
A Recyclable Inorganic Lanthanide Cluster Catalyst for Chemoselective Aerobic Oxidation of Thiols
 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Calculation of the Yield by Internal Standard Using 1H NMR
3.3. Optimization Studies for the Oxidative Coupling of Thiols by Sm-OC (Table 1)
3.4. General Procedure for the Oxidation of Thiols
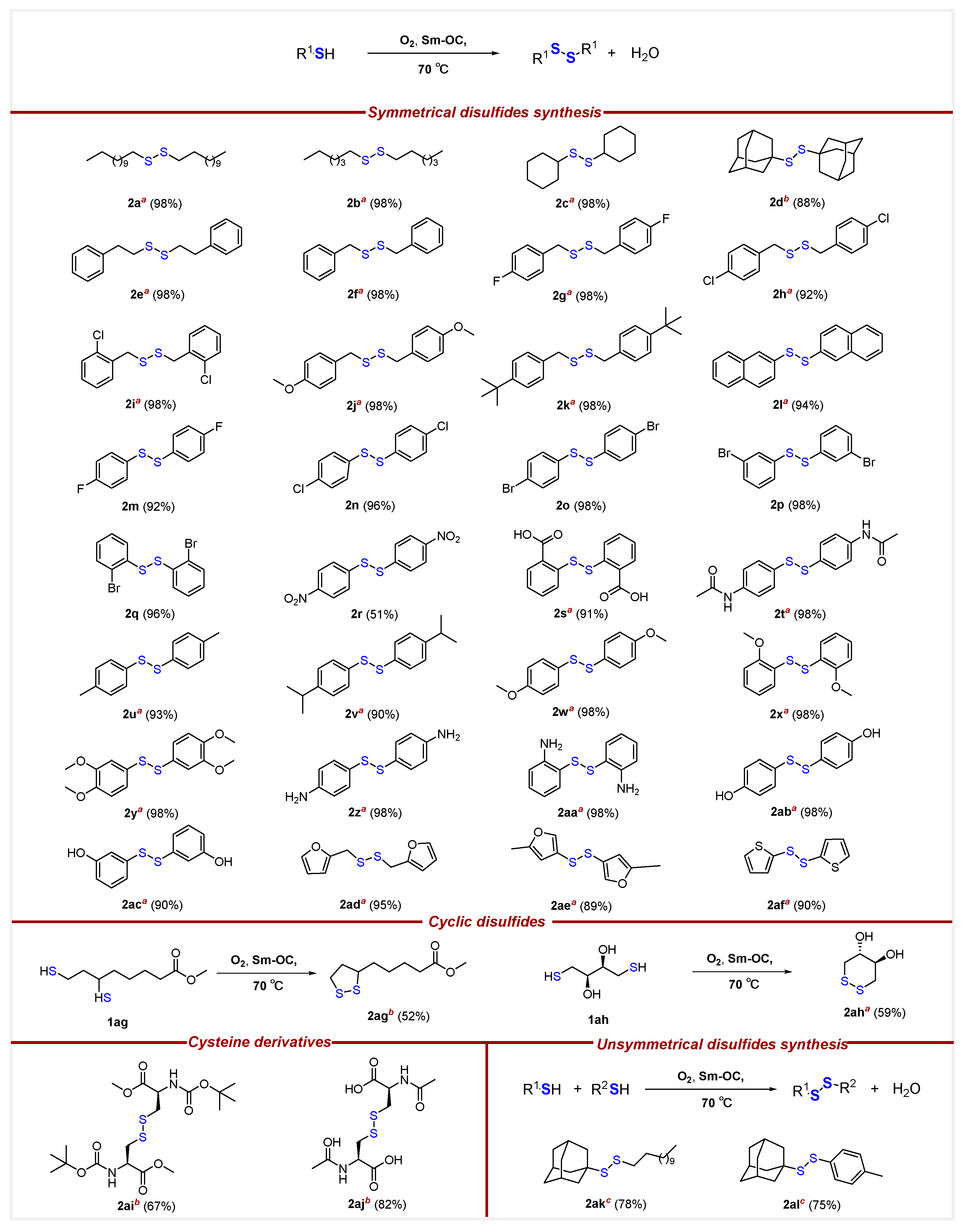
- 1,2-Didodecyldisulfane (2a) [37]. According to the general procedure, the reaction of Sm-OC (9.10 mg, 1.15 mol%) and dodecane-1-thiol (60.7 mg, 0.300 mmol) afforded 59.2 mg of 2a in 98% yield as a white solid. 1H NMR (500 MHz, CDCl3) δ 2.69 (t, J = 7.4 Hz, 4H), 1.68 (m, 4H), 1.39 (m, 4H), 1.35–1.21 (m, 32H), 0.89 (t, J = 6.9 Hz, 6H); 13C{1H} NMR (126 MHz, CDCl3) δ 39.3, 32.0, 29.8, 29.7 (×2), 29.6, 29.5, 29.4, 29.3, 28.6, 22.8, 14.2.
- 1,2-Dihexyldisulfane (2b) [40]. According to the general procedure, the reaction of Sm-OC (9.10 mg, 1.15 mol%) and hexane-1-thiol (35.5 mg, 0.300 mmol) afforded 34.5 mg of 2b in 98% yield as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 2.69 (t, J = 7.4 Hz, 4H), 1.68 (m, 4H), 1.39 (m, 4H), 1.36–1.24 (m, 8H), 0.90 (t, J = 6.9 Hz, 6H); 13C{1H} NMR (126 MHz, CDCl3) δ 39.3, 31.5, 29.3, 28.3, 22.6, 14.1.
- 1,2-Dicyclohexyldisulfane (2c) [40]. To a round-bottom flask in oil-bath, Sm-OC (9.10 mg, 1.15 mol%), cyclohexanethiol (58.1 mg, 0.500 mmol), and extra-dry EtOAc (8.00 mL) were added. The flask was filled with oxygen balloon (0.3 Mpa) and the reaction mixture was stirred at 70 °C for a duration of 16 h. Subsequently, the reaction mixture was cooled by removing from the oil-bath. The reaction mixture was diluted with EtOAc (10 mL) and HCl (15.0 mL, 0.100 M, aq). The aqueous layer was extracted with EtOAc (3 × 15.0 mL). Organic layers were combined, dried over MgSO4, filtered, and concentrated, affording 56.5 mg of 2c in 98% yield as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 2.69 (m, 2H), 2.05 (m, 4H), 1.79 (m, 4H), 1.61 (m, 2H), 1.39–1.17 (m, 10H); 13C{1H} NMR (126 MHz, CDCl3) δ 50.1, 33.0, 26.2, 25.8.
- 1,2-Di((3S,5S,7S)-adamantan-1-yl)disulfane (2d) [41]. To a round-bottom flask in oil-bath, Sm-OC (18.2 mg, 2.30 mol%), (3s,5s,7s)-adamantane-1-thiol (50.5 mg, 0.300 mmol), and extra-dry EtOAc (16.0 mL) were added. The flask was filled with oxygen balloon (0.3 MPa) and the reaction mixture was stirred at 70 °C for a duration of 16 h. Subsequently, the reaction mixture was cooled by removing from the oil-bath. The reaction mixture was diluted with EtOAc (10.0 mL) and NaOH solution (15.0 mL, 1.00 M, aq). The aqueous layer was extracted with EtOAc (3 × 15.0 mL). Organic layers were combined, dried over MgSO4, filtered, and concentrated, affording 44.2 mg of 2d in 88% yield as a white solid. 1H NMR (500 MHz, CDCl3) δ 2.07 (m, 6H), 1.82 (m, 12H), 1.67 (m, 12H); 13C{1H} NMR (126 MHz, CDCl3) δ 47.4, 43.2, 36.2, 30.1.
- 1,2-Diphenethyldisulfane (2e) [42]. To a round-bottom flask in oil-bath, Sm-OC (9.10 mg, 1.15 mol%), 2-phenylethane-1-thiol (69.1 mg, 0.500 mmol), and extra-dry EtOAc (8.00 mL) were added. The flask was filled with oxygen balloon (0.3 MPa) and the reaction mixture was stirred at 70 °C for a duration of 16 h. Subsequently, the reaction mixture was cooled by removing from the oil-bath. The reaction mixture was diluted with EtOAc (10.0 mL) and HCl (15.0 mL, 0.100 M, aq). The aqueous layer was extracted with EtOAc (3 × 15.0 mL). Organic layers were combined, dried over MgSO4, filtered, and concentrated, affording 67.2 mg of 2e in 98% yield as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.30–7.27 (m, 4H), 7.23–7.16 (m, 6H), 2.98 (m, 4H), 2.92 (m, 4H); 13C{1H} NMR (126 MHz, CDCl3) δ 140.1, 128.7, 128.6, 126.5, 40.3, 35.8.
- 1,2-Dibenzyldisulfane (2f) [43]. According to the general procedure, the reaction of Sm-OC (9.10 mg, 1.15 mol%) and phenylmethanethiol (37.3 mg, 0.300 mmol) afforded 36.2 mg of 2f in 98% yield as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.25–7.11 (m, 10H), 3.49 (s, 4H); 13C{1H} NMR (126 MHz, CDCl3) δ 137.4, 129.4, 128.5, 127.4, 43.3.
- 1,2-Bis(4-fluorobenzyl)disulfane (2g) [44]. According to the general procedure, the reaction of Sm-OC (9.10 mg, 1.15 mol%) and (4-fluorophenyl)methanethiol (42.7 mg, 0.300 mmol) afforded 41.5 mg of 2g in 98% yield as a yellow solid. 1H NMR (500 MHz, CDCl3) δ 7.21 (m, 4H), 7.02 (m, 4H), 3.60 (s, 4H); 13C{1H} NMR (126 MHz, CDCl3) δ 162.3 (d, JC-F = 246.4 Hz), 133.2 (d, JC-F = 2.8 Hz), 131.0 (d, JC-F = 8.5 Hz), 115.5 (d, JC-F = 21.5 Hz), 42.5.
- 1,2-Bis(4-chlorobenzyl)disulfane (2h) [45]. According to the general procedure, the reaction of Sm-OC (9.10 mg, 1.15 mol%) and (4-chlorophenyl)methanethiol (47.6 mg, 0.300 mmol) afforded 43.5 mg of 2h in 92% yield as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.31 (m, 4H), 7.17 (m, 4H), 3.59 (s, 4H); 13C{1H} NMR (126 MHz, CDCl3) δ 135.9, 133.5, 130.7, 128.8, 42.6.
- 1,2-Bis(2-chlorobenzyl)disulfane (2i) [46]. According to the general procedure, the reaction of Sm-OC (9.10 mg, 1.15 mol%) and (2-chlorophenyl)methanethiol (47.6 mg, 0.300 mmol) afforded 46.3 mg of 2i in 98% yield as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.29 (m, 2H), 7.18 (m, 2H), 7.16–7.11 (m, 4H), 3.70 (s, 4H); 13C{1H} NMR (126 MHz, CDCl3) δ 135.1, 134.2, 131.6, 129.8, 129.0, 126.8, 41.2.
- 1,2-Bis(4-methoxybenzyl)disulfane (2j) [36]. According to the general procedure, the reaction of Sm-OC (9.10 mg, 1.15 mol%) and (4-methoxyphenyl)methanethiol (46.3 mg, 0.300 mmol) afforded 45.0 mg of 2j in 98% yield as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.19 (m, 4H), 6.87 (m, 4H), 3.81 (s, 6H), 3.61 (s, 4H); 13C{1H} NMR (126 MHz, CDCl3) δ 159.1, 130.6, 129.5, 114.0, 55.4, 42.8.
- 1,2-Bis(4-(tert-butyl)benzyl)disulfane (2k) [44]. According to the general procedure, the reaction of Sm-OC (9.10 mg, 1.15 mol%) and (4-(tert-butyl)phenyl)methanethiol (54.1 mg, 0.300 mmol) afforded 52.7 mg of 2k in 98% yield as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.37 (m, 4H), 7.21 (m, 4H), 3.63 (s, 4H), 1.34 (s, 18H); 13C{1H} NMR (126 MHz, CDCl3) δ 150.5, 134.3, 129.2, 125.5, 43.1, 34.6, 31.4.
- 1,2-Di(naphthalen-2-yl)disulfane (2l) [43]. According to the general procedure, the reaction of Sm-OC (9.10 mg, 1.15 mol%) and naphthalene-2-thiol (48.1 mg, 0.300 mmol) afforded 44.9 mg of 2l in 94% yield as a white solid. 1H NMR (500 MHz, CDCl3) δ 8.01 (m, 2H), 7.85–7.78 (m, 4H), 7.75 (m, 2H), 7.65 (m, 2H), 7.52–7.43 (m, 4H); 13C{1H} NMR (126 MHz, CDCl3) δ 134.3, 133.5, 132.6, 129.1, 127.8, 127.5, 126.8, 126.6, 126.3, 125.7.
- 1,2-Bis(4-fluorophenyl)disulfane (2m) [43]. To a round-bottom flask in oil-bath, Sm-OC (9.10 mg, 1.15 mol%), 4-fluorobenzenethiol (64.1 mg, 0.500 mmol), and extra-dry EtOAc (8.00 mL) were added. The flask was filled with oxygen balloon (0.3 MPa) and the reaction mixture was stirred at 70 °C for a duration of 16 h. Subsequently, the reaction mixture was cooled by removing from the oil-bath. The reaction mixture was diluted with EtOAc (10.0 mL) and HCl (15.0 mL, 0.100 M, aq). The aqueous layer was extracted with EtOAc (3 × 15.0 mL). Organic layers were combined, dried over MgSO4, filtered, and concentrated, affording 58.5 mg of 2m in 92% yield as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.46 (m, 4H), 7.02 (m, 4H); 13C{1H} NMR (126 MHz, CDCl3) δ 162.7 (d, JC-F = 248.1 Hz), 132.3 (d, JC-F = 2.8 Hz), 131.4 (d, JC-F = 8.5 Hz), 116.4 (d, JC-F = 22.0 Hz).
- 1,2-Bis(4-chlorophenyl)disulfane (2n) [43]. According to the general procedure, the reaction of Sm-OC (9.10 mg, 1.15 mol%) and 4-chlorobenzenethiol (72.3 mg, 0.500 mmol) afforded 68.9 mg of 2n in 96% yield as a white solid: 1H NMR (500 MHz, CDCl3) δ 7.39 (m, 4H), 7.26 (m, 4H); 13C{1H} NMR (126 MHz, CDCl3) δ 135.2, 133.7, 129.4 (×2).
- 1,2-Bis(4-bromophenyl)disulfane (2o) [43]. According to the general procedure, the reaction of Sm-OC (9.10 mg, 1.15 mol%) and 4-bromobenzenethiol (56.7 mg, 0.300 mmol), after chromatography (silica, 100% Hexane) afforded 55.3 mg of 2o in 98% yield as a white solid: 1H NMR (500 MHz, CDCl3) δ 7.44 (m, 4H), 7.35 (m, 4H); 13C{1H} NMR (126 MHz, CDCl3) δ 135.8, 132.3, 129.5, 121.6.
- 1,2-Bis(3-bromophenyl)disulfane (2p) [42]. According to the general procedure, the reaction of Sm-OC (9.10 mg, 1.15 mol%) and 3-bromobenzenethiol (56.7 mg, 0.300 mmol) afforded 55.3 mg of 2p in 98% yield as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.64 (m, 2H), 7.44–7.35 (m, 4H), 7.19 (m, 2H); 13C{1H} NMR (126 MHz, CDCl3) δ 138.7, 130.6 (×2), 130.0, 126.0, 123.2.
- 1,2-Bis(2-bromophenyl)disulfane (2q) [40]. According to the general procedure, the reaction of Sm-OC (9.10 mg, 1.15 mol%) and 2-bromobenzenethiol (56.7 mg, 0.300 mmol) afforded 54.2 mg of 2q in 96% yield as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.49–7.41 (m, 4H), 7.18 (m, 2H), 6.99 (m, 2H); 13C{1H} NMR (126 MHz, CDCl3) δ 136.2, 133.0, 128.3, 128.0, 127.0, 121.1.
- 1,2-Bis(4-nitrophenyl)disulfane (2r) [40]. According to the general procedure, the reaction of Sm-OC (9.10 mg, 1.15 mol%) and 4-nitrobenzenethiol (46.6 mg, 0.300 mmol) afforded 23.6mg of 2r in 51% yield as a yellow solid. 1H NMR (500 MHz, DMSO-d6) δ 8.25 (m, 4H), 7.81 (m, 4H); 13C{1H} NMR (126 MHz, DMSO-d6) δ 146.6, 143.6, 126.7, 124.6.
- 2,2′-Disulfanediyldibenzoic acid (2s) [40]. To a round-bottom flask in oil-bath, Sm-OC (9.10 mg, 1.15 mol%), 2-mercaptobenzoic acid (46.3 mg, 0.300 mmol), and extra-dry EtOAc (8.0 mL) were added. The flask was filled with oxygen balloon (0.3 MPa) and the reaction mixture was stirred at 70 °C for a duration of 16 h. Subsequently, the reaction mixture was cooled by removing from the oil-bath. The reaction mixture was diluted with EtOAc (10.0 mL) and HCl (15.0 mL, 0.100 M, aq). The aqueous layer was extracted with EtOAc (3 × 15.0 mL), organic layers were combined, dried over MgSO4, filtered, and concentrated, after chromatography (0–10% MeOH/EtOAc), affording 41.8 mg of 2s in 91% yield a white solid. 1H NMR (500 MHz, DMSO-d6) δ 8.02 (m, 2H), 7.61 (m, 2H), 7.54 (m, 2H), 7.32 (m, 2H); 13C{1H} NMR (126 MHz, DMSO-d6) δ 167.8, 138.9, 133.0, 131.5, 128.7, 125.9, 124.9.
- N,N’-(disulfanediylbis(4,1-phenylene))diacetamide (2t) [40]. To a round-bottom flask in oil-bath, Sm-OC (9.10 mg, 1.15 mol%), N-(4-mercaptophenyl)acetamide (83.6 mg, 0.500 mmol), and extra-dry EtOAc (8.0 mL) were added. The flask was filled with oxygen balloon (0.3 MPa) and the reaction mixture was stirred at 70 °C for a duration of 16 h. Subsequently, the reaction mixture was cooled by removing from the oil-bath. The reaction mixture was diluted with EtOAc (10.0 mL) and HCl (15.0 mL, 0.100 M, aq). The aqueous layer was extracted with EtOAc (3 × 15.0 mL). Organic layers were combined, dried over MgSO4, filtered, and concentrated, affording 81.4 mg of 2t in 98% yield as a white solid. 1H NMR (500 MHz, DMSO-d6) δ 10.07 (s, 2H), 7.59 (m, 4H), 7.42 (m, 4H), 2.04 (s, 6H); 13C{1H} NMR (126 MHz, DMSO-d6) δ 168.5, 139.5, 130.1, 129.4, 119.7, 24.0.
- 1,2-Di-p-tolyldisulfane (2u) [43]. According to the general procedure, the reaction of Sm-OC (9.10 mg, 1.15 mol%) and 4-methylbenzenethiol (37.3 mg, 0.300 mmol) afforded 34.4 mg of 2u in 93% yield as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.54 (m, 4H), 7.26 (m, 4H), 2.48 (s, 6H); 13C{1H} NMR (126 MHz, CDCl3) δ 137.5, 134.0, 129.9, 128.6, 21.1.
- 1,2-Bis(4-isopropylphenyl)disulfane (2v) [46]. To a round bottom flask in oil-bath, Sm-OC (9.10 mg, 1.15 mol%), 4-isopropylbenzenethiol (76.1 mg, 0.500 mmol), and extra-dry EtOAc (8.0 mL) were added. The flask was filled with oxygen balloon (0.3 MPa) and the reaction mixture was stirred at 70 °C for a duration of 16 h. Subsequently, the reaction mixture was cooled by removing from the oil-bath. The reaction mixture was diluted with EtOAc (10.0 mL) and HCl (15.0 mL, 0.100 M, aq). The aqueous layer was extracted with EtOAc (3 × 15.0 mL). Organic layers were combined, dried over MgSO4, filtered, and concentrated afforded 68.1 mg of 2v in 90% yield as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.47 (m, 4H), 7.20 (m, 4H), 2.91 m, 2H), 1.26 (d, J = 7.0 Hz, 12H); 13C{1H} NMR (126 MHz, CDCl3) δ 148.4, 134.4, 128.3, 127.3, 33.8, 24.0.
- 1,2-Bis(4-methoxyphenyl)disulfane (2w) [43]. According to the general procedure, the reaction of Sm-OC (9.10 mg, 1.15 mol%) and 4-methoxybenzenethiol (42.1 mg, 0.300 mmol) afforded 40.9 mg of 2w in 98% yield as a yellow oil. 1H NMR (500 MHz, CDCl3) δ 7.41 (m, 4H), 6.85 (m, 4H), 3.81 (s, 6H); 13C{1H} NMR (126 MHz, CDCl3) δ 160.0, 132.7, 128.5, 114.7, 55.4.
- 1,2-Bis(2-methoxybenzyl)disulfane (2x) [43]. According to the general procedure, the reaction of Sm-OC (9.10 mg, 1.15 mol%) and (2-methoxyphenyl)methanethiol (42.1 mg, 0.300 mmol) afforded 40.9 mg of 2x in 98% yield as a white solid: 1H NMR (500 MHz, CDCl3) δ 7.55 (m, 2H), 7.20 (m, 2H), 6.93 (m, 2H), 6.87 (m, 2H), 3.91 (s, 6H); 13C{1H} NMR (126 MHz, CDCl3) δ 156.6, 127.8, 127.6, 124.6, 121.4, 110.5, 55.9.
- 1,2-Bis(3,4-dimethoxyphenyl)disulfane (2y) [43]. According to the general procedure, the reaction of Sm-OC (9.10 mg, 1.15 mol%) and 3,4-dimethoxybenzenethiol (51.1 mg, 0.300 mmol) afforded 49.8 mg of 2y in 98% yield as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.06 (d, J = 2.1 Hz, 1H), 7.04 (d, J = 2.1 Hz, 1H), 7.01 (m, 2H), 6.79 (s, 1H), 6.78 (s, 1H), 3.87 (s, 6H), 3.83 (s, 6H); 13C{1H} NMR (126 MHz, CDCl3) δ 149.6, 149.2, 128.7, 123.9, 114.1, 111.3, 56.0 (×2).
- 4,4′-Disulfanediyldianiline (2z) [40]. To a round-bottom flask in oil-bath, Sm-OC (9.10 mg, 1.15 mol%), 4-aminobenzenethiol (37.6 mg, 0.300 mmol), and extra-dry EtOAc (8.0 mL) were added. The flask was filled with oxygen balloon (0.3 MPa) and the reaction mixture was stirred at 70 °C for a duration of 16 h. Subsequently, the reaction mixture was cooled by removing from the oil-bath. The reaction mixture was diluted with EtOAc (10.0 mL) and washed with NaOH solution (15.0 mL, 1.00 M, aq). The aqueous layer was extracted with EtOAc (3 × 15.0 mL). Organic layers were combined, dried over MgSO4, filtered, and concentratedd afforded 36.5 mg of 2z in 98% yield as a yellow solid. 1H NMR (500 MHz, CDCl3) δ 7.16 (m, 4H), 6.47 (m, 4H), 3.69 (s, 4H); 13C{1H} NMR (126 MHz, CDCl3) δ 147.2, 133.9, 125.5, 115.4.
- 2,2′-Disulfanediyldianiline (2aa) [40]. To a round-bottom flask in oil-bath, Sm-OC (9.10 mg, 1.15 mol%), 2-aminobenzenethiol (37.6 mg, 0.300 mmol), and extra-dry EtOAc (8.0 mL) were added. The flask was filled with oxygen balloon (0.3 MPa) and the reaction mixture was stirred at 70 °C for a duration of 16 h. Subsequently, the reaction mixture was cooled by removing from the oil-bath. The reaction mixture was diluted with EtOAc (10.0 mL) and washed with NaOH solution (15.0 mL, 1.00 M, aq). The aqueous layer was extracted with EtOAc (3 × 15.0 mL). Organic layers were combined, dried over MgSO4, filtered, and concentrated, affording 36.5 mg of 2aa in 98% yield as a yellow solid. 1H NMR (500 MHz, CDCl3) δ 7.22 –7.13 (m, 4H), 6.72 (m, 2H), 6.60 (m, 2H), 4.35 (s, 4H); 13C{1H} NMR (126 MHz, CDCl3) δ 148.7, 136.8, 131.6, 118.8, 118.3, 115.3.
- 4,4′-Disulfanediyldiphenol (2ab) [47]. According to the general procedure, the reaction of Sm-OC (9.10 mg, 1.15 mol%) and 4-mercaptophenol (37.9 mg, 0.300 mmol) afforded 36.8 mg of 2ab in 98% yield as a yellow solid. 1H NMR (500 MHz, DMSO-d6) δ 9.85 (s, 2H), 7.28 (m, 4H), 6.77 (m, 4H); 13C{1H} NMR (126 MHz, DMSO-d6) δ 158.3, 133.0, 125.2, 116.3.
- 3,3′-Disulfanediyldiphenol (2ac) [48]. To a round-bottom flask in oil-bath, Sm-OC (9.10 mg, 1.15 mol%), 3-mercaptophenol (37.9 mg, 0.300 mmol), and extra-dry EtOAc (8.0 mL) were added. The flask was filled with oxygen balloon (0.3 MPa) and the reaction mixture was stirred at 70 °C for a duration of 16 h. Subsequently, the reaction mixture was cooled by removing from the oil-bath. The reaction mixture was diluted with EtOAc (10.0 mL) and HCl (15.0 mL, 0.100 M, aq). The aqueous layer was extracted with EtOAc (3 × 15.0 mL). Organic layers were combined, dried over MgSO4, filtered, and concentrated, after chromatography (25–50% EtOAc/Hexane), affording 33.8 mg of 2ac in 90% yield as a white solid. 1H NMR (500 MHz, DMSO-d6) δ 9.77 (s, 2H), 7.18 (m, 2H), 6.96–6.88 (m, 4H), 6.67 (m, 2H); 13C{1H} NMR (126 MHz, DMSO-d6) δ 158.1, 136.7, 130.3, 117.2, 114.6, 113.0.
- 1,2-Bis(furan-2-ylmethyl)disulfane (2ad) [42]. To a round-bottom flask in oil-bath, Sm-OC (9.10 mg, 1.15 mol%), thiophene-2-thiol (57.1 mg, 0.500 mmol), and extra-dry EtOAc (8.0 mL) were added. The flask was filled with oxygen balloon (0.3 MPa) and the reaction mixture was stirred at 70 °C for a duration of 16 h. Subsequently, the reaction mixture was cooled by removing from the oil-bath. The reaction mixture was diluted with EtOAc (10.0 mL) and HCl (15.0 mL, 0.100 M, aq). The aqueous layer was extracted with EtOAc (3 × 15.0 mL). Organic layers were combined, dried over MgSO4, filtered, and concentrated, after chromatography (0–12.5% EtOAc/Hexane), affording 53.7 mg of 2ad in 95% yield as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.40 (m, 2H), 6.35 (m, 2H), 6.24 (m, 2H), 3.70 (s, 4H); 13C{1H} NMR (126 MHz, CDCl3) δ 150.3, 142.5, 110.8, 109.0, 35.7.
- 1,2-Bis(2-methylfuran-3-yl)disulfane (2ae) [49]. To a round-bottom flask in oil-bath, Sm-OC (9.10 mg, 1.15 mol%), 2-methylfuran-3-thiol (34.2 mg, 0.300 mmol), and extra-dry EtOAc (8.0 mL) were added. The flask was filled with oxygen balloon (0.3 MPa) and the reaction mixture was stirred at 70 °C for a duration of 16 h. Subsequently, the reaction mixture was cooled by removing from the oil-bath. The reaction mixture was diluted with Hexane (10.0 mL) and NaOH (15 mL, 1.00 M, aq). The aqueous layer was extracted with Hexane (3 × 15.0 mL). Organic layers were combined, dried over MgSO4, filtered, and concentrated, affording 30.2 mg of 2ae in 89% yield as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.27 (m, 2H), 6.38 (m, 2H), 2.10 (m, 6H); 13C{1H} NMR (126 MHz, CDCl3) δ 157.1, 140.9, 114.8, 112.8, 11.5.
- 1,2-Di(thiophen-2-yl)disulfane (2af) [41]. To a round-bottom flask in oil-bath, Sm-OC (9.10 mg, 1.15 mol%), thiophene-2-thiol (58.1 mg, 0.500 mmol), and extra-dry EtOAc (8.0 mL) were added. The flask was filled with oxygen balloon (0.3 MPa) and the reaction mixture was stirred at 70 °C for a duration of 16 h. Subsequently, the reaction mixture was cooled by removing from the oil-bath. The reaction mixture was diluted with EtOAc (10.0 mL) and HCl (15 mL, 0.100 M, aq). The aqueous layer was extracted with EtOAc (3 × 15.0 mL). Organic layers were combined, dried over MgSO4, filtered, and concentrated, after chromatography (100% Hexane), affording 51.8 mg of 2af in 90% yield as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.51 (dd, J = 5.3, 1.4 Hz, 2H), 7.17 (dd, J = 3.7, 1.4 Hz, 2H), 7.02 (dd, J = 5.3, 3.7 Hz, 2H); 13C{1H} NMR (126 MHz, CDCl3) δ 135.8, 135.7, 132.3, 127.8.
- Methyl5-(1,2-dithiolan-3-yl)pentanoate (2ag) [50]. To a round-bottom flask in oil-bath, Sm-OC (18.2 mg, 2.30 mol%), methyl -6,8-dimercaptooctanoate (66.7 mg, 0.300 mmol), and extra-dry EtOAc (16.0 mL) were added. The flask was filled with oxygen balloon (0.3 MPa) and the reaction mixture was stirred at 70 °C for a duration of 16 h. Subsequently, the reaction mixture was cooled by removing from the oil-bath. The reaction mixture was diluted with EtOAc (10.0 mL) and HCl (15.0 mL, 0.100 M, aq). The aqueous layer was extracted with EtOAc (3 × 15.0 mL). Organic layers were combined, dried over MgSO4, filtered, and concentrated, after chromatography (0–12.5% EtOAc/Hexane), affording 34.4 mg of 2ag in 52% yield as a yellow oil. 1H NMR (500 MHz, CDCl3) δ 3.68 (s, 3H), 3.57 (m, 1H), 3.19 (m, 1H), 3.12 (m, 1H), 2.47 (m, 1H), 2.33 (t, J = 7.5 Hz, 2H), 1.92 (m, 1H), 1.72–1.64 (m, 4H), 1.52–1.44 (m, 2H); 13C{1H} NMR (126 MHz, CDCl3) δ 174.0, 56.4, 51.6, 40.3, 38.6, 34.7, 33.9, 28.8, 24.7.
- (4R,5R)-1,2-dithiane-4,5-diol (2ah) [51]. To a round-bottom flask in oil-bath, Sm-OC (9.10 mg, 1.15 mol%), (2R,3R)-1,4-dimercaptobutane-2,3-diol (46.3 mg, 0.300 mmol), and extra-dry EtOAc (8.0 mL) were added. The flask was filled with oxygen balloon (0.3 MPa) and the reaction mixture was stirred at 70 °C for a duration of 16 h. Subsequently, the reaction mixture was cooled by removing from the oil-bath. The reaction mixture was diluted with EtOAc (10.0 mL) and HCl (15 mL, 0.100 M, aq). The aqueous layer was extracted with EtOAc (3 × 15.0 mL). Organic layers were combined, dried over MgSO4, filtered, and concentrated, after chromatography (0–10% EtOAc/Hexane), affording 26.9 mg of 2ah in 59% yield as a white solid. 1H NMR (500 MHz, Methanol-d4) δ 3.58–3.42 (m, 2H), 3.12–2.96 (m, 2H), 2.93–2.84 (m, 2H); 13C{1H} NMR (126 MHz, Methanol-d4) δ 75.6, 41.7.
- Dimethyl 3,3′-disulfanediyl(2R,2′R)-bis(2-((tert-butoxycarbonyl)amino)propanoate) (2ai) [21]. To a round-bottom flask in oil-bath, Sm-OC (18.2 mg, 2.30 mol%), methyl (tert-butoxycarbonyl)-L-cysteinate (70.6 mg, 0.300 mmol), and extra-dry EtOAc (16.0 mL) were added. The flask was filled with oxygen balloon (0.3 MPa) and the reaction mixture was stirred at 70 °C for a duration of 16 h. Subsequently, the reaction mixture was cooled by removing from the oil-bath. The reaction mixture was diluted with EtOAc (10.0 mL) and HCl (15.0 mL, 0.100 M, aq). The aqueous layer was extracted with EtOAc (3 × 15.0 mL). Organic layers were combined, dried over MgSO4, filtered, and concentrated, after chromatography (20–30% EtOAc/Hexane), affording 47.1 mg of 2ai in 67% yield as a white solid. 1H NMR (500 MHz, DMSO-d6) δ 7.38 (d, J = 8.2 Hz, 2H), 4.26 (m, 2H), 3.64 (s, 6H), 3.08 (m, 2H), 2.90 (m, 2H), 1.38 (s, 18H); 13C{1H} NMR (126 MHz, DMSO-d6) δ 171.4, 155.3, 78.5, 52.7, 52.1, 39.1, 28.1.
- (2R,2′R)-3,3′-disulfanediylbis(2-acetamidopropanoic acid) (2aj) [52]. To a round-bottom flask in oil-bath, Sm-OC (18.2 mg, 2.30 mol%), acetyl-L-cysteine (49.3 mg, 0.300 mmol), and extra-dry EtOAc (16.0 mL) were added. The flask was filled with oxygen balloon (0.3 MPa) and the reaction mixture was stirred at 70 °C for a duration of 16 h. Subsequently, the reaction mixture was cooled by removing from the oil-bath. The reaction mixture was diluted with EtOAc (10.0 mL) and HCl (15.0 mL, 0.100 M, aq). The aqueous layer was extracted with EtOAc (3 × 15.0 mL). Organic layers were combined, dried over MgSO4, filtered, and concentrated, affording 40.0 mg of 2aj in 82% yield as a white solid. 1H NMR (500 MHz, D2O) δ 4.68 (dd, J = 8.6, 4.3 Hz, 2H), 3.38 (dd, J = 14.1, 4.3 Hz, 2H), 3.03 (dd, J = 14.1, 8.6 Hz, 2H), 2.04 (s, 6H); 13C{1H} NMR (126 MHz, D2O) δ 177.7, 174.1, 53.2, 39.4, 21.8. HRMS (ESI-TOF) calcd. For C10H16N2O6S2 [M + H]+ 325.0522, found: 325.0521.
- 1-((3s,5s,7s)-adamantan-1-yl)-2-dodecyldisulfane (2ak) [20]. To a round-bottom flask in oil-bath, Sm-OC (9.10 mg, 1.15 mol%), dodecane-1-thiol (40.5 mg, 0.200 mmol), (3s,5s,7s)-adamantane-1-thiol (101.0 mg, 0.600 mmol), and extra-dry EtOAc (8.0 mL) were added. The flask was filled with oxygen balloon (0.3 MPa) and the reaction mixture was stirred at 70 °C for a duration of 16 h. Subsequently, the reaction mixture was cooled by removing from the oil-bath. The reaction mixture was diluted with EtOAc (10.0 mL) and HCl (15.0 mL, 0.100 M, aq). The aqueous layer was extracted with EtOAc (3 × 15.0 mL). Organic layers were combined, dried over MgSO4, filtered, and concentrated, after chromatography (100% Hexane), affording 57.5mg of 2ak in 78% yield as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 2.67 (t, J = 7.0 Hz, 2H), 2.12–2.03 (m, 3H), 1.89–1.82 (m, 6H), 1.74–1.61 (m, 8H), 1.42–1.23 (m, 18H), 0.89 (t, J = 7.0 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 49.3, 42.7, 41.6, 36.3, 32.0, 29.9, 29.7(×3), 29.6, 29.4(×2), 29.3, 28.7, 22.8, 14.2.
- 1-((3s,5s,7s)-adamantan-1-yl)-2-(p-tolyl)disulfane (2al) [53]. To a round-bottom flask in oil-bath, Sm-OC (9.10 mg, 1.15 mol%), 4-methylbenzenethiol (24.8 mg, 0.200 mmol), (3s,5s,7s)-adamantane-1-thiol (101.0 mg, 0.600 mmol), and extra-dry EtOAc (8.0 mL) were added. The flask was filled with oxygen balloon (0.3 MPa) and the reaction mixture was stirred at 70 °C for a duration of 16 h. Subsequently, the reaction mixture was cooled by removing from the oil-bath. The reaction mixture was diluted with EtOAc (10.0 mL) and HCl (15.0 mL, 0.100 M, aq). The aqueous layer was extracted with EtOAc (3 × 15.0 mL). Organic layers were combined, dried over MgSO4, filtered, and concentrated, after chromatography (100% Hexane), affording 43.6 mg of 2al in 75% yield as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.46 (m, 2H), 7.11 (m, 2H), 2.33 (s, 3H), 2.06–2.02 (m, 3H), 1.87–1.84 (m, 6H), 1.68–1.62 (m, 6H); 13C NMR{1H} (126 MHz, CDCl3) δ 136.1, 135.9, 129.5, 127.1, 50.7, 42.6, 36.2, 30.0, 21.1.
3.5. Procedure of Control Experiments (Table 2)
3.6. Synthesis of Methyl 6,8-Dimercaptooctanoate [54]
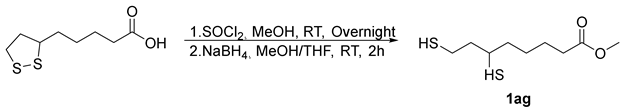
3.7. Recycle Experiment of Sm-OC
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jiang, H.Q.; Wang, Q.Y.; Zang, S.Y.; Li, J.S.; Wang, Q.F. Enhanced Photoactivity of Sm, N, P-tridoped Anatase-TiO2 nano-photocatalyst for 4-chlorophenol degradation under sunlight irradiation. J. Hazard. Mater. 2013, 261, 44–54. [Google Scholar] [CrossRef]
- Prieto, A.; Jaroschik, F. Recent Applications of Rare Earth Complexes in Photoredox Catalysis for Organic Synthesis. Curr. Org. Chem. 2022, 26, 6–41. [Google Scholar] [CrossRef]
- Zheng, X.Y.; Xie, J.; Kong, X.J.; Long, L.S.; Zheng, L.S. Recent Advances in the Assembly of High-nuclearity Lanthanide Clusters. Coord. Chem. Rev. 2019, 378, 222–236. [Google Scholar] [CrossRef]
- Cotton, S.; Raithby, P. Systematics and Surprises in Lanthanide Coordination Chemistry. Coord. Chem. Rev. 2017, 340, 220–231. [Google Scholar] [CrossRef]
- Blais, C.; Morvan, T.; Daiguebonne, C.; Suffren, Y.; Calvez, G.; Bernot, K.; Guillou, O. Lanthanide-Based Coordination Polymers Molecular Alloys Stability: A Thermochemical Approach. Inorg. Chem. 2023, 62, 4495–4502. [Google Scholar] [CrossRef]
- Teng, H.L.; Luo, Y.; Wang, B.L.; Zhang, L.; Nishiura, M.; Hou, Z.M. Synthesis of Chiral Aminocyclopropanes by Rare-Earth-Metal-Catalyzed Cyclopropene Hydroamination. Angew. Chem., Int. Ed. 2016, 55, 15406–15410. [Google Scholar] [CrossRef]
- Teng, H.-L.; Luo, Y.; Nishiura, M.; Hou, Z.-M. Diastereodivergent Asymmetric Carboamina-tion/Annulation of Cyclopropenes with Aminoalkenes by Chiral Lanthanum Catalysts. J. Am. Chem. Soc. 2017, 139, 16506–16509. [Google Scholar] [CrossRef]
- Song, G.; Wylie, W.; Hou, Z. Enantioselective C-H Bond Addition of Pyridines to Alkenes Catalyzed by Chiral Half-Sandwich Rare-Earth Complexes. J. Am. Chem. 2014, 136, 12209–12212. [Google Scholar] [CrossRef]
- Newton, C.; Kossler, D.; Cramer, N. Asymmetric Catalysis Powered by Chiral Cyclopentadienyl Lig-ands. J. Am. Chem. Soc. 2016, 138, 3935–3941. [Google Scholar] [CrossRef]
- Shan, H.; Ling, L.; Hu, J.; Zhang, H. Application in the Asymmetric Catalytic Reactions of Chiral Cyclopentadienyl-Transition-Metal Complexes. Chin. J. Org. Chem. 2019, 39, 1548–1556. [Google Scholar]
- Luo, Y.; Teng, H.L.; Xue, C.; Nishiura, M.; Hou, Z. Yttrium-Catalyzed Regioselective α-C–H Silylation of Methyl Sulfides with Hydrosilanes. ACS Catal. 2018, 8, 8027–8032. [Google Scholar] [CrossRef]
- Guan, Y.; Chang, K.; Sun, Q.; Xu, X. Progress in Rare-Earth Metal-Based Lewis Pair Chemistry. Chin. J. Org. Chem. 2022, 42, 1326–1335. [Google Scholar] [CrossRef]
- Sun, Q.; Xu, X.; Xu, X. Recent Advances in Rare-Earth Metal-Catalyzed C−H Functionalization Reactions. ChemCatChem 2022, 14, e202201083. [Google Scholar] [CrossRef]
- Xue, C.; Luo, Y.; Teng, H.; Ma, Y.; Nishiura, M.; Hou, Z. Ortho-Selective C-H Borylation of Aromatic Ethers with Pinacol-borane by Organo Rare-Earth Catalysts. ACS Catal. 2018, 8, 5017–5022. [Google Scholar] [CrossRef]
- Barger, C.J.; Dicken, R.D.; Weidner, V.L.; Motta, A.; Lohr, T.L.; Marks, T.J. La[N(SiMe3)2]3-Catalyzed Deoxygenative Reduction of Amides with Pinacolborane. Scope and Mechanism. J. Am. Chem. Soc. 2020, 142, 8019–8028. [Google Scholar] [CrossRef]
- Kaufmann, S.; Roesky, P.W. Investigating a Redox Active Samarium Complex in Catalytic Reactions. Eur. J. Inorg. Chem. 2021, 2021, 2899–2905. [Google Scholar] [CrossRef]
- Tamang, S.R.; Singh, A.; Bedi, D.; Bazkiaei, A.R.; Warner, A.A.; Glogau, K.; McDonald, C.; Unruh, D.K.; Findlater, M. Poly-nuclear Lanthanide-diketonato Clusters for the Catalytic Hydroboration of Carboxamides and Esters. Nat. Catal. 2020, 3, 154–162. [Google Scholar] [CrossRef]
- Ouyang, T.; Huang, H.H.; Wang, J.W.; Zhong, D.C.; Lu, T.B. A Dinuclear Cobalt Cryptate as a Homogeneous Photocatalyst for Highly Selective and Efficient Visible-Light Driven CO2 Reduction to CO in CH3CN/H2O Solution. Angew. Chem.-Int. Edit. 2017, 56, 738–743. [Google Scholar] [CrossRef]
- Zheng, Z.P. Recent Development in Clusters of Rare Earths and Actinides: Chemistry and Materials; Springer: Berlin/Heidelberg, Germany, 2017; pp. 1–49. [Google Scholar]
- Wang, L.J.; Chen, L.X.; Qin, Z.X.; Zhao, B.H.; Ni, K.; Li, H.Z.; Li, J.Y.; Duan, H.X.; Ren, F.Z.; An, J. Samarium-Oxo/Hydroxy Cluster: A Solar Photocatalyst for Chemoselective Aerobic Oxidation of Thiols for Disulfide Synthesis. J. Org. Chem. 2024, 89, 8357–8362. [Google Scholar] [CrossRef]
- Guo, J.M.; Zha, J.J.; Zhang, T.; Ding, C.H.; Tan, Q.T.; Xu, B. PdCl/DMSO-Catalyzed Thiol-Disulfide Exchange: Synthesis of Unsymmetrical Disulfide. Org. Lett. 2021, 23, 3167–3172. [Google Scholar] [CrossRef]
- Bottec-chia, C.; Erdmann, N.; Tijssen, P.M.A.; Milroy, L.G.; Brunsveld, L.; Hessel, V.; Noël, T. Batch and Flow Synthesis of Disulfides by Visible-Light-Induced TiO2 Photocatalysis. Chemsuschem 2016, 9, 1781–1785. [Google Scholar] [CrossRef]
- Wang, L.J.; Chen, L.X.; Qin, Z.X.; Ni, K.; Li, X.; Yu, Z.; Kuang, Z.C.; Qin, X.S.; Duan, H.X.; An, J. Application of Iodine as a Catalyst in Aerobic Oxidations: A Sustainable Approach for Thiol Oxidations. Molecules 2023, 28, 6789. [Google Scholar] [CrossRef]
- Chen, L.X.; Li, J.Y.; Ni, K.; Qin, X.S.; Wang, L.J.; Hou, J.M.; Wang, C.; Li, X.; Wang, M.L.; An, J. Innovative Application of Polyether Amine as a Recyclable Catalyst in Aerobic Thiophenol Oxidation. Organics 2024, 5, 59–70. [Google Scholar] [CrossRef]
- Bang, E.-K.; Lista, M.; Sforazzini, G.; Sakai, N.; Matile, S. Poly(disulfide)s. Chem. Sci. 2012, 3, 1752–1763. [Google Scholar] [CrossRef]
- Bargh, J.; Isidro-Llobet, A.; Parker, J.; Spring, D. Cleavable linkers in antibody-drug conjugates. Chem. Soc. Rev. 2019, 48, 4361–4374. [Google Scholar] [CrossRef]
- Li, H.; Peng, M.; Li, J.; Do, H.; Ni, K.; Wang, M.; Yuan, Z.; Wang, L.; Zhao, T.; Zhang, X.; et al. Redox-Click Chemistry for Disulfide Formation from Thiols. ChemRxiv 2023. preprint. [Google Scholar] [CrossRef]
- Zhang, R.; Nie, T.; Fang, Y.; Huang, H.; Wu, J. Poly(disulfide)s: From Synthesis to Drug Delivery. Biomacromolecules 2022, 23, 1–19. [Google Scholar] [CrossRef]
- Laurent, Q.; Martinent, R.; Lim, B.; Pham, A.; Kato, T.; Lopez-Andarias, J.; Sakai, N.; Matile, S. Thiol-Mediated Uptake. JACS Au 2021, 1, 710–728. [Google Scholar] [CrossRef]
- Fass, D.; Thorpe, C. Chemistry and Enzymology of Disulfide Cross-Linking in Proteins. Chem. Rev. 2018, 118, 1169–1198. [Google Scholar] [CrossRef]
- Wang, M.; Jiang, X. Sulfur-Sulfur Bond Construction. Top. Curr. Chem. 2018, 376, 14–53. [Google Scholar] [CrossRef]
- Ghosh, I.; Khamrai, J.; Savateev, A.; Shlapakov, N.; Antonietti, M.; Konig, B. Organic Semiconductor Photocatalyst Can Bifunctionalize Arenes and Heteroarenes. Science 2019, 365, 360–366. [Google Scholar] [CrossRef]
- Yuan, T.; Sun, L.; Wu, Z.; Wang, R.; Cai, X.; Lin, W.; Zheng, M.; Wang, X. Mild and Metal-free Birch-type Hydrogenation of (hetero)arenes with Boron Carbonitride in Water. Nat. Catal. 2022, 5, 1157–1168. [Google Scholar] [CrossRef]
- Zhang, Z.; Qiu, C.; Xu, Y.; Han, Q.; Tang, J.; Loh, K.P.; Su, C. Semiconductor Photocatalysis to Engineering Deuterated n-Alkyl Pharmaceuticals Enabled by Synergistic Activation of Water and Alkanols. Nat. Commun. 2020, 11, 4722. [Google Scholar] [CrossRef]
- Zhang, S.; Ge, Q.; Guo, D.; Hu, W.; Liu, H. Synthesis and Anti-cancer Evaluation of α-lipoic acid Derivatives. Bioorg. Med. Chem. Lett. 2010, 20, 3078–3083. [Google Scholar] [CrossRef]
- Galanis, A.S.; Albericio, F.; Grotli, M. Solid-Phase Peptide Synthesis in Water Using Microwave-Assisted Heating. Org. Lett. 2009, 11, 4488–4491. [Google Scholar] [CrossRef]
- Spiliopoulou, N.; Kokotos, C.G. Photochemical Metal-Free Aerobic Oxidation of Thiols to Disulfides. Green Chem. 2021, 23, 546–551. [Google Scholar] [CrossRef]
- Primas, N.; Lano, G.; Brun, D.; Curti, C.; Sallee, M.; Sampol-Manos, E.; Lamy, E.; Bornet, C.; Burtey, S.; Vanelle, P. Stability Study of Parenteral N-Acetylcysteine, and Chemical Inhibition of Its Dimerization. Pharmaceuticals 2023, 16, 72. [Google Scholar] [CrossRef]
- Musiejuk, M.; Witt, D. Recent Developments in the Synthesis of Unsymmetrical Disulfanes (Disulfides). A Review. Org. Prep. Proced. Int. 2015, 47, 95–131. [Google Scholar] [CrossRef]
- Song, L.; Li, W.; Duan, W.; An, J.; Tang, S.; Li, L.; Yang, G. Natural Gallic Acid Catalyzed Aerobic Oxidative Coupling with the Assistance of MnCO3 for Synthesis of Disulfanes in Water. Green Chem. 2019, 21, 1432–1438. [Google Scholar] [CrossRef]
- Arisawa, M.; Sugata, C.; Yamaguchi, M. Oxidation/Reduction Interconversion of Thiols and Disulfides Using Hydrogen and Oxygen Catalyzed by a Rhodium Complex. Tetrahedron Lett. 2005, 46, 6097–6099. [Google Scholar] [CrossRef]
- Yue, H.; Wang, J.; Xie, Z.; Tian, J.; Sang, D.; Liu, S. 1,3-Diisopropylcarbodiimide-Mediated Synthesis of Disulfides from Thiols. ChemistrySelect 2020, 5, 4273–4277. [Google Scholar] [CrossRef]
- Xu, H.; Zhang, Y.-F.; Lang, X. TEMPO Visible Light Photocatalysis: The Selective Aerobic Oxidation of Thiols to Disulfides. Chin. Chem. Lett. 2020, 31, 1520–1524. [Google Scholar] [CrossRef]
- Bhattacherjee, D.; Sufian, A.; Mahato, S.K.; Begum, S.; Banerjee, K.; De, S.; Srivastava, H.K.; Bhabak, K.P. Trisulfides over Disulfides: Highly Selective Synthetic Strategies, Anti-Proliferative Activities and Sustained H2S Release Profiles. Chem. Commun. 2019, 55, 13534–13537. [Google Scholar] [CrossRef]
- Howard, J.L.; Schotten, C.; Alston, S.T.; Browne, D.L. Preparation of Difluoromethylthioethers through Difluoromethylation of Disulfides Using TMS-CF2H. Chem. Commun. 2016, 52, 8448–8451. [Google Scholar] [CrossRef]
- Hayashi, M.; Okunaga, K.; Nishida, S.; Kawamura, K.; Eda, K. Oxidative Transformation of Thiols to Disulfides Promoted by Activated Carbon–Air System. Tetrahedron Lett. 2010, 51, 6734–6736. [Google Scholar] [CrossRef]
- Bartolozzi, A.; Foudoulakis, H.M.; Cole, B.M. Development of a Tandem Base-Catalyzed, Triphenylphosphine-Mediated Disulfide Reduction-Michael Addition. Synthesis 2008, 2008, 2023–2032. [Google Scholar] [CrossRef]
- Li, X.; Fan, J.; Cui, D.; Yan, H.; Shan, S.; Lu, Y.; Cheng, X.; Loh, T.-P. Catalyst- and Metal-Free Photo-Oxidative Coupling of Thiols with BrCCl3. Eur. J. Org. Chem. 2022, 2022, e202200340. [Google Scholar] [CrossRef]
- Ryu, S.H.; Ra, J.; Ko, H.M. Efficient Synthesis of Sulfenamides through Mitsunobu-Type Coupling Reaction of Thiols with Amines Using Dibenzyl Azodicarboxylate. Asian J. Org. Chem. 2020, 9, 933–938. [Google Scholar] [CrossRef]
- Sarbova, V.; Koschella, A.; Cheng, F.; Kelly, S.M.; Heinze, T. Studies on the Sulfation of Cellulose α-Lipoate and Ability of the Sulfated Product to Stabilize Colloidal Suspensions of Gold Nanoparticles. Carbohydr. Polym. 2015, 124, 117–123. [Google Scholar] [CrossRef]
- Calandra, N.A.; Cheng, Y.L.; Kocak, K.A.; Miller, J.S. Total Synthesis of Spiruchostatin A via Chemoselective Macrocyclization Using an Accessible Enantiomerically Pure Latent Thioester. Org. Lett. 2009, 11, 1971–1974. [Google Scholar] [CrossRef]
- Oba, M.; Tanaka, K.; Nishiyama, K.; Ando, W. Aerobic Oxidation of Thiols to Disulfides Catalyzed by Diaryl Tellurides under Photosensitized Conditions. J. Org. Chem. 2011, 76, 4173–4177. [Google Scholar] [CrossRef] [PubMed]
- Delarue Bizzini, L.; Zwick, P.; Mayor, M. Preparation of Unsymmetrical Disulfides from Thioacetates and Thiosulfonates. Eur. J. Org. Chem. 2019, 2019, 6956–6960. [Google Scholar] [CrossRef]
- Cravero, R.M.; Luna, L.E.; Barboza, A.V. A Novel Method for the Synthesis of (1,8-Dioxo-2,3,4,5,6,7,8,9-Octahydro-1H-Xanthen-9-Yl)Acetic Acids on Solid Phase. Synthesis 2011, 2011, 4027–4032. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Entry | Solvent | Catalyst (mol%) | Temperature (°C) | Time (h) | Yield a (%) |
| 1 | AcOEt | 10 | 70 | 16 | >98 |
| 2 | AcOEt | 5 | 70 | 16 | >98 |
| 3 | AcOEt | 2 | 70 | 16 | >98 |
| 4 | AcOEt | 1 | 70 | 16 | >98 |
| 5 | AcOEt | 0.2 | 70 | 16 | 4 |
| 6 | MeOH | 10 | rt | 16 | 22 |
| 7 | EtOH | 10 | rt | 16 | 26 |
| 8 | MeCN | 10 | rt | 16 | 36 |
| 9 | THF | 10 | rt | 16 | 44 |
| 10 | AcOEt | 10 | rt | 16 | 42 |
| 11 | AcOEt | 1 | 70 | 4 | 28 |
| 12 | AcOEt | 1 | 70 | 1 | 13 |
 | |||
|---|---|---|---|
| Entry | Solvent | Catalyst | Yield c (%) |
| 1 | EtOAc | / | 10 |
| 2 | EtOAc | n-Bu4NI a | 4 |
| 3 | EtOAc | NaI a | 9 |
| 4 | EtOAc | SmCl3 b | 7 |
| 5 | EtOAc | Sm2O3 b | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Qin, Z.; Chen, L.; Qin, X.; Hou, J.; Wang, C.; Li, X.; Duan, H.; Fang, B.; Wang, M.; et al. A Recyclable Inorganic Lanthanide Cluster Catalyst for Chemoselective Aerobic Oxidation of Thiols. Molecules 2024, 29, 3361. https://doi.org/10.3390/molecules29143361
Wang L, Qin Z, Chen L, Qin X, Hou J, Wang C, Li X, Duan H, Fang B, Wang M, et al. A Recyclable Inorganic Lanthanide Cluster Catalyst for Chemoselective Aerobic Oxidation of Thiols. Molecules. 2024; 29(14):3361. https://doi.org/10.3390/molecules29143361
Chicago/Turabian StyleWang, Lijun, Zixuan Qin, Lingxia Chen, Xinshu Qin, Jiaman Hou, Chao Wang, Xuan Li, Hongxia Duan, Bing Fang, Minlong Wang, and et al. 2024. "A Recyclable Inorganic Lanthanide Cluster Catalyst for Chemoselective Aerobic Oxidation of Thiols" Molecules 29, no. 14: 3361. https://doi.org/10.3390/molecules29143361
APA StyleWang, L., Qin, Z., Chen, L., Qin, X., Hou, J., Wang, C., Li, X., Duan, H., Fang, B., Wang, M., & An, J. (2024). A Recyclable Inorganic Lanthanide Cluster Catalyst for Chemoselective Aerobic Oxidation of Thiols. Molecules, 29(14), 3361. https://doi.org/10.3390/molecules29143361