 7, 10900. [Google Scholar] [CrossRef] [PubMed]
- Osmulski, P.A.; Hochstrasser, M.; Gaczynska, M. A Tetrahedral Transition State at the Active Sites of the 20S Proteasome Is Coupled to Opening of the α-Ring Channel. Structure 2009, 17, 1137–1147. [Google Scholar] [CrossRef] [PubMed]
- Sahu, I.; Mali, S.M.; Sulkshane, P.; Rozenberg, A.; Xu, C.; Morag, R.; Sahoo, M.P.; Singh, S.K.; Ding, Z.; Wang, Y.; et al. Signature Activities of 20S Proteasome Include Degradation of the Ubiquitin-Tag with the Protein under Hypoxia. bioRxiv 2019, bioRxiv:2019.12.20.883942. [Google Scholar] [CrossRef]
- Dick, T.P.; Nussbaum, A.K.; Deeg, M.; Heinemeyer, W.; Groll, M.; Schirle, M.; Keilholz, W.; Stevanović, S.; Wolf, D.H.; Huber, R.; et al. Contribution of Proteasomal β-Subunits to the Cleavage of Peptide Substrates Analyzed with Yeast Mutants. J. Biol. Chem. 1998, 273, 25637–25646. [Google Scholar] [CrossRef] [PubMed]
- Heinemeyer, W.; Fischer, M.; Krimmer, T.; Stachon, U.; Wolf, D.H. The Active Sites of the Eukaryotic 20 S Proteasome and Their Involvement in Subunit Precursor Processing. J. Biol. Chem. 1997, 272, 25200–25209. [Google Scholar] [CrossRef]
- Huber, E.M.; Basler, M.; Schwab, R.; Heinemeyer, W.; Kirk, C.J.; Groettrup, M.; Groll, M. Immuno- and Constitutive Proteasome Crystal Structures Reveal Differences in Substrate and Inhibitor Specificity. Cell 2012, 148, 727–738. [Google Scholar] [CrossRef]
- Groll, M.; Larionov, O.V.; Huber, R.; de Meijere, A. Inhibitor-Binding Mode of Homobelactosin C to Proteasomes: New Insights into Class I MHC Ligand Generation. Proc. Natl. Acad. Sci. USA 2006, 103, 4576–4579. [Google Scholar] [CrossRef] [PubMed]
- Cardozo, C.; Kohanski, R.A. Altered Properties of the Branched Chain Amino Acid-Preferring Activity Contribute to Increased Cleavages after Branched Chain Residues by the “Immunoproteasome”. J. Biol. Chem. 1998, 273, 16764–16770. [Google Scholar] [CrossRef] [PubMed]
- Orlowski, M.; Cardozo, C.; Michaud, C. Evidence for the Presence of Five Distinct Proteolytic Components in the Pituitary Multicatalytic Proteinase Complex. Properties of Two Components Cleaving Bonds on the Carboxyl Side of Branched Chain and Small Neutral Amino Acids. Biochemistry 1993, 32, 1563–1572. [Google Scholar] [CrossRef] [PubMed]
- Romero, P.; Corradin, G.; Luescher, I.F.; Maryanski, J.L. H-2Kd-Restricted Antigenic Peptides Share a Simple Binding Motif. J. Exp. Med. 1991, 174, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Full Article: Trial Watch: Proteasomal Inhibitors for Anticancer Therapy. Available online: https://www.tandfonline.com/doi/full/10.4161/23723556.2014.974463 (accessed on 9 April 2024).
- Cromm, P.M.; Crews, C.M. The Proteasome in Modern Drug Discovery: Second Life of a Highly Valuable Drug Target. ACS Cent. Sci. 2017, 3, 830–838. [Google Scholar] [CrossRef]
- Kisselev, A.F.; Goldberg, A.L. Proteasome Inhibitors: From Research Tools to Drug Candidates. Chem. Biol. 2001, 8, 739–758. [Google Scholar] [CrossRef] [PubMed]
- Dou, Q.; Zonder, J. Overview of Proteasome Inhibitor-Based Anti-Cancer Therapies: Perspective on Bortezomib and Second Generation Proteasome Inhibitors versus Future Generation Inhibitors of Ubiquitin-Proteasome System. Curr. Cancer Drug Targets 2014, 14, 517–536. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Frezza, M.; Schmitt, S.; Kanwar, J.; Dou, Q.P. Bortezomib as the First Proteasome Inhibitor Anticancer Drug: Current Status and Future Perspectives. Curr. Cancer Drug Targets 2011, 11, 239–253. [Google Scholar] [CrossRef] [PubMed]
- 21602_Velcade_Approv.Pdf. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2003/21602_Velcade_Approv.pdf (accessed on 15 July 2024).
- Borissenko, L.; Groll, M. 20S Proteasome and Its Inhibitors: Crystallographic Knowledge for Drug Development. Chem. Rev. 2007, 107, 687–717. [Google Scholar] [CrossRef]
- Demo, S.D.; Kirk, C.J.; Aujay, M.A.; Buchholz, T.J.; Dajee, M.; Ho, M.N.; Jiang, J.; Laidig, G.J.; Lewis, E.R.; Parlati, F.; et al. Antitumor Activity of PR-171, a Novel Irreversible Inhibitor of the Proteasome. Cancer Res. 2007, 67, 6383–6391. [Google Scholar] [CrossRef]
- Schrader, J.; Henneberg, F.; Mata, R.A.; Tittmann, K.; Schneider, T.R.; Stark, H.; Bourenkov, G.; Chari, A. The Inhibition Mechanism of Human 20S Proteasomes Enables Next-Generation Inhibitor Design. Science 2016, 353, 594–598. [Google Scholar] [CrossRef] [PubMed]
- Ruschak, A.M.; Slassi, M.; Kay, L.E.; Schimmer, A.D. Novel Proteasome Inhibitors to Overcome Bortezomib Resistance. JNCI J. Natl. Cancer Inst. 2011, 103, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- 208462_Ninlaro_Approv.Pdf. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/208462Orig1s000Approv.pdf (accessed on 15 July 2024).
- Kupperman, E.; Lee, E.C.; Cao, Y.; Bannerman, B.; Fitzgerald, M.; Berger, A.; Yu, J.; Yang, Y.; Hales, P.; Bruzzese, F.; et al. Evaluation of the Proteasome Inhibitor MLN9708 in Preclinical Models of Human Cancer. Cancer Res. 2010, 70, 1970–1980. [Google Scholar] [CrossRef] [PubMed]
- Offidani, M.; Corvatta, L.; Caraffa, P.; Gentili, S.; Maracci, L.; Leoni, P. An Evidence-Based Review of Ixazomib Citrate and Its Potential in the Treatment of Newly Diagnosed Multiple Myeloma. OncoTargets Ther. 2014, 7, 1793–1800. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Kwok, B.H.; Sin, N.; Crews, C.M. Eponemycin Exerts Its Antitumor Effect through the Inhibition of Proteasome Function. Cancer Res. 1999, 59, 2798–2801. [Google Scholar] [PubMed]
- Meng, L.; Mohan, R.; Kwok, B.H.B.; Elofsson, M.; Sin, N.; Crews, C.M. Epoxomicin, a Potent and Selective Proteasome Inhibitor, Exhibits in Vivo Antiinflammatory Activity. Proc. Natl. Acad. Sci. USA 1999, 96, 10403–10408. [Google Scholar] [CrossRef] [PubMed]
- Groll, M.; Kim, K.B.; Kairies, N.; Huber, R.; Crews, C.M. Crystal Structure of Epoxomicin:20S Proteasome Reveals a Molecular Basis for Selectivity of α′,β′-Epoxyketone Proteasome Inhibitors. J. Am. Chem. Soc. 2000, 122, 1237–1238. [Google Scholar] [CrossRef]
- Crawford, L.J.; Walker, B.; Irvine, A.E. Proteasome Inhibitors in Cancer Therapy. J. Cell Commun. Signal. 2011, 5, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Muchamuel, T.; Basler, M.; Aujay, M.A.; Suzuki, E.; Kalim, K.W.; Lauer, C.; Sylvain, C.; Ring, E.R.; Shields, J.; Jiang, J.; et al. A Selective Inhibitor of the Immunoproteasome Subunit LMP7 Blocks Cytokine Production and Attenuates Progression of Experimental Arthritis. Nat. Med. 2009, 15, 781–787. [Google Scholar] [CrossRef]
- Parlati, F.; Lee, S.J.; Aujay, M.; Suzuki, E.; Levitsky, K.; Lorens, J.B.; Micklem, D.R.; Ruurs, P.; Sylvain, C.; Lu, Y.; et al. Carfilzomib Can Induce Tumor Cell Death through Selective Inhibition of the Chymotrypsin-like Activity of the Proteasome. Blood 2009, 114, 3439–3447. [Google Scholar] [CrossRef]
- Lickliter, J.; Bomba, D.; Anderl, J.; Fan, A.; Kirk, C.J.; Wang, J. AB0509 Kzr-616, a Selective Inhibitor of the Immunoproteasome, Shows a Promising Safety and Target Inhibition Profile in a Phase i, Double-Blind, Single (SAD) and Multiple Ascending Dose (MAD) Study in Healthy Volunteers. Ann. Rheum. Dis. 2018, 77, 1413–1414. [Google Scholar] [CrossRef]
- Kuhn, D.J.; Hunsucker, S.A.; Chen, Q.; Voorhees, P.M.; Orlowski, M.; Orlowski, R.Z. Targeted Inhibition of the Immunoproteasome Is a Potent Strategy against Models of Multiple Myeloma That Overcomes Resistance to Conventional Drugs and Nonspecific Proteasome Inhibitors. Blood 2009, 113, 4667–4676. [Google Scholar] [CrossRef] [PubMed]
- Kirk, C.J.; Muchamuel, T.; Wang, J.; Fan, R.A. Discovery and Early Clinical Development of Selective Immunoproteasome Inhibitors. Cells 2021, 11, 9. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, R.; Catley, L.P.; Hideshima, T.; Lentzsch, S.; Mitsiades, C.S.; Mitsiades, N.; Neuberg, D.; Goloubeva, O.; Pien, C.S.; Adams, J.; et al. Proteasome Inhibitor PS-341 Inhibits Human Myeloma Cell Growth in Vivo and Prolongs Survival in a Murine Model. Cancer Res. 2002, 62, 4996–5000. [Google Scholar] [PubMed]
- Chauhan, D.; Catley, L.; Li, G.; Podar, K.; Hideshima, T.; Velankar, M.; Mitsiades, C.; Mitsiades, N.; Yasui, H.; Letai, A.; et al. A Novel Orally Active Proteasome Inhibitor Induces Apoptosis in Multiple Myeloma Cells with Mechanisms Distinct from Bortezomib. Cancer Cell 2005, 8, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Seavey, M.M.; Lu, L.D.; Stump, K.L.; Wallace, N.H.; Ruggeri, B.A. Novel, Orally Active, Proteasome Inhibitor, Delanzomib (CEP-18770), Ameliorates Disease Symptoms and Glomerulonephritis in Two Preclinical Mouse Models of SLE. Int. Immunopharmacol. 2012, 12, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Dang, L.; Wen, F.; Yang, Y.; Liu, D.; Wu, K.; Qi, Y.; Li, X.; Zhao, J.; Zhu, D.; Zhang, C.; et al. Proteasome Inhibitor MG132 Inhibits the Proliferation and Promotes the Cisplatin-Induced Apoptosis of Human Esophageal Squamous Cell Carcinoma Cells. Int. J. Mol. Med. 2014, 33, 1083–1088. [Google Scholar] [CrossRef] [PubMed]
- Hines, J.; Groll, M.; Fahnestock, M.; Crews, C.M. Proteasome Inhibition by Fellutamide B Induces Nerve Growth Factor Synthesis. Chem. Biol. 2008, 15, 501–512. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, M.; Chatterjee, S.; Kauer, J.C.; Das, M.; Messina, P.; Freed, B.; Biazzo, W.; Siman, R. Potent Inhibitors of Proteasome. J. Med. Chem. 1995, 38, 2276–2277. [Google Scholar] [CrossRef]
- Kuhn, D.J.; Chen, Q.; Voorhees, P.M.; Strader, J.S.; Shenk, K.D.; Sun, C.M.; Demo, S.D.; Bennett, M.K.; van Leeuwen, F.W.B.; Chanan-Khan, A.A.; et al. Potent Activity of Carfilzomib, a Novel, Irreversible Inhibitor of the Ubiquitin-Proteasome Pathway, against Preclinical Models of Multiple Myeloma. Blood 2007, 110, 3281–3290. [Google Scholar] [CrossRef]
- Varga, C.; Laubach, J.; Hideshima, T.; Chauhan, D.; Anderson, K.C.; Richardson, P.G. Novel Targeted Agents in the Treatment of Multiple Myeloma. Hematol. Oncol. Clin. N. Am. 2014, 28, 903–925. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.V.; Bandi, M.; Aujay, M.A.; Kirk, C.J.; Hark, D.E.; Raje, N.; Chauhan, D.; Anderson, K.C. PR-924, a Selective Inhibitor of the Immunoproteasome Subunit LMP-7, Blocks Multiple Myeloma Cell Growth Both in Vitro and in Vivo. Br. J. Haematol. 2011, 152, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Xin, B.-T.; Huber, E.M.; de Bruin, G.; Heinemeyer, W.; Maurits, E.; Espinal, C.; Du, Y.; Janssens, M.; Weyburne, E.S.; Kisselev, A.F.; et al. Structure-Based Design of Inhibitors Selective for Human Proteasome Β2c or Β2i Subunits. J. Med. Chem. 2019, 62, 1626–1642. [Google Scholar] [CrossRef]
- Princiotta, M.F.; Schubert, U.; Chen, W.; Bennink, J.R.; Myung, J.; Crews, C.M.; Yewdell, J.W. Cells Adapted to the Proteasome Inhibitor 4-Hydroxy- 5-Iodo-3-Nitrophenylacetyl-Leu-Leu-Leucinal-Vinyl Sulfone Require Enzymatically Active Proteasomes for Continued Survival. Proc. Natl. Acad. Sci. USA 2001, 98, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Marastoni, M.; Baldisserotto, A.; Trapella, C.; Gavioli, R.; Tomatis, R. Synthesis and Biological Evaluation of New Vinyl Ester Pseudotripeptide Proteasome Inhibitors. Eur. J. Med. Chem. 2006, 41, 978–984. [Google Scholar] [CrossRef] [PubMed]
- Baldisserotto, A.; Destro, F.; Vertuani, G.; Marastoni, M.; Gavioli, R.; Tomatis, R. N-Terminal-Prolonged Vinyl Ester-Based Peptides as Selective Proteasome Β1 Subunit Inhibitors. Bioorg. Med. Chem. 2009, 17, 5535–5540. [Google Scholar] [CrossRef] [PubMed]
- Asai, A.; Hasegawa, A.; Ochiai, K.; Yamashita, Y.; Mizukami, T. Belactosin A, a Novel Antituor Antibiotic Acting on Cyclin/CDK Mediated Cell Cycle Regulation, Produced by Streptomyces sp. J. Antibiot. 2000, 53, 81–83. [Google Scholar] [CrossRef] [PubMed]
- Asai, A.; Tsujita, T.; Sharma, S.V.; Yamashita, Y.; Akinaga, S.; Funakoshi, M.; Kobayashi, H.; Mizukami, T. A New Structural Class of Proteasome Inhibitors Identified by Microbial Screening Using Yeast-Based Assay. Biochem. Pharmacol. 2004, 67, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Groll, M.; Balskus, E.P.; Jacobsen, E.N. Structural Analysis of Spiro β-Lactone Proteasome Inhibitors. J. Am. Chem. Soc. 2008, 130, 14981–14983. [Google Scholar] [CrossRef]
- Dick, L.R.; Cruikshank, A.A.; Grenier, L.; Melandri, F.D.; Nunes, S.L.; Stein, R.L. Mechanistic Studies on the Inactivation of the Proteasome by Lactacystin: A CENTRAL ROLE FOR Clasto-LACTACYSTIN β-LACTONE. J. Biol. Chem. 1996, 271, 7273–7276. [Google Scholar] [CrossRef]
- Wehenkel, M.; Ban, J.-O.; Ho, Y.-K.; Carmony, K.C.; Hong, J.T.; Kim, K.B. A Selective Inhibitor of the Immunoproteasome Subunit LMP2 Induces Apoptosis in PC-3 Cells and Suppresses Tumour Growth in Nude Mice. Br. J. Cancer 2012, 107, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Bhattarai, D.; Yoo, J.; Miller, Z.; Park, J.E.; Lee, S.; Lee, W.; Driscoll, J.J.; Kim, K.B. Development of Novel Epoxyketone-Based Proteasome Inhibitors as a Strategy To Overcome Cancer Resistance to Carfilzomib and Bortezomib. J. Med. Chem. 2019, 62, 4444–4455. [Google Scholar] [CrossRef] [PubMed]
- Voss, C.; Scholz, C.; Knorr, S.; Beck, P.; Stein, M.L.; Zall, A.; Kuckelkorn, U.; Kloetzel, P.-M.; Groll, M.; Hamacher, K.; et al. α-Keto Phenylamides as P1′-Extended Proteasome Inhibitors. ChemMedChem 2014, 9, 2557–2564. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P.; Buzder-Lantos, P.; Tantos, A.; Farkas, A.; Szilágyi, A.; Bánóczi, Z.; Hudecz, F.; Friedrich, P. On the Sequential Determinants of Calpain Cleavage. J. Biol. Chem. 2004, 279, 20775–20785. [Google Scholar] [CrossRef] [PubMed]
- Sorimachi, H.; Mamitsuka, H.; Ono, Y. Understanding the Substrate Specificity of Conventional Calpains. Biol. Chem. 2012, 393, 853–871. [Google Scholar] [CrossRef] [PubMed]
- Cuerrier, D.; Moldoveanu, T.; Davies, P.L. Determination of Peptide Substrate Specificity for μ-Calpain by a Peptide Library-Based Approach: The Importance of Primed Side Interactions. J. Biol. Chem. 2005, 280, 40632–40641. [Google Scholar] [CrossRef] [PubMed]
- Craiu, A.; Gaczynska, M.; Akopian, T.; Gramm, C.F.; Fenteany, G.; Goldberg, A.L.; Rock, K.L. Lactacystin and Clasto-Lactacystin β-Lactone Modify Multiple Proteasome β-Subunits and Inhibit Intracellular Protein Degradation and Major Histocompatibility Complex Class I Antigen Presentation. J. Biol. Chem. 1997, 272, 13437–13445. [Google Scholar] [CrossRef] [PubMed]
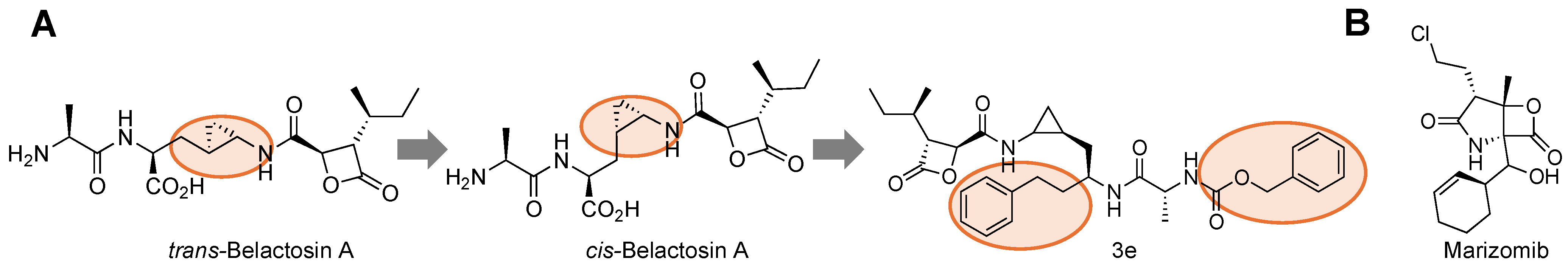
- Kawamura, S.; Unno, Y.; List, A.; Mizuno, A.; Tanaka, M.; Sasaki, T.; Arisawa, M.; Asai, A.; Groll, M.; Shuto, S. Potent Proteasome Inhibitors Derived from the Unnatural Cis-Cyclopropane Isomer of Belactosin A: Synthesis, Biological Activity, and Mode of Action. J. Med. Chem. 2013, 56, 3689–3700. [Google Scholar] [CrossRef]
- Nakamura, H.; Watanabe, M.; Ban, H.S.; Nabeyama, W.; Asai, A. Synthesis and Biological Evaluation of Boron Peptide Analogues of Belactosin C as Proteasome Inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 3220–3224. [Google Scholar] [CrossRef]
- Yoshida, K.; Yamaguchi, K.; Sone, T.; Unno, Y.; Asai, A.; Yokosawa, H.; Matsuda, A.; Arisawa, M.; Shuto, S. Synthesis of 2,3- and 3,4-Methanoamino Acid Equivalents with Stereochemical Diversity and Their Conversion into the Tripeptide Proteasome Inhibitor Belactosin A and Its Highly Potent Cis-Cyclopropane Stereoisomer. Org. Lett. 2008, 10, 3571–3574. [Google Scholar] [CrossRef]
- Warren, K. Phase 1 Trial of Marizomib Alone and in Combination with Panobinostat for Children with Diffuse Intrinsic Pontine Glioma. Clinical Trial Registration NCT04341311. 2024. Available online: https://clinicaltrials.gov/study/NCT04341311 (accessed on 31 December 2023).
- Aadi Bioscience, Inc. A Phase 2, Open-Label Study of ABI-009 (Nab-Rapamycin) in Patients with Recurrent High-Grade Glioma and Patients with Newly Diagnosed Glioblastoma; Clinical Trial Registration NCT03463265; clinicaltrials.gov. 2023. Available online: https://clinicaltrials.gov/study/NCT03463265 (accessed on 31 December 2023).
- European Organisation for Research and Treatment of Cancer—EORTC. A Phase III Trial of Marizomib in Combination with Standard Temozolomide-Based Radiochemotherapy versus Standard Temozolomide-Based Radiochemotherapy Alone in Patients with Newly Diagnosed Glioblastoma; Clinical Trial Registration NCT03345095; clinicaltrials.gov. 2024. Available online: https://clinicaltrials.gov/study/NCT03345095 (accessed on 31 December 2023).
- Stubba, D.; Bensinger, D.; Steinbacher, J.; Proskurjakov, L.; Salcedo Gómez, Á.; Schmidt, U.; Roth, S.; Schmitz, K.; Schmidt, B. Cell-Based Optimization of Covalent Reversible Ketoamide Inhibitors Bridging the Unprimed to the Primed Site of the Proteasome Β5 Subunit. ChemMedChem 2019, 14, 2005–2022. [Google Scholar] [CrossRef] [PubMed]
- Braun, H.A.; Umbreen, S.; Groll, M.; Kuckelkorn, U.; Mlynarczuk, I.; Wigand, M.E.; Drung, I.; Kloetzel, P.-M.; Schmidt, B. Tripeptide Mimetics Inhibit the 20 S Proteasome by Covalent Bonding to the Active Threonines. J. Biol. Chem. 2005, 280, 28394–28401. [Google Scholar] [CrossRef] [PubMed]
- Stein, M.L.; Cui, H.; Beck, P.; Dubiella, C.; Voss, C.; Krüger, A.; Schmidt, B.; Groll, M. Systematic Comparison of Peptidic Proteasome Inhibitors Highlights the α-Ketoamide Electrophile as an Auspicious Reversible Lead Motif. Angew. Chem. Int. Ed. 2014, 53, 1679–1683. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Sang, X.; Wang, J.; Xu, Y.; An, J.; Chu, Z.T.; Saha, A.; Warshel, A.; Huang, Z. Elucidation of the α-Ketoamide Inhibition Mechanism: Revealing the Critical Role of the Electrostatic Reorganization Effect of Asp17 in the Active Site of the 20S Proteasome. ACS Catal. 2023, 13, 14368–14376. [Google Scholar] [CrossRef]
- Piccinini, M.; Mostert, M.; Croce, S.; Baldovino, S.; Papotti, M.; Rinaudo, M.T. Interferon-γ-Inducible Subunits Are Incorporated in Human Brain 20S Proteasome. J. Neuroimmunol. 2003, 135, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Mishto, M.; Bellavista, E.; Santoro, A.; Stolzing, A.; Ligorio, C.; Nacmias, B.; Spazzafumo, L.; Chiappelli, M.; Licastro, F.; Sorbi, S.; et al. Immunoproteasome and LMP2 Polymorphism in Aged and Alzheimer’s Disease Brains. Neurobiol. Aging 2006, 27, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Hideshima, T.; Chauhan, D.; Hayashi, T.; Akiyama, M.; Mitsiades, N.; Mitsiades, C.; Podar, K.; Munshi, N.C.; Richardson, P.G.; Anderson, K.C. Proteasome Inhibitor PS-341 Abrogates IL-6 Triggered Signaling Cascades via Caspase-Dependent Downregulation of Gp130 in Multiple Myeloma. Oncogene 2003, 22, 8386–8393. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.K.; Bargagna-Mohan, P.; Wehenkel, M.; Mohan, R.; Kim, K.-B. LMP2-Specific Inhibitors: Chemical Genetic Tools for Proteasome Biology. Chem. Biol. 2007, 14, 419–430. [Google Scholar] [CrossRef]
- Ivancsits, D.; Nimmanapali, R.; Sun, M.; Shenk, K.; Demo, S.D.; Bennett, M.K.; Dalton, W.S.; Alsina, M. The Proteasome Inhibitor PR-171 Inhibits Cell Growth, Induces Apoptosis, and Overcomes De Novo and Acquired Drug Resistance in Human Multiple Myeloma Cells. Blood 2005, 106, 1575. [Google Scholar] [CrossRef]
- O’Connor, O.A.; Stewart, A.K.; Vallone, M.; Molineaux, C.J.; Kunkel, L.A.; Gerecitano, J.F.; Orlowski, R.Z. A Phase 1 Dose Escalation Study of the Safety and Pharmacokinetics of the Novel Proteasome Inhibitor Carfilzomib (PR-171) in Patients with Hematologic Malignancies. Clin. Cancer Res. 2009, 15, 7085–7091. [Google Scholar] [CrossRef]
- Berenson, J.R.; Hilger, J.D.; Yellin, O.; Dichmann, R.; Patel-Donnelly, D.; Boccia, R.V.; Bessudo, A.; Stampleman, L.; Gravenor, D.; Eshaghian, S.; et al. Replacement of Bortezomib with Carfilzomib for Multiple Myeloma Patients Progressing from Bortezomib Combination Therapy. Leukemia 2014, 28, 1529–1536. [Google Scholar] [CrossRef] [PubMed]
- Carmony, K.C.; Kim, K.B. Activity-Based Imaging Probes of the Proteasome. Cell Biochem. Biophys. 2013, 67, 91–101. [Google Scholar] [CrossRef] [PubMed]
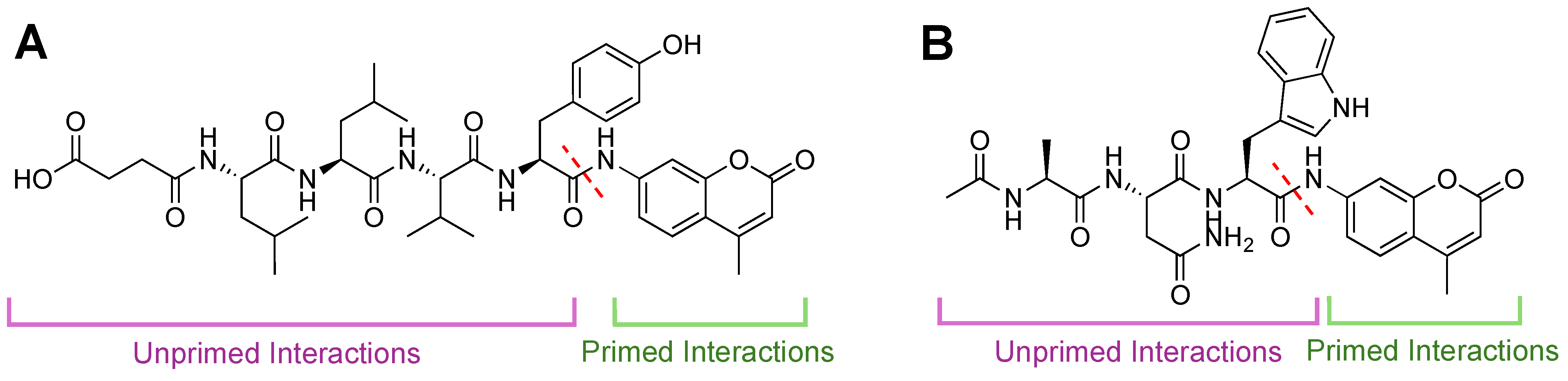
- Kisselev, A.F.; Goldberg, A.L. Monitoring Activity and Inhibition of 26S Proteasomes with Fluorogenic Peptide Substrates. Methods Enzymol. 2005, 398, 364–378. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.A.; Trader, D.J. Methods to Discover and Evaluate Proteasome Small Molecule Stimulators. Molecules 2019, 24, 2341. [Google Scholar] [CrossRef]
- Miller, Z.; Kim, K.-S.; Lee, D.-M.; Kasam, V.; Baek, S.E.; Lee, K.H.; Zhang, Y.-Y.; Ao, L.; Carmony, K.; Lee, N.-R.; et al. Proteasome Inhibitors with Pyrazole Scaffolds from Structure-Based Virtual Screening. J. Med. Chem. 2015, 58, 2036–2041. [Google Scholar] [CrossRef] [PubMed]
- Rožman, K.; Alexander, E.M.; Ogorevc, E.; Bozovičar, K.; Sosič, I.; Aldrich, C.C.; Gobec, S. Psoralen Derivatives as Inhibitors of Mycobacterium Tuberculosis Proteasome. Molecules 2020, 25, 1305. [Google Scholar] [CrossRef] [PubMed]
- Gulder, T.A.M.; Moore, B.S. Salinosporamide Natural Products: Potent 20 S Proteasome Inhibitors as Promising Cancer Chemotherapeutics. Angew. Chem. Int. Ed. 2010, 49, 9346–9367. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.A.; Trader, D.J. Development and Application of a Sensitive Peptide Reporter to Discover 20S Proteasome Stimulators. ACS Comb. Sci. 2018, 20, 269–276. [Google Scholar] [CrossRef]
- Coleman, R.A.; Trader, D.J. A Sensitive High-Throughput Screening Method for Identifying Small Molecule Stimulators of the Core Particle of the Proteasome. Curr. Protoc. Chem. Biol. 2018, 10, e52. [Google Scholar] [CrossRef]
- Green, M.; Loewenstein, P.M. Autonomous Functional Domains of Chemically Synthesized Human Immunodeficiency Virus Tat Trans-Activator Protein. Cell 1988, 55, 1179–1188. [Google Scholar] [CrossRef]
- Frankel, A.D.; Pabo, C.O. Cellular Uptake of the Tat Protein from Human Immunodeficiency Virus. Cell 1988, 55, 1189–1193. [Google Scholar] [CrossRef] [PubMed]
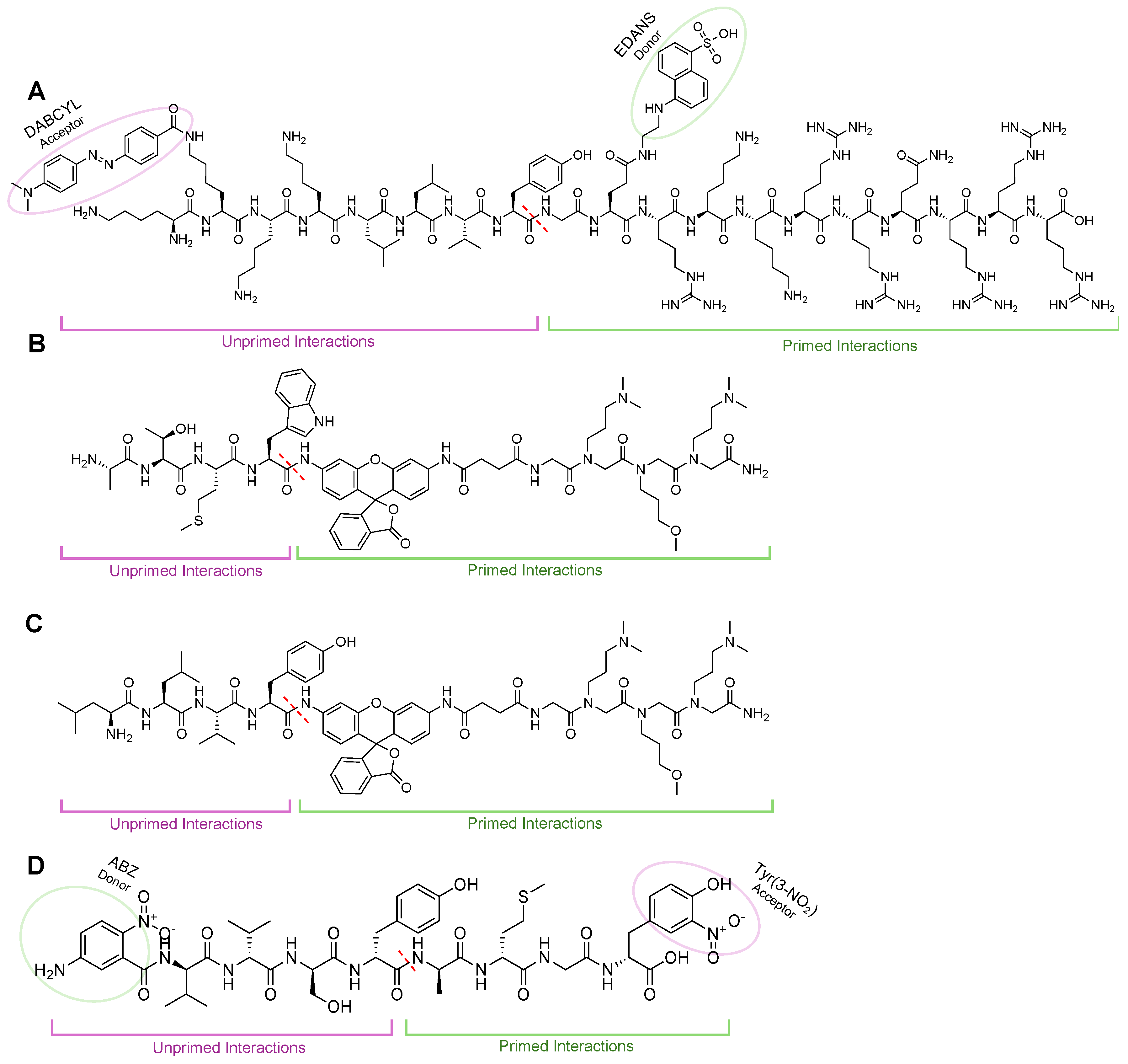
- Zerfas, B.L.; Trader, D.J. Synthesis and Application of an Activity-Based Peptide-Peptoid Hybrid Probe for the Immunoproteasome. Curr. Protoc. Chem. Biol. 2019, 11, e76. [Google Scholar] [CrossRef] [PubMed]
- Zerfas, B.L.; Trader, D.J. Monitoring the Immunoproteasome in Live Cells Using an Activity-Based Peptide–Peptoid Hybrid Probe. J. Am. Chem. Soc. 2019, 141, 5252–5260. [Google Scholar] [CrossRef] [PubMed]
- Zerfas, B.L.; Coleman, R.A.; Salazar-Chaparro, A.F.; Macatangay, N.J.; Trader, D.J. Fluorescent Probes with Unnatural Amino Acids to Monitor Proteasome Activity in Real-Time. ACS Chem. Biol. 2020, 15, 2588–2596. [Google Scholar] [CrossRef] [PubMed]
- Urru, S.A.M.; Veglianese, P.; De Luigi, A.; Fumagalli, E.; Erba, E.; Gonella Diaza, R.; Carrà, A.; Davoli, E.; Borsello, T.; Forloni, G.; et al. A New Fluorogenic Peptide Determines Proteasome Activity in Single Cells. J. Med. Chem. 2010, 53, 7452–7460. [Google Scholar] [CrossRef] [PubMed]
- Gruba, N.; Wysocka, M.; Brzezińska, M.; Dębowski, D.; Sieńczyk, M.; Gorodkiewicz, E.; Guszcz, T.; Czaplewski, C.; Rolka, K.; Lesner, A. Bladder Cancer Detection Using a Peptide Substrate of the 20S Proteasome. FEBS J. 2016, 283, 2929–2948. [Google Scholar] [CrossRef] [PubMed]
- Wakata, A.; Lee, H.-M.; Rommel, P.; Toutchkine, A.; Schmidt, M.; Lawrence, D.S. Simultaneous Fluorescent Monitoring of Proteasomal Subunit Catalysis. J. Am. Chem. Soc. 2010, 132, 1578–1582. [Google Scholar] [CrossRef] [PubMed]
- Hewings, D.S.; Flygare, J.A.; Wertz, I.E.; Bogyo, M. Activity-Based Probes for the Multicatalytic Proteasome. FEBS J. 2017, 284, 1540–1554. [Google Scholar] [CrossRef] [PubMed]
- Verdoes, M.; Florea, B.I.; Menendez-Benito, V.; Maynard, C.J.; Witte, M.D.; van der Linden, W.A.; van den Nieuwendijk, A.M.C.H.; Hofmann, T.; Berkers, C.R.; van Leeuwen, F.W.B.; et al. A Fluorescent Broad-Spectrum Proteasome Inhibitor for Labeling Proteasomes In Vitro and In Vivo. Chem. Biol. 2006, 13, 1217–1226. [Google Scholar] [CrossRef]
- Verdoes, M.; Hillaert, U.; Florea, B.I.; Sae-Heng, M.; Risseeuw, M.D.P.; Filippov, D.V.; van der Marel, G.A.; Overkleeft, H.S. Acetylene Functionalized BODIPY Dyes and Their Application in the Synthesis of Activity Based Proteasome Probes. Bioorg. Med. Chem. Lett. 2007, 17, 6169–6171. [Google Scholar] [CrossRef]
- Muli, C.S.; Trader, D.J. 20S Proteasome Hydrolysis of LLVY Substrates to Determine Preferences for Moieties in Its Primed Substrate Channel. Bioorg. Med. Chem. Lett. 2023, 85, 129233. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Class | Substrate Channel | Subunit | Stage | Ref |
|---|---|---|---|---|---|
| Bortezomib | Boronic Acid | Non-primed | β5/β1 | FDA Approved | [65,79] |
| Ixazomib | Boronic Acid | Non-primed | β5 | FDA Approved | [80] |
| Delanzomib | Boronic Acid | Non-primed | β5 | Clinical | [81] |
| MG-132 | Aldehyde | Non-primed | β5 | Research Tool | [82] |
| PSI | Aldehyde | Non-primed | β5 β2, β1 | Pre-Clinical | [59] |
| Felutamide B | Aldehyde | Non-primed | β5, β2, β1 | Pre-Clinical | [83] |
| CEP1612 | Aldehyde | Non-primed | β5 | Pre-Clinical | [84] |
| ISPI-001 | Aldehyde | Non-primed | β5i/β1i | Pre-Clinical | [77] |
| Epoxomicin | Epoxide | Non-primed | β5 | Research Tool | [71] |
| Carfilzomib | Epoxide | Non-primed | β5 | FDA Approved | [85] |
| Oprozomib | Epoxide | Non-primed | β5, β5i | Clinical | [86] |
| ONX-0914 | Epoxide | Non-primed | β5i | Research Tool | [74] |
| PR-924 | Epoxide | Non-primed | β5i | Pre-Clinical | [87] |
| KZR-616 | Epoxide | Non-primed | β5i | Clinical | [76] |
| LU-002i | Epoxide | Non-primed | β2i | Pre-Clinical | [88] |
| NLVS | Vinyl-Sulfone | Non-primed | β5 | Pre-Clinical | [89] |
| HMB-Val-Ser-Leu-VE | Vinyl-Sulfone | Non-primed | β2 | Pre-Clinical | [90] |
| Z-NH-(CH2)5-CO-Leu-Leu-Leu-VE | Vinyl-Sulfone | Non-primed | β1 | Research Tool | [91] |
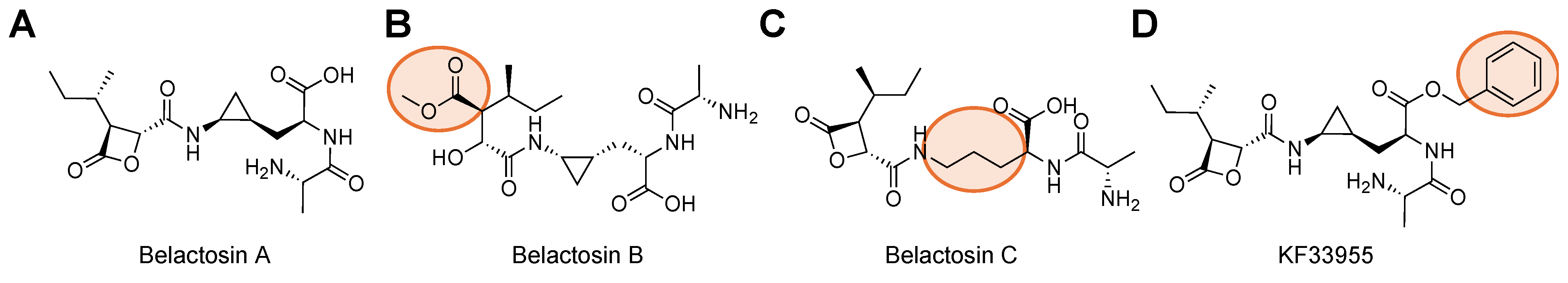
| Belactosin A | β-lactone | Primed | β5 | Pre-Clinical | [92] |
| Belactosin C | β-lactone | Primed | β5 | Pre-Clinical | [53] |
| KF33955 | β-lactone | Primed | β5 | Pre-Clinical | [93] |
| Marizomib | β-lactone | Primed | β5, β2, β1 | Clinical | [73,94] |
| Omuralide | β-lactone | Primed | β5 | Pre-Clinical | [92,95] |
| UK-101 | Epoxide | Primed | β1i | Pre-Clinical | [96] |
| Carfilzomib-P1′ | Epoxide | Primed | β5 | Pre-Clinical | [97] |
| B-Sc2189 | α-ketoamide | Primed | β5 | Pre-Clinical | [98] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loy, C.A.; Trader, D.J. Primed for Interactions: Investigating the Primed Substrate Channel of the Proteasome for Improved Molecular Engagement. Molecules 2024, 29, 3356. https://doi.org/10.3390/molecules29143356
Loy CA, Trader DJ. Primed for Interactions: Investigating the Primed Substrate Channel of the Proteasome for Improved Molecular Engagement. Molecules. 2024; 29(14):3356. https://doi.org/10.3390/molecules29143356
Chicago/Turabian StyleLoy, Cody A., and Darci J. Trader. 2024. "Primed for Interactions: Investigating the Primed Substrate Channel of the Proteasome for Improved Molecular Engagement" Molecules 29, no. 14: 3356. https://doi.org/10.3390/molecules29143356
APA StyleLoy, C. A., & Trader, D. J. (2024). Primed for Interactions: Investigating the Primed Substrate Channel of the Proteasome for Improved Molecular Engagement. Molecules, 29(14), 3356. https://doi.org/10.3390/molecules29143356