
Structural Evolution of Small-Sized Phosphorus-Doped Boron Clusters: A Half-Sandwich-Structured PB15 Cluster
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
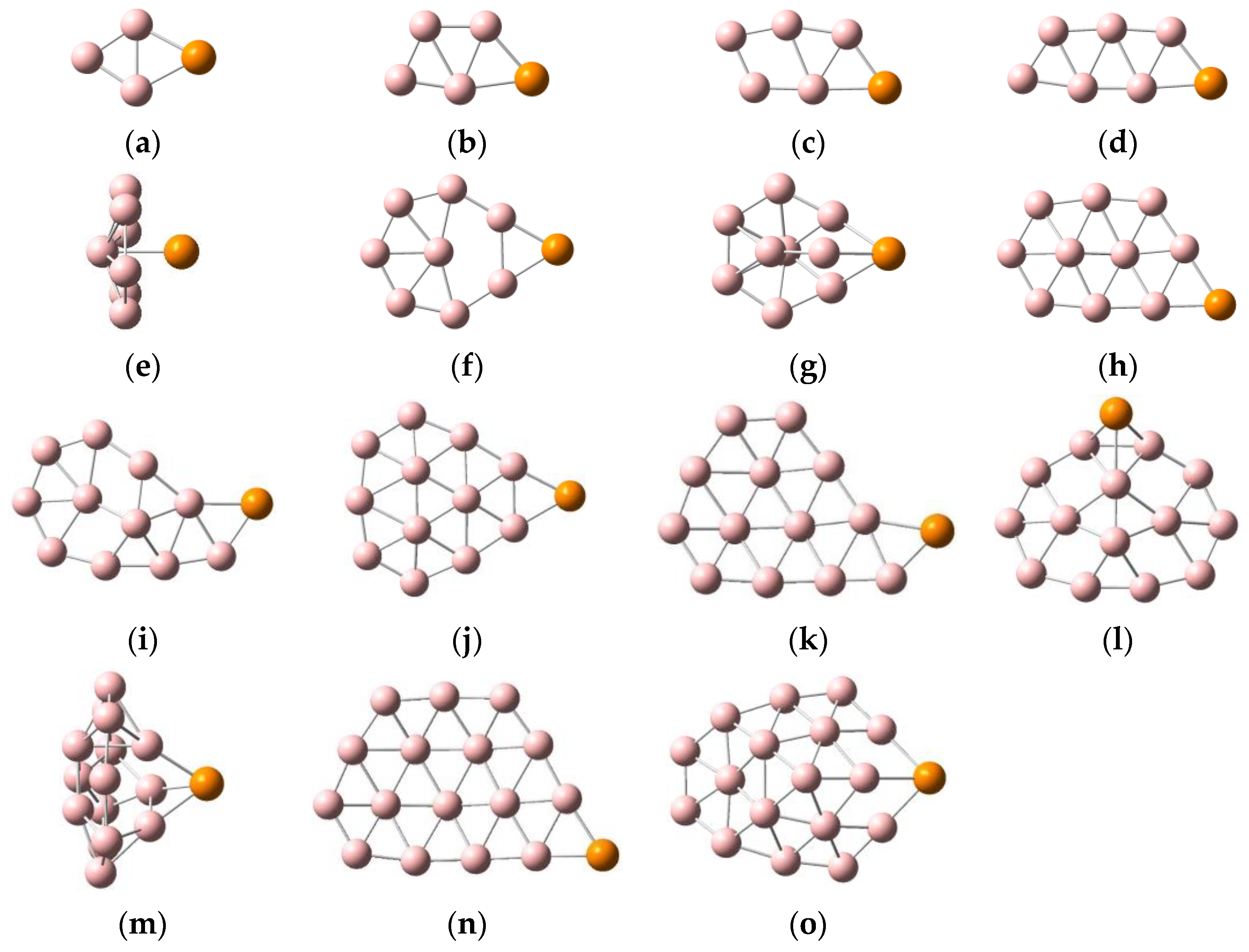
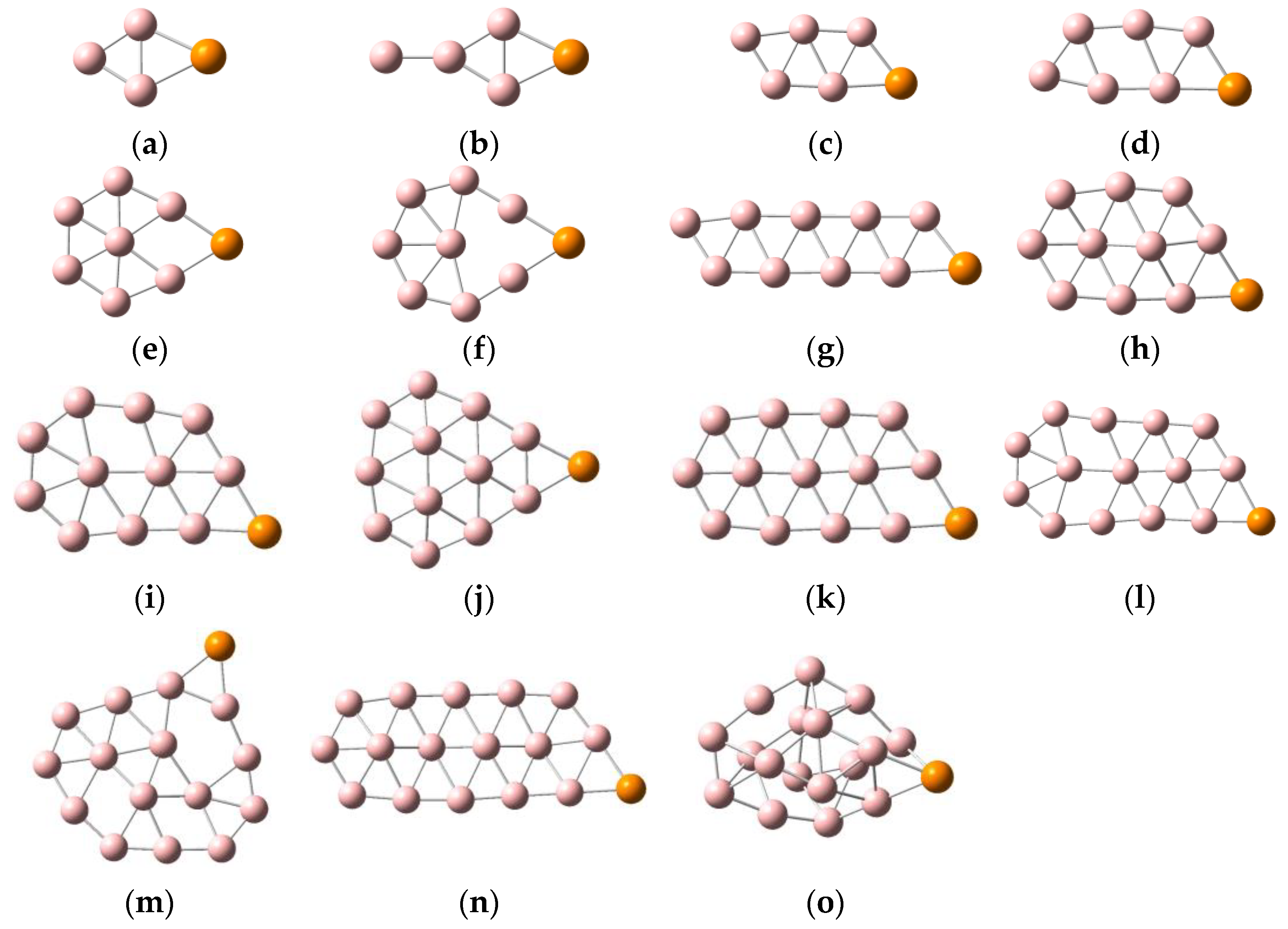
2.1. Geometric Configurations
2.2. Relative Stabilities
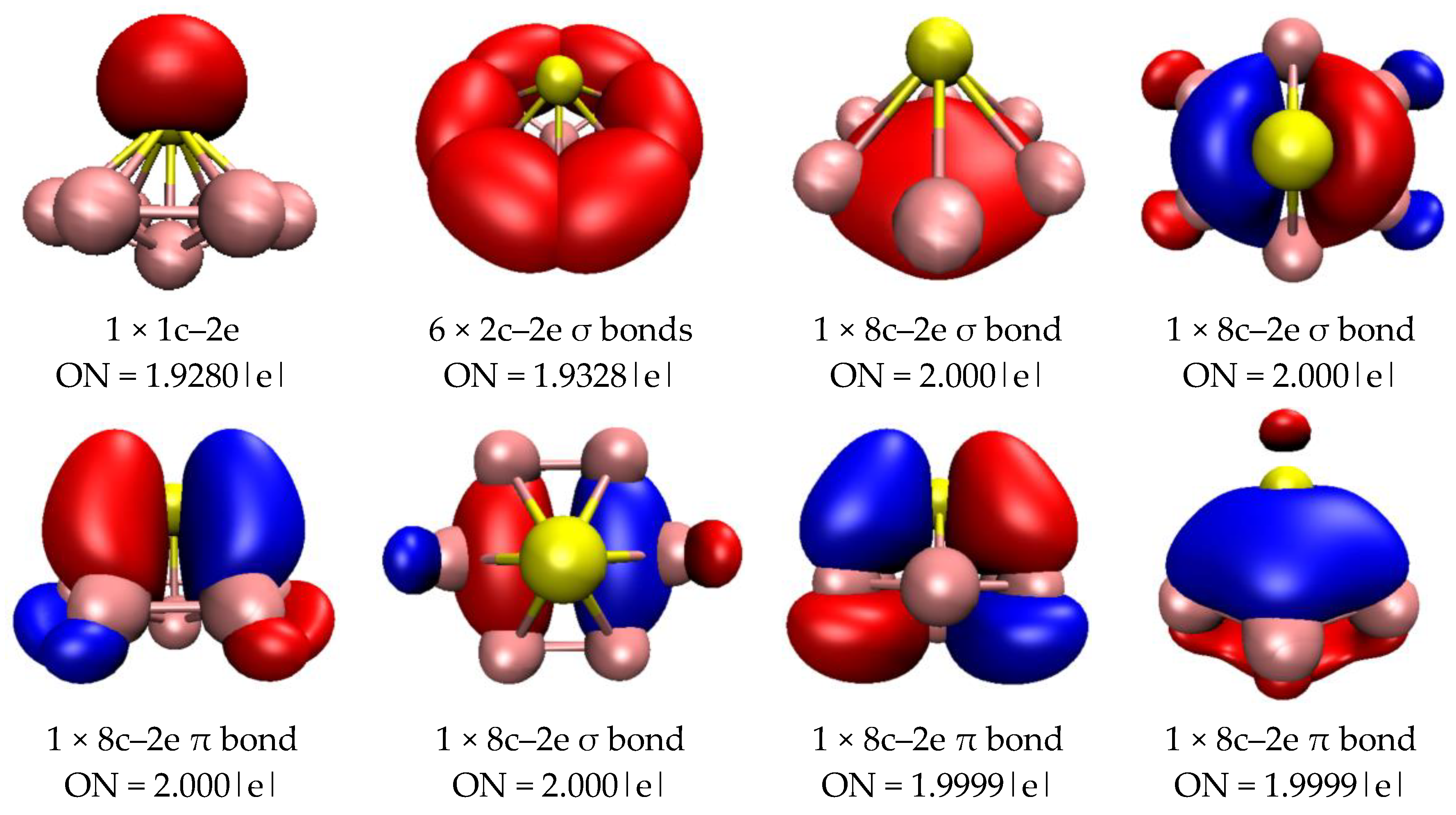
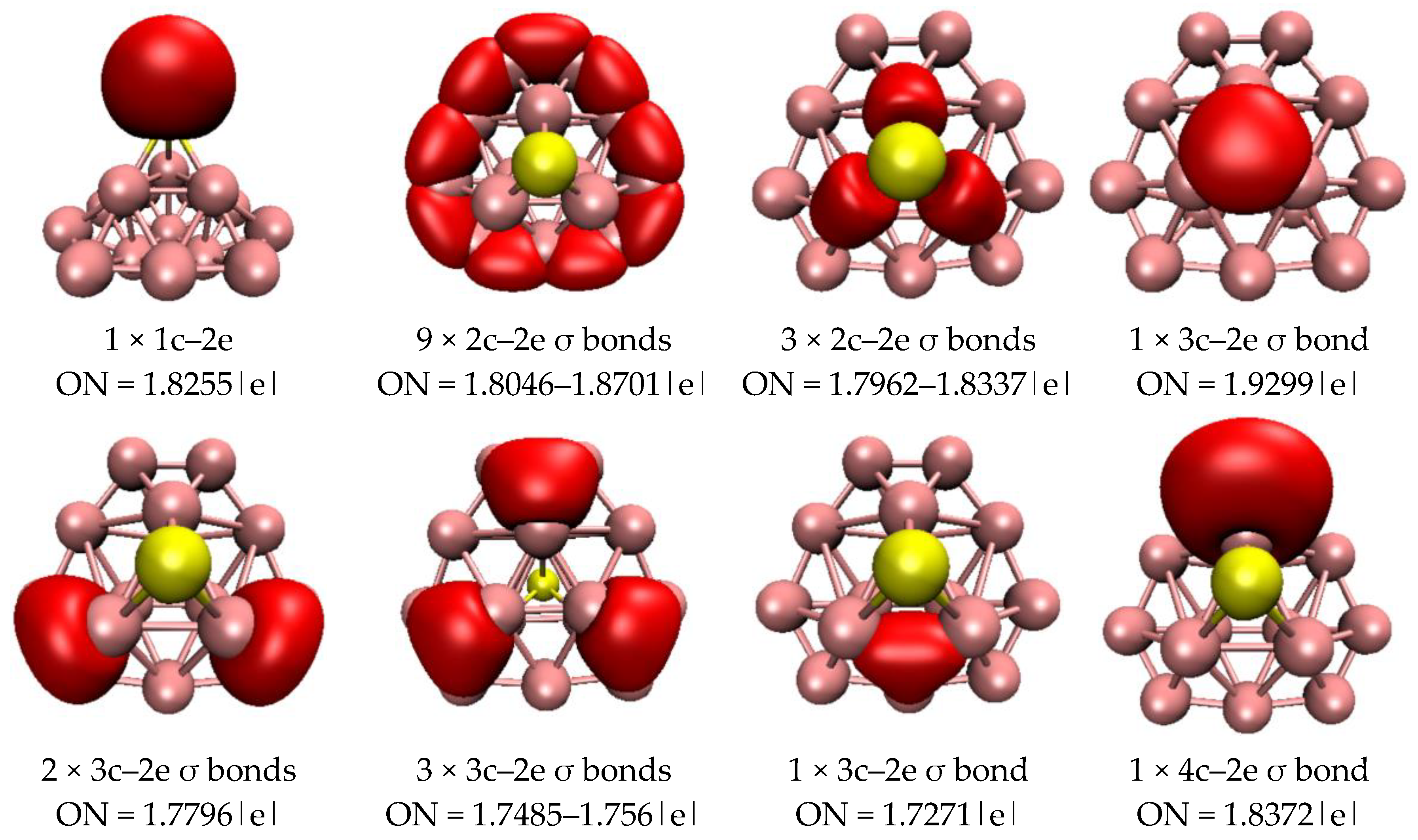
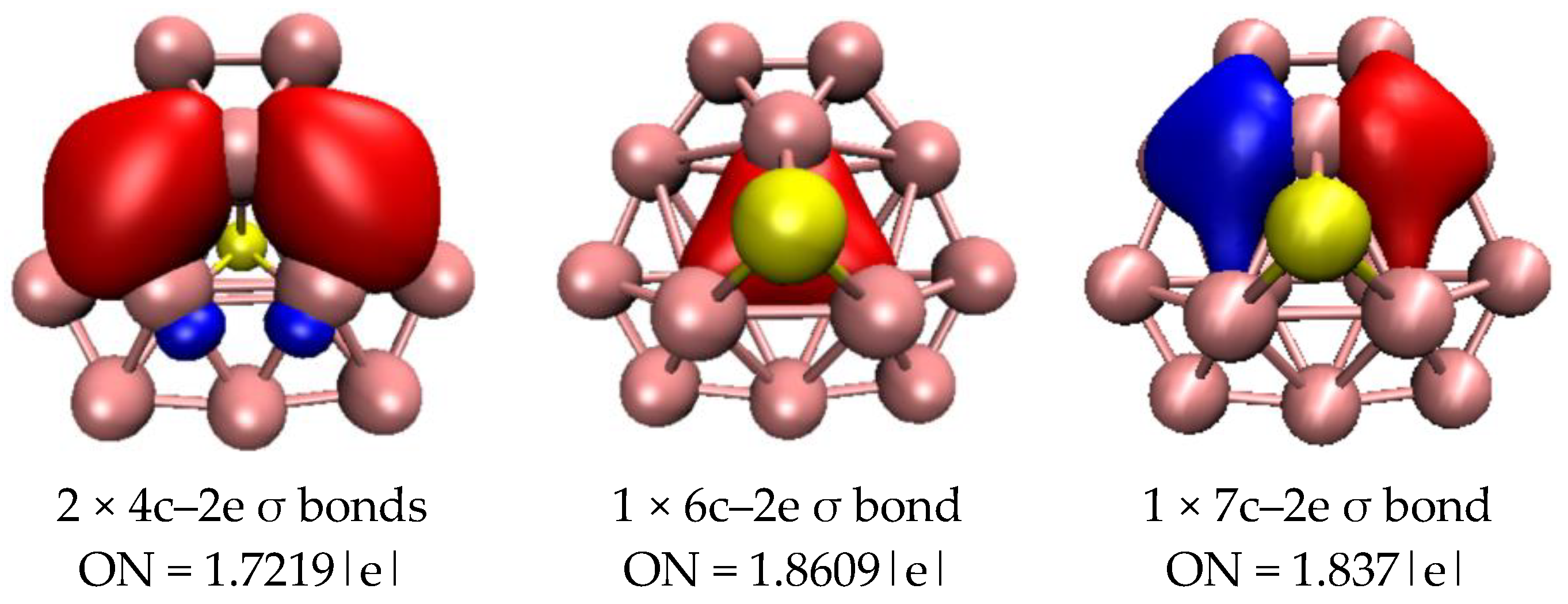
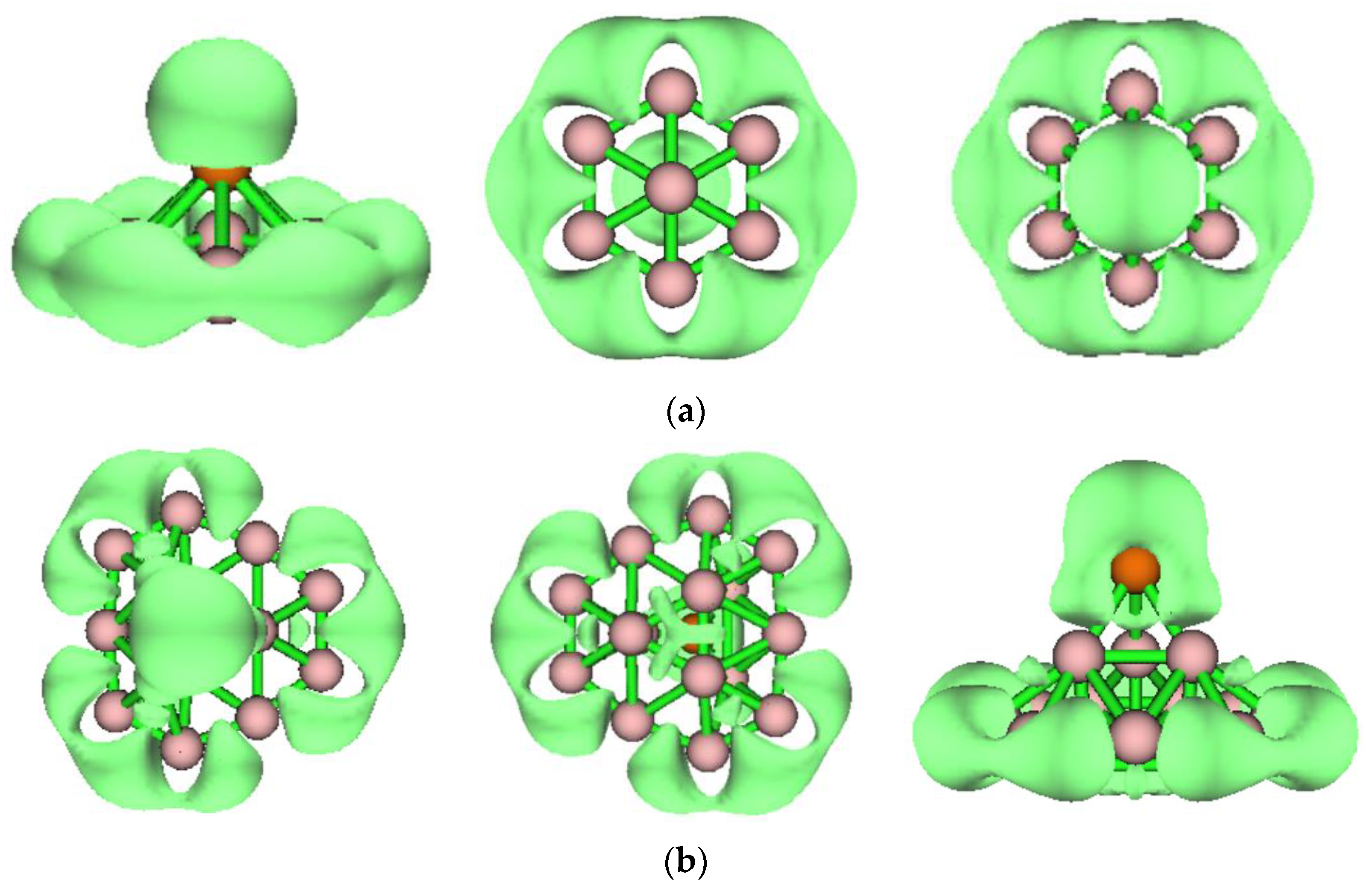
2.3. Chemical Bonding Analysis
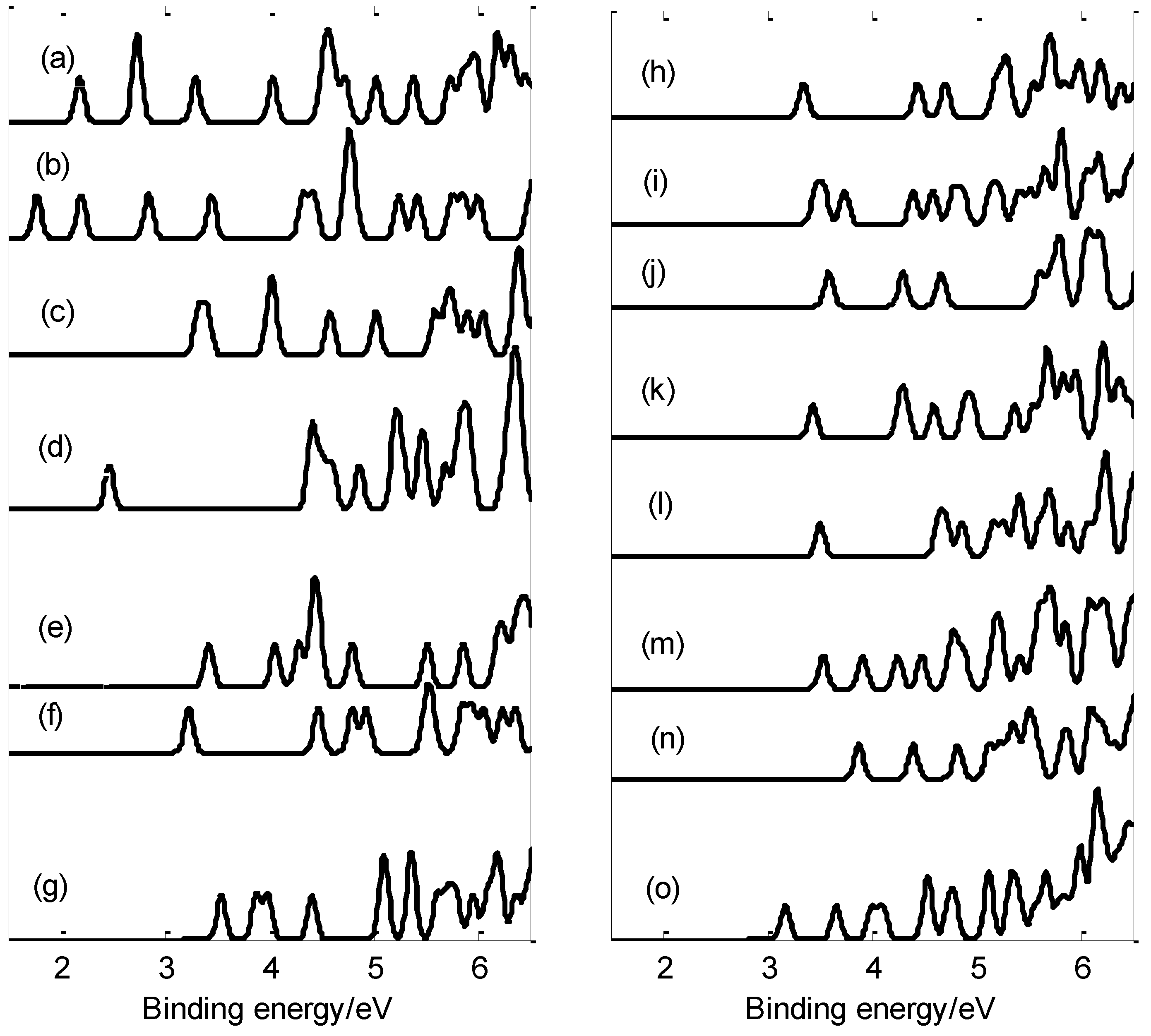
2.4. Photoelectron Spectra
3. Computation Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Harmgarth, N.; Liebing, P.; Lorenz, V.; Engelhardt, F.; Hilfert, L.; Busse, S.; Goldhahn, R.; Edelmann, F.T. Synthesis and Structural Characterization of p-Carboranylamidine Derivatives. Molecules 2023, 28, 3837. [Google Scholar] [CrossRef] [PubMed]
- Diaw-Ndiaye, F.; Sanz Miguel, P.J.; Rodríguez, R.; Macías, R. The Synthesis, Characterization, and Fluxional Behavior of a Hydridorhodatetraborane. Molecules 2023, 28, 6462. [Google Scholar] [CrossRef]
- Das, S.; Shareef, M.A.; Das, B.C. Design and Synthesis of New Boron-Based Benzo[c][1,2,5] oxadiazoles and Benzo[c][1,2,5] thiadiazoles as Potential Hypoxia Inhibitors. Inorganics 2023, 11, 34. [Google Scholar] [CrossRef]
- Teixidor, F.; Núñez, R.; Viñas, C. Towards the Application of Purely Inorganic Icosahedral Boron Clusters in Emerging Nanomedicine. Molecules 2023, 28, 4449. [Google Scholar] [CrossRef]
- Sivaev, I.B.; Bregadze, V.I. Borane, Carborane and Metallacarborane Anions for Stabilization of Transient and Highly Reactive Intermediates; World Scientific: Singapore, 2018; Volume 1, pp. 147–203. [Google Scholar]
- Iijima, S. Helical microtubules of graphitic carbon. Nature 1991, 354, 56–58. [Google Scholar] [CrossRef]
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric Field Effect in Atomically Thin Carbon Films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [PubMed]
- Kroto, H.W.; Heath, J.R.; O’Brien, S.C.; Curl, R.F.; Smalley, R.E. C60: Buckminsterfullerene. Nature 1985, 318, 162–163. [Google Scholar] [CrossRef]
- Zhai, H.; Wang, L.; Alexandrova, A.N.; Boldyrev, A.I. Electronic structure and chemical bonding of B5− and B5 by photoelectron spectroscopy and ab initio calculations. J. Chem. Phys. 2002, 117, 7917–7924. [Google Scholar] [CrossRef]
- Zhai, H.; Kiran, B.; Li, J.; Wang, L.-S. Hydrocarbon analogues of boron clusters--planarity, aromaticity and antiaromaticity. Nat. Mater. 2003, 2, 827–833. [Google Scholar] [CrossRef]
- Chen, Q.; Wei, G.F.; Tian, W.J.; Bai, H.; Liu, Z.P.; Zhai, H.J.; Li, S.D. Quasi-planar aromatic B36 and B36− clusters: All-boron analogues of coronene. Phys. Chem. Chem. Phys. 2014, 16, 18282–18287. [Google Scholar] [CrossRef]
- Boustani, I. Systematic ab initio investigation of bare boron clusters: Determination of the geometryand electronic structures of Bn (n = 2–14). Phys. Rev. B 1997, 55, 16426–16438. [Google Scholar] [CrossRef]
- Kiran, B.; Bulusu, S.; Zhai, H.J.; Yoo, S.; Zeng, X.C.; Wang, L.S. Planar-to-tubular structural transition in boron clusters: B20 as the embryo of single-walled boron nanotubes. Proc. Natl. Acad. Sci. USA 2005, 102, 961–964. [Google Scholar] [CrossRef] [PubMed]
- Bean, D.E.; Fowler, P.W. Double Aromaticity in “Boron Toroids”. J. Phys. Chem. C 2009, 113, 15569–15575. [Google Scholar] [CrossRef]
- Zhai, H.J.; Zhao, Y.F.; Li, W.L.; Chen, Q.; Bai, H.; Hu, H.S.; Piazza, Z.A.; Tian, W.J.; Lu, H.G.; Wu, Y.B.; et al. Observation of an all-boron fullerene. Nat. Chem. 2014, 6, 727–731. [Google Scholar] [CrossRef] [PubMed]
- Bai, H.; Chen, Q.; Zhai, H.J.; Li, S.D. Endohedral and exohedral metalloborospherenes: M@B40 (M = Ca, Sr) and M&B40 (M = Be, Mg). Angew. Chem. Int. Ed. 2014, 54, 941–945. [Google Scholar]
- Shakerzadeh, E.; Biglari, Z.; Tahmasebi, E. M@B40 (M = Li, Na, K) serving as a potential promising novel NLO nanomaterial. Chem. Phys. Lett. 2016, 654, 76–80. [Google Scholar] [CrossRef]
- An, Y.; Zhang, M.; Wu, D.; Fu, Z.; Wang, T.; Xia, C. Electronic transport properties of the first all-boron fullerene B40 and its metallofullerene Sr@B40. Phys. Chem. Chem. Phys. 2016, 18, 12024–12028. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Zhang, X. The hydrogen storage capacity of Sc atoms decorated porous boron fullerene B40: A DFT study. Int. J. Hydrogen Energy 2016, 41, 16992–16999. [Google Scholar] [CrossRef]
- Bai, H.; Bai, B.; Zhang, L.; Huang, W.; Mu, Y.W.; Zhai, H.J.; Li, S.D. Lithium-Decorated Borospherene B40: A Promising Hydrogen Storage Medium. Sci. Rep. 2016, 6, 35518. [Google Scholar] [CrossRef]
- Li, S.; Zhang, Z.; Long, Z.; Chen, D. Structures, Stabilities, and Spectral Properties of Endohedral Borospherenes M@B40 0/− (M = H2, HF, and H2O). ACS Omega 2019, 4, 5705–5713. [Google Scholar] [CrossRef]
- Dong, H.; Hou, T.; Lee, S.T.; Li, Y. New Ti-decorated B40 fullerene as a promising hydrogen storage material. Sci. Rep. 2015, 5, 9952. [Google Scholar] [CrossRef] [PubMed]
- Sergeeva, A.P.; Zubarev, D.Y.; Zhai, H.J.; Boldyrev, A.I.; Wang, L.S. A Photoelectron Spectroscopic and Theoretical Study of B16− and B162−: An All-Boron Naphthalene. J. Am. Chem. Soc. 2008, 130, 7244–7246. [Google Scholar] [CrossRef] [PubMed]
- Piazza, Z.A.; Hu, H.S.; Li, W.L.; Zhao, Y.F.; Li, J.; Wang, L.S. Planar hexagonal B36 as a potential basis for extended single-atom layer boron sheets. Nat. Commun. 2014, 5, 3113. [Google Scholar] [CrossRef]
- Ren, M.; Jin, S.; Wei, D.; Jin, Y.; Tian, Y.; Lu, C.; Gutsev, G.L. NbB12−: A new member of half-sandwich type doped boron clusters with high stability. Phys. Chem. Chem. Phys. 2019, 21, 21746–21752. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Chen, B.; Kuang, X.; Lu, C.; Gutsev, G.L. Structural evolution and electronic properties of medium-sized boron clusters doped with scandium. J. Phys. Condens. Matter 2019, 31, 485302. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.; Ren, M.; Lu, C.; Bi, J.; Maroulis, G. A quasi-plane IrB18− cluster with high stability. Phys. Chem. Chem. Phys. 2020, 22, 5942–5948. [Google Scholar] [CrossRef]
- Chen, T.T.; Li, W.L.; Chen, W.J.; Yu, X.H.; Dong, X.R.; Li, J.; Wang, L.S. Spherical trihedral metallo-borospherenes. Nat. Commun. 2020, 11, 2766. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.J.; Kulichenko, M.; Choi, H.W.; Cavanagh, J.; Yuan, D.F.; Boldyrev, A.I.; Wang, L.S. Photoelectron Spectroscopy of Size-Selected Bismuth-Boron Clusters: BiBn− (n = 6–8). J. Phys. Chem. A 2021, 125, 6751–6760. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.T.; Li, W.L.; Bai, H.; Chen, W.J.; Dong, X.R.; Li, J.; Wang, L.S. ReB8− and ReB9−: New Members of the Transition-Metal-Centered Borometallic Molecular Wheel Family. J. Phys. Chem. A 2019, 123, 5317–5324. [Google Scholar] [CrossRef]
- Cheung, L.F.; Czekner, J.; Kocheril, G.S.; Wang, L.S. ReB6−: A Metallaboron Analog of Metallabenzenes. J. Am. Chem. Soc. 2019, 141, 17854–17860. [Google Scholar] [CrossRef]
- Cheung, L.F.; Kocheril, G.S.; Czekner, J.; Wang, L.-S. Observation of Möbius Aromatic Planar Metallaborocycles. J. Am. Chem. Soc. 2020, 142, 3356–3360. [Google Scholar] [CrossRef]
- Liang, W.Y.; Das, A.; Dong, X.; Cui, Z.H. Lithium doped tubular structure in LiB20 and LiB20−: A viable global minimum. Phys. Chem. Chem. Phys. 2018, 20, 16202–16208. [Google Scholar] [CrossRef]
- Saha, R.; Kar, S.; Pan, S.; Martinez-Guajardo, G.; Merino, G.; Chattaraj, P.K. A Spinning Umbrella: Carbon Monoxide and Dinitrogen Bound MB12− Clusters (M = Co, Rh, Ir). J. Phys. Chem. A 2017, 121, 2971–2979. [Google Scholar] [CrossRef]
- Sergeeva, A.P.; Popov, I.A.; Piazza, Z.A.; Li, W.L.; Romanescu, C.; Wang, L.S.; Boldyrev, A.I. Understanding boron through size-selected clusters: Structure, chemical bonding, and fluxionality. Acc. Chem. Res. 2014, 47, 1349–1358. [Google Scholar] [CrossRef]
- Liang, W.; Das, A.; Dong, X.; Wang, M.; Cui, Z. Structural and electronic properties of MB22− (M = Na, K) clusters: Tubular boron versus quasi-planar boron forms. New J. Chem. 2019, 43, 6507–6512. [Google Scholar] [CrossRef]
- Popov, I.A.; Li, W.L.; Piazza, Z.A.; Boldyrev, A.I.; Wang, L.S. Complexes between planar boron clusters and transition metals: A photoelectron spectroscopy and ab initio study of CoB12− and RhB12−. J. Phys. Chem. A 2014, 118, 8098–8105. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.L.; Sun, W.G.; Kuang, X.Y.; Lu, C.; Xia, X.X.; Shi, H.X.; Maroulis, G. Structural Stability and Evolution of Medium-Sized Tantalum-Doped Boron Clusters: A Half-Sandwich-Structured TaB12− Cluster. Inorg. Chem. 2018, 57, 343–350. [Google Scholar] [CrossRef]
- Chacko, S.; Kanhere, D.G.; Boustani, I. Ab initiodensity functional investigation of B24 clusters: Rings, tubes, planes, and cages. Phys. Rev. B 2003, 68, 035414. [Google Scholar] [CrossRef]
- Lv, J.; Wang, Y.; Zhang, L.; Lin, H.; Zhao, J.; Ma, Y. Stabilization of fullerene-like boron cages by transition metal encapsulation. Nanoscale 2015, 7, 10482–10489. [Google Scholar] [CrossRef]
- Yang, Y.J.; Li, S.X.; Chen, D.L.; Long, Z.W. Structural and Electronic Properties of Single-Atom Transition Metal-Doped Boron Clusters MB24 (M = Sc, V, and Mn). ACS Omega 2021, 6, 30442–30450. [Google Scholar] [CrossRef]
- Li, S.X.; Yang, Y.J.; Chen, D.L.; Long, Z.W. Structures, and electronic and spectral properties of single-atom transition metal-doped boron clusters MB24− (M = Sc, Ti, V, Cr, Mn, Fe, Co, and Ni). RSC Adv. 2022, 12, 16706–16716. [Google Scholar] [CrossRef] [PubMed]
- Tuyet Mai, D.T.; Van Duong, L.; Pham-Ho, M.P.; Nguyen, M.T. Electronic Structure and Properties of Silicon-Doped Boron Clusters BnSi with n = 15–24 and Their Anions. J. Phys. Chem. C 2020, 124, 6770–6783. [Google Scholar] [CrossRef]
- Van Duong, L.; Nguyen, M.T. Electronic structure of the boron fullerene B14 and its silicon derivatives B13Si+, B13Si− and B12Si2: A rationalization using a cylinder model. Phys. Chem. Chem. Phys. 2016, 18, 17619–17626. [Google Scholar] [CrossRef]
- Li, P.F.; Zhai, H.J. Structures and chemical bonding of boron-based B12O and B11Au clusters. A counterexample in boronyl chemistry. Phys. Chem. Chem. Phys. 2022, 24, 10952–10961. [Google Scholar] [CrossRef]
- Li, S.X.; Yang, Y.J.; Chen, D.L. PB12+ and P2B12+/0/–: The Novel B12 Cage Doped by Nonmetallic P Atoms. ACS Omega 2023, 8, 44831–44838. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.J.; Li, S.X.; Chen, D.L.; Long, Z.W. Structural Evolution and Electronic Properties of Selenium-Doped Boron Clusters SeBn0/− (n = 3–16). Molecules 2023, 28, 357. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; Wang, Y.; Zhu, L.; Ma, Y. Particle-swarm structure prediction on clusters. J. Chem. Phys. 2012, 137, 084104. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Alexandrova, A.N.; Boldyrev, A.I.; Zhai, H.J.; Wang, L.S. All-boron aromatic clusters as potential new inorganic ligands and building blocks in chemistry. Coord. Chem. Rev. 2006, 250, 2811–2866. [Google Scholar] [CrossRef]
- Barroso, J.; Pan, S.; Merino, G. Structural transformations in boron clusters induced by metal doping. Chem. Soc. Rev. 2022, 51, 1098–1123. [Google Scholar] [CrossRef]
- Gong, L.F.; Guo, W.L.; Wu, X.M.; Li, Q.S. B7– as a novel ligand: Theoretical investigations on structures and chemical bonding of LiB7 and BeB7+. Chem. Phys. Lett. 2006, 429, 326–334. [Google Scholar] [CrossRef]
- Kheshti, T.; Mahdavifar, Z.; Noorizadeh, S. Umbrella-shaped vs planar; evolutionary search for BnQ, Be©BnQ (n = 6–12, Q = 0, −1) clusters. J. Mol. Liq. 2021, 328, 115389. [Google Scholar] [CrossRef]
- Đorđević, S.; Radenković, S. Spatial and Electronic Structures of BeB8 and MgB8: How far Does the Analogy Go? Chem. Phys. Chem. 2022, 23, e202200070. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Wang, C.; Wu, Q.; Lan, J.; Chai, Z.; Shi, W. Highly stable actinide(iii) complexes supported by doubly aromatic ligands. Phys. Chem. Chem. Phys. 2022, 24, 5921–5928. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; Wang, Y.; Zhu, L.; Ma, Y. B38: An all-boron fullerene analogue. Nanoscale 2014, 6, 11692–11696. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Jalife, S.; Vasquez-Espinal, A.; Barroso, J.; Orozco-Ic, M.; Ravell, E.; Cabellos, J.L.; Liang, W.Y.; Cui, Z.H.; Merino, G. Li2B24: The simplest combination for a three-ring boron tube. Nanoscale 2019, 11, 2143–2147. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Liu, Y.Q.; Liu, X.B.; Pan, S.; Cui, Z.H.; Merino, G. Be4B12 +: A Covalently Bonded Archimedean Beryllo-Borospherene. Angew. Chem. Int. Ed. 2022, 61, e202208152. [Google Scholar] [CrossRef]
- Tian, Y.; Wei, D.; Jin, Y.; Barroso, J.; Lu, C.; Merino, G. Exhaustive exploration of MgBn (n = 10–20) clusters and their anions. Phys. Chem. Chem. Phys. 2019, 21, 6935–6941. [Google Scholar] [CrossRef]
- Li, P.; Du, X.; Wang, J.J.; Lu, C.; Chen, H. Probing the Structural Evolution and Stabilities of Medium-Sized MoBn0/– Clusters. J. Phys. Chem. C 2018, 122, 20000–20005. [Google Scholar] [CrossRef]
- Jin, S.; Chen, B.; Kuang, X.; Lu, C.; Sun, W.; Xia, X.; Gutsev, G.L. Structural and Electronic Properties of Medium-Sized Aluminum-Doped Boron Clusters AlBn and Their Anions. J. Phys. Chem. C 2019, 123, 6276–6283. [Google Scholar] [CrossRef]
- Bartlett, R.J.; Musiał, M. Coupled-cluster theory in quantum chemistry. Rev. Mod. Phys. 2007, 79, 291–352. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput.Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, D.; Yang, Y.; Li, S.; Chen, D. Structural Evolution of Small-Sized Phosphorus-Doped Boron Clusters: A Half-Sandwich-Structured PB15 Cluster. Molecules 2024, 29, 3384. https://doi.org/10.3390/molecules29143384
Wang D, Yang Y, Li S, Chen D. Structural Evolution of Small-Sized Phosphorus-Doped Boron Clusters: A Half-Sandwich-Structured PB15 Cluster. Molecules. 2024; 29(14):3384. https://doi.org/10.3390/molecules29143384
Chicago/Turabian StyleWang, Danyu, Yueju Yang, Shixiong Li, and Deliang Chen. 2024. "Structural Evolution of Small-Sized Phosphorus-Doped Boron Clusters: A Half-Sandwich-Structured PB15 Cluster" Molecules 29, no. 14: 3384. https://doi.org/10.3390/molecules29143384