
Ru(II)-Catalyzed Asymmetric Transfer Hydrogenation of α-Alkyl-β-Ketoaldehydes via Dynamic Kinetic Resolution
 , ,
, ,  and
and 
Abstract
1. Introduction
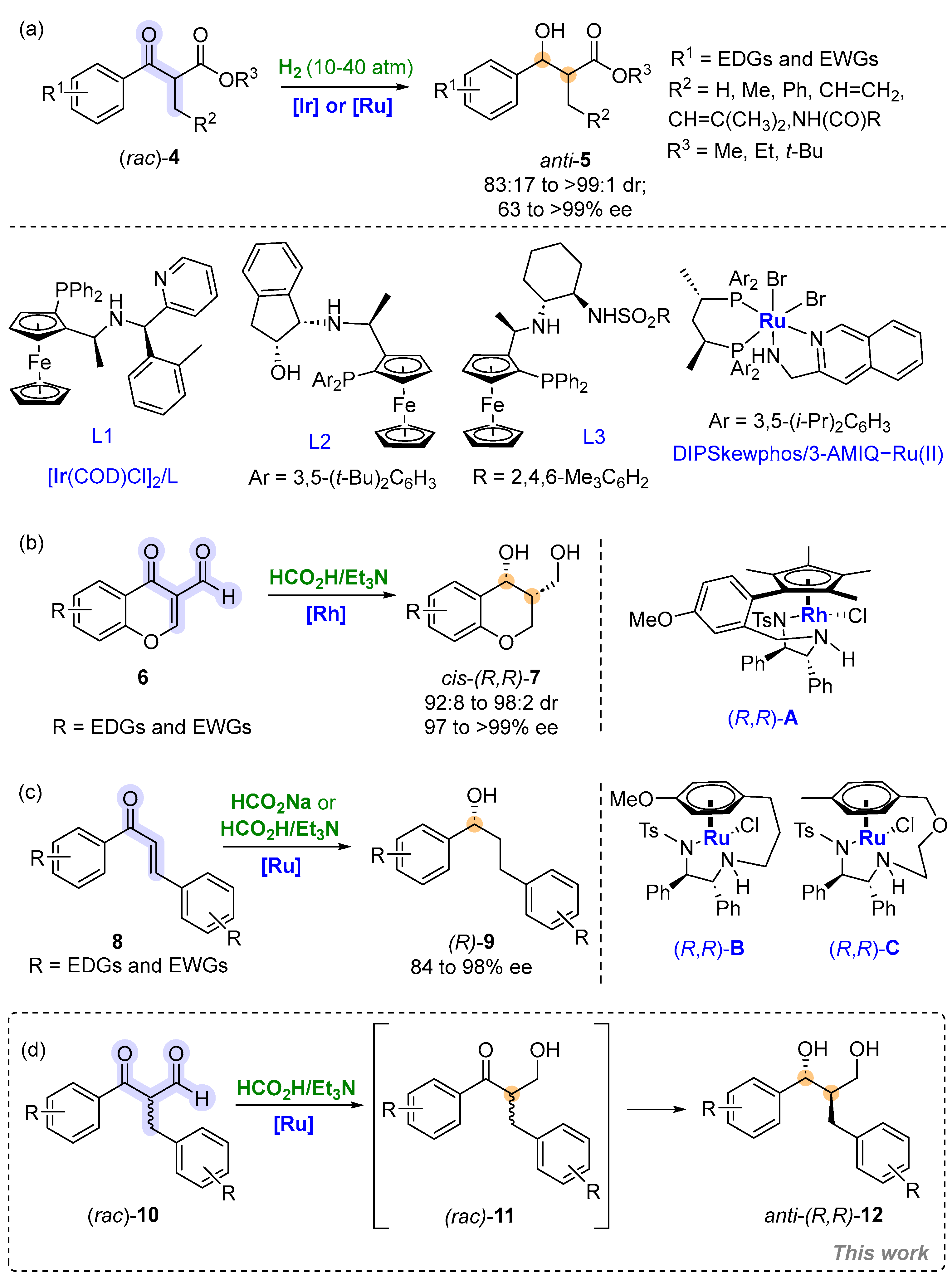
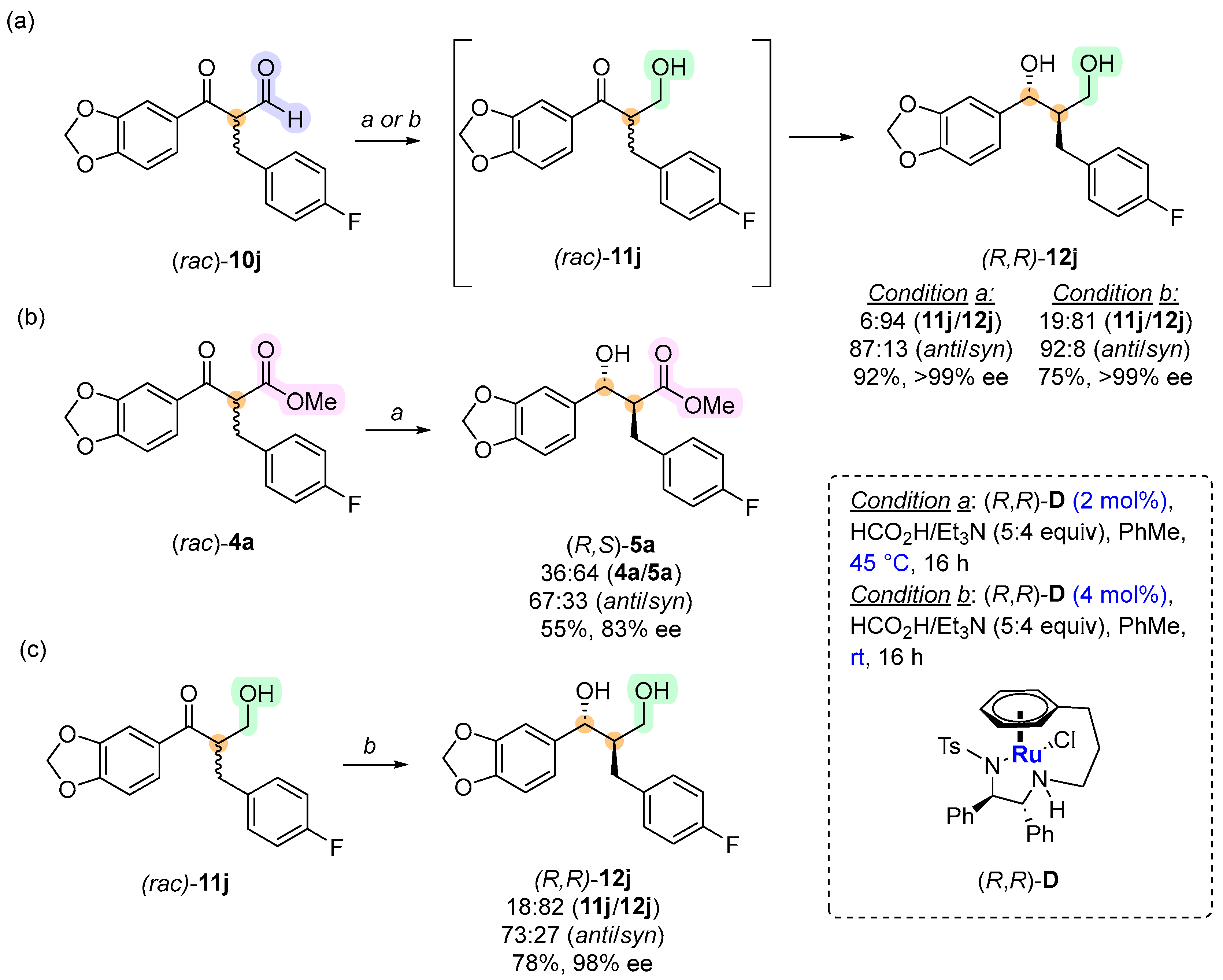
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. General Procedure for the Synthesis of Compounds 8a–j
3.3. General Procedure for the Synthesis of Compounds 13a–j
3.4. Method for the Synthesis of Compound 4a
3.5. General Procedure for the Synthesis of Compounds 10/15a–j
3.6. General Procedure for the Synthesis of Compounds 12a–j and 5a
3.7. Method for the Synthesis of Compound 16
3.8. Method for the Synthesis of Compound 17
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Teponno, R.B.; Kusari, S.; Spiteller, M. Recent Advances in Research on Lignans and Neolignans. Nat. Prod. Rep. 2016, 33, 1044–1092. [Google Scholar] [CrossRef] [PubMed]
- Zálešák, F.; Bon, D.J.Y.D.; Pospíšil, J. Lignans and Neolignans: Plant Secondary Metabolites as a Reservoir of Biologically Active Substances. Pharmacol. Res. 2019, 146, 104284. [Google Scholar] [CrossRef] [PubMed]
- Abegaz, B.M.; Kinfe, H.H. Naturally Occurring Homoisoflavonoids: Phytochemistry, Biological Activities, and Synthesis (Part II). Nat. Prod. Commun. 2019, 14, 1–20. [Google Scholar] [CrossRef]
- Ayad, T.; Phansavath, P.; Ratovelomanana-Vidal, V. Transition-Metal-Catalyzed Asymmetric Hydrogenation and Transfer Hydrogenation: Sustainable Chemistry to Access Bioactive Molecules. Chem. Rec. 2016, 16, 2754–2771. [Google Scholar] [CrossRef] [PubMed]
- Bhat, V.; Welin, E.R.; Guo, X.; Stoltz, B.M. Advances in Stereoconvergent Catalysis from 2005 to 2015: Transition-Metal-Mediated Stereoablative Reactions, Dynamic Kinetic Resolutions, and Dynamic Kinetic Asymmetric Transformations. Chem. Rev. 2017, 117, 4528–4561. [Google Scholar] [CrossRef] [PubMed]
- Molina Betancourt, R.; Echeverria, P.-G.; Ayad, T.; Phansavath, P.; Ratovelomanana-Vidal, V. Recent Progress and Applications of Transition-Metal-Catalyzed Asymmetric Hydrogenation and Transfer Hydrogenation of Ketones and Imines through Dynamic Kinetic Resolution. Synthesis 2021, 53, 30–50. [Google Scholar] [CrossRef]
- Hou, C.-J.; Hu, X.-P. Sterically Hindered Chiral Ferrocenyl P,N,N-Ligands for Highly Diastereo-/Enantioselective Ir-Catalyzed Hydrogenation of α-Alkyl-β-Ketoesters via Dynamic Kinetic Resolution. Org. Lett. 2016, 18, 5592–5595. [Google Scholar] [CrossRef]
- Gu, G.; Lu, J.; Yu, O.; Wen, J.; Yin, Q.; Zhang, X. Enantioselective and Diastereoselective Ir-Catalyzed Hydrogenation of α-Substituted β-Ketoesters via Dynamic Kinetic Resolution. Org. Lett. 2018, 20, 1888–1892. [Google Scholar] [CrossRef]
- Ling, F.; Wang, Y.; Huang, A.; Wang, Z.; Wang, S.; He, J.; Zhao, X.; Zhong, W. Iridium-Catalyzed Enantioselective and Diastereoselective Hydrogenation of Racemic β’-Keto-β-Amino Esters via Dynamic Kinetic Resolution. Adv. Synth. Catal. 2021, 363, 4714–4719. [Google Scholar] [CrossRef]
- Yurino, T.; Nishihara, R.; Yasuda, T.; Yang, S.; Utsumi, N.; Katayama, T.; Arai, N.; Ohkuma, T. Asymmetric Hydrogenation of α-Alkyl-Substituted β-Keto Esters and Amides through Dynamic Kinetic Resolution. Org. Lett. 2024, 26, 2872–2876. [Google Scholar] [CrossRef]
- Yang, H.; Yu, H.; Stolarzewicz, I.A.; Tang, W. Enantioselective Transformations in the Synthesis of Therapeutic Agents. Chem. Rev. 2023, 123, 9397–9446. [Google Scholar] [CrossRef] [PubMed]
- Caleffi, G.S.; Demidoff, F.C.; Nájera, C.; Costa, P.R.R. Asymmetric Hydrogenation and Transfer Hydrogenation in the Enantioselective Synthesis of Flavonoids. Org. Chem. Front. 2022, 9, 1165–1194. [Google Scholar] [CrossRef]
- Pereira, A.M.; Cidade, H.; Tiritan, M.E. Stereoselective Synthesis of Flavonoids: A Brief Overview. Molecules 2023, 28, 426. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Astruc, D. The Golden Age of Transfer Hydrogenation. Chem. Rev. 2015, 115, 6621–6686. [Google Scholar] [CrossRef]
- Cotman, A.E. Escaping from Flatland: Stereoconvergent Synthesis of Three-Dimensional Scaffolds via Ruthenium(II)-Catalyzed Noyori–Ikariya Transfer Hydrogenation. Chem. Eur. J. 2021, 27, 39–53. [Google Scholar] [CrossRef]
- Matsunami, A.; Kayaki, Y. Upgrading and Expanding the Scope of Homogeneous Transfer Hydrogenation. Tetrahedron Lett. 2018, 59, 504–513. [Google Scholar] [CrossRef]
- Nedden, H.G.; Zanotti-Gerosa, A.; Wills, M. The Development of Phosphine-Free “Tethered” Ruthenium(II) Catalysts for the Asymmetric Reduction of Ketones and Imines. Chem. Rec. 2016, 16, 2623–2643. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Phansavath, P.; Ratovelomanana-Vidal, V. Rh-Mediated Asymmetric-Transfer Hydrogenation of 3-Substituted Chromones: A Route to Enantioenriched Cis-3-(Hydroxymethyl)Chroman-4-ol Derivatives through Dynamic Kinetic Resolution. Org. Lett. 2019, 21, 3276–3280. [Google Scholar] [CrossRef]
- Echeverria, P.-G.; Zheng, L.-S.; Llopis, Q.; He, B.; Westermeyer, A.; Molina Betancourt, R.; Phansavath, P.; Ratovelomanana-Vidal, V. Tethered Rh(III)-N-(p-Tolylsulfonyl)-1,2-Diphenylethylene-1,2-Diamine Complexes: Efficient Catalysts for Asymmetric Transfer Hydrogenation. SynOpen 2022, 6, 75–79. [Google Scholar] [CrossRef]
- Hall, T.H.; Adams, H.; Vyas, V.K.; Michael Chu, K.L.; Wills, M. Asymmetric Transfer Hydrogenation of Unsaturated Ketones; Factors Influencing 1,4- vs 1,2- Regio- and Enantioselectivity, and Alkene vs Alkyne Directing Effects. Tetrahedron 2021, 77, 131771. [Google Scholar] [CrossRef]
- Demidoff, F.C.; Caleffi, G.S.; Figueiredo, M.; Costa, P.R.R. Ru(II)-Catalyzed Asymmetric Transfer Hydrogenation of Chalcones in Water: Application to the Enantioselective Synthesis of Flavans BW683C and Tephrowatsin E. J. Org. Chem. 2022, 87, 14208–14222. [Google Scholar] [CrossRef] [PubMed]
- Sterle, M.; Huš, M.; Lozinšek, M.; Zega, A.; Cotman, A.E. Hydrogen-Bonding Ability of Noyori–Ikariya Catalysts Enables Stereoselective Access to CF3 -Substituted Syn-1,2-Diols via Dynamic Kinetic Resolution. ACS Catal. 2023, 13, 6242–6248. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, F.V.; Caleffi, G.S.; Costa-Júnior, P.C.T.; Costa, P.R.R. Enantioselective Synthesis of Isoflavanones and Pterocarpans through a Ru II -Catalyzed ATH-DKR of Isoflavones. ChemCatChem 2021, 13, 5097–5108. [Google Scholar] [CrossRef]
- Gediya, S.K.; Clarkson, G.J.; Wills, M. Asymmetric Transfer Hydrogenation: Dynamic Kinetic Resolution of α-Amino Ketones. J. Org. Chem. 2020, 85, 11309–11330. [Google Scholar] [CrossRef] [PubMed]
- Rolt, A.; O’Neill, P.M.; Liang, T.J.; Stachulski, A.V. Synthesis of MeBmt and Related Derivatives via Syn-Selective ATH-DKR. RSC Adv. 2019, 9, 40336–40339. [Google Scholar] [CrossRef] [PubMed]
- Cotman, A.E.; Lozinšek, M.; Wang, B.; Stephan, M.; Mohar, B. Trans-Diastereoselective Ru(II)-Catalyzed Asymmetric Transfer Hydrogenation of α-Acetamido Benzocyclic Ketones via Dynamic Kinetic Resolution. Org. Lett. 2019, 21, 3644–3648. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.-S.; Férard, C.; Phansavath, P.; Ratovelomanana-Vidal, V. Rhodium-Mediated Asymmetric Transfer Hydrogenation: A Diastereo- and Enantioselective Synthesis of Syn-α-Amido β-Hydroxy Esters. Chem. Commun. 2018, 54, 283–286. [Google Scholar] [CrossRef] [PubMed]
- Limanto, J.; Krska, S.W.; Dorner, B.T.; Vazquez, E.; Yoshikawa, N.; Tan, L. Dynamic Kinetic Resolution: Asymmetric Transfer Hydrogenation of α-Alkyl-Substituted β-Ketoamides. Org. Lett. 2010, 12, 512–515. [Google Scholar] [CrossRef] [PubMed]
- Seashore-Ludlow, B.; Villo, P.; Häcker, C.; Somfai, P. Enantioselective Synthesis of Anti-β-Hydroxy-α-Amido Esters via Transfer Hydrogenation. Org. Lett. 2010, 12, 5274–5277. [Google Scholar] [CrossRef]
- Caleffi, G.S.; Rosa, A.S.; de Souza, L.G.; Avelar, J.L.S.; Nascimento, S.M.R.; de Almeida, V.M.; Tucci, A.R.; Ferreira, V.N.; da Silva, A.J.M.; Santos-Filho, O.A.; et al. Aurones: A Promising Scaffold to Inhibit SARS-CoV-2 Replication. J. Nat. Prod. 2023, 86, 1536–1549. [Google Scholar] [CrossRef]
- Soto, M.; Soengas, R.G.; Rodríguez-Solla, H. Solvent-Controlled Hydrogenation of 2′-Hydroxychalcones: A Simple Solution to the Total Synthesis of Bussealins. Adv. Synth. Catal. 2020, 362, 5422–5431. [Google Scholar] [CrossRef]
- Gümüş, M.; Koca, İ. Enamines and Dimethylamino Imines as Building Blocks in Heterocyclic Synthesis: Reactions of DMF-DMA Reagent with Different Functional Groups. ChemistrySelect 2020, 5, 12377–12397. [Google Scholar] [CrossRef]
- Zhao, Y.; Yuan, Y.; Kong, L.; Zhang, F.; Li, Y. Synthesis of 1-Alkyl-3-(2-Oxo-2-Aryl/Alkyl-Ethyl)Indolin-2-Ones through Gold/Brønsted Acid Relay Actions: Observation of Selective C=C Bond Cleavage of Enaminones. Synthesis 2017, 49, 3609–3618. [Google Scholar] [CrossRef]
- Haara, S.; Oka, K. Synthesis and Characters of l-Substituted A-Norsteroids. Tetrahedron Lett. 1966, 7, 1057–1061. [Google Scholar] [CrossRef]
- Eicher, T.; Graf, R.; Konzmann, H.; Pick, R. Synthese Und Reaktionen von 2,3-Diaryl- Und 2,3-Dialkylcyclopropenoniminen. Synthesis 1987, 1987, 887–892. [Google Scholar] [CrossRef]
- Caleffi, G.S.; Brum, J.d.O.C.; Costa, A.T.; Domingos, J.L.O.; Costa, P.R.R. Asymmetric Transfer Hydrogenation of Arylidene-Substituted Chromanones and Tetralones Catalyzed by Noyori–Ikariya Ru(II) Complexes: One-Pot Reduction of C═C and C═O Bonds. J. Org. Chem. 2021, 86, 4849–4858. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Liu, J.; Di Tommaso, D.; Iggo, J.A.; Catlow, C.R.A.; Bacsa, J.; Xiao, J. A Multilateral Mechanistic Study into Asymmetric Transfer Hydrogenation in Water. Chem. Eur. J. 2008, 14, 7699–7715. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Xiao, J. Aqueous-Phase Asymmetric Transfer Hydrogenation of Ketones—A Greener Approach to Chiral Alcohols. Chem. Commun. 2007, 2449–2466. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Li, X.; King, F.; Xiao, J. Insight into and Practical Application of PH-Controlled Asymmetric Transfer Hydrogenation of Aromatic Ketones in Water. Angew. Chem. Int. Ed. 2005, 44, 3407–3411. [Google Scholar] [CrossRef]
- Wu, X.; Li, X.; Hems, W.; King, F.; Xiao, J. Accelerated Asymmetric Transfer Hydrogenation of Aromatic Ketones in Water. Org. Biomol. Chem. 2004, 2, 1818. [Google Scholar] [CrossRef]
- Hayes, A.M.; Morris, D.J.; Clarkson, G.J.; Wills, M. A Class of Ruthenium(II) Catalyst for Asymmetric Transfer Hydrogenations of Ketones. J. Am. Chem. Soc. 2005, 127, 7318–7319. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wang, J.; Hu, X.; Shen, K.; Wang, W.; Chu, Y.; Lin, L.; Liu, X.; Feng, X. Catalytic Asymmetric Roskamp Reaction of α-Alkyl-α-Diazoesters with Aromatic Aldehydes: Highly Enantioselective Synthesis of α-Alkyl-β-Keto Esters. J. Am. Chem. Soc. 2010, 132, 8532–8533. [Google Scholar] [CrossRef] [PubMed]
- Bull, J.A.; Croft, R.A.; Davis, O.A.; Doran, R.; Morgan, K.F. Oxetanes: Recent Advances in Synthesis, Reactivity, and Medicinal Chemistry. Chem. Rev. 2016, 116, 12150–12233. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Jia, J.; Xu, B.; Zhao, A.; Guo, C.C. Iron-Facilitated Oxidative Radical Decarboxylative Cross-Coupling between α-Oxocarboxylic Acids and Acrylic Acids: An Approach to α,β-Unsaturated Carbonyls. J. Org. Chem. 2015, 80, 3586–3596. [Google Scholar] [CrossRef] [PubMed]
- Stroba, A.; Schaeffer, F.; Hindie, V.; Lopez-Garcia, L.; Adrian, I.; Fröhner, W.; Hartmann, R.W.; Biondi, R.M.; Engel, M. 3,5-Diphenylpent-2-Enoic Acids as Allosteric Activators of the Protein Kinase PDK1: Structure-Activity Relationships and Thermodynamic Characterization of Binding as Paradigms for PIF-Binding Pocket-Targeting Compounds. J. Med. Chem. 2009, 52, 4683–4693. [Google Scholar] [CrossRef] [PubMed]
- Steinbach, T.; Wahlen, C.; Wurm, F.R. Poly(Phosphonate)-Mediated Horner-Wadsworth-Emmons Reactions. Polym. Chem. 2015, 6, 1192–1202. [Google Scholar] [CrossRef]
- Sheshenev, A.E.; Boltukhina, E.V.; White, A.J.P.; Hii, K.K. Methylene-Bridged Bis(Imidazoline)-Derived 2-Oxopyrimidinium Salts as Catalysts for Asymmetric Michael Reactions. Angew. Chem. Int. Ed. 2013, 52, 6988–6991. [Google Scholar] [CrossRef]
- Hodgson, G.K.; Scaiano, J.C. Heterogeneous Dual Photoredox-Lewis Acid Catalysis Using a Single Bifunctional Nanomaterial. ACS Catal. 2018, 8, 2914–2922. [Google Scholar] [CrossRef]
- Chiaradia, L.D.; Martins, P.G.A.; Cordeiro, M.N.S.; Guido, R.V.C.; Ecco, G.; Andricopulo, A.D.; Yunes, R.A.; Vernal, J.; Nunes, R.J.; Terenzi, H. Synthesis, Biological Evaluation, and Molecular Modeling of Chalcone Derivatives as Potent Inhibitors of Mycobacterium Tuberculosis Protein Tyrosine Phosphatases (PtpA and PtpB). J. Med. Chem. 2012, 55, 390–402. [Google Scholar] [CrossRef]
- Li, H.C.; An, C.; Wu, G.; Li, G.X.; Huang, X.B.; Gao, W.X.; Ding, J.C.; Zhou, Y.B.; Liu, M.C.; Wu, H.Y. Transition-Metal-Free Highly Chemoselective and Stereoselective Reduction with Se/DMF/H2O System. Org. Lett. 2018, 20, 5573–5577. [Google Scholar] [CrossRef]
- Liu, P.; Liang, R.; Lu, L.; Yu, Z.; Li, F. Use of a Cyclometalated Iridium(III) Complex Containing a N∧C∧N-Coordinating Terdentate Ligand as a Catalyst for the α-Alkylation of Ketones and N-Alkylation of Amines with Alcohols. J. Org. Chem. 2017, 82, 1943–1950. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Daw, P.; Ben David, Y.; Milstein, D. Manganese-Catalyzed α-Alkylation of Ketones, Esters, and Amides Using Alcohols. ACS Catal. 2018, 8, 10300–10305. [Google Scholar] [CrossRef] [PubMed]
- Pandey, B.; Xu, S.; Ding, K. Selective Ketone Formations via Cobalt-Catalyzed β-Alkylation of Secondary Alcohols with Primary Alcohols. Org. Lett. 2019, 21, 7420–7423. [Google Scholar] [CrossRef] [PubMed]
- Elangovan, S.; Sortais, J.; Beller, M.; Darcel, C. Iron-Catalyzed α-Alkylation of Ketones with Alcohols. Angew. Chem. Int. Ed. 2015, 54, 14483–14486. [Google Scholar] [CrossRef]
- Lakshminarayana, B.; Mahendar, L.; Ghosal, P.; Sreedhar, B.; Satyanarayana, G.; Subrahmanyam, C. Fabrication of Pd/CuFe2O4 Hybrid Nanowires: A Heterogeneous Catalyst for Heck Couplings. New J. Chem. 2018, 42, 1646–1654. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Structure | 8 a | R1 | R2 | R3 | 13 b | Yield (%) c | 10 d | 10:15 e | Yield (%) f |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 |  | 8a | H | H | H | 13a | 87 | 10a | 4:96 | 70 |
| 2 | 8b | H | H | OMe | 13b | 53 | 10b | 48:52 | 79 | |
| 3 | 8c | H | H | CF3 | 13c | 67 | 10c | 0:100 | 71 | |
| 4 | 8d | H | H | F | 13d | 65 | 10d | 15:85 | 49 | |
| 5 | 8e | H | F | H | 13e | 79 | 10e | 12:88 | 65 | |
| 6 | 8f | F | H | H | 13f | 37 | 10f | 24:76 | 57 | |
| 7 | 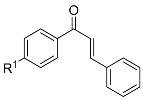 | 8g | OMe | - | - | 13g | 92 | 10g | 33:67 | 58 |
| 8 | 8h | Br | - | - | 13h | 91 | 10h | 37:63 | 68 | |
| 9 | 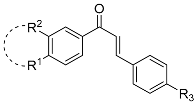 | 8i | Cl | H | F | 13i | 77 | 10i | 53:47 | 58 |
| 10 | 8j | OCH2O | F | 13j | 88 | 10j | 50:50 | 91 | ||
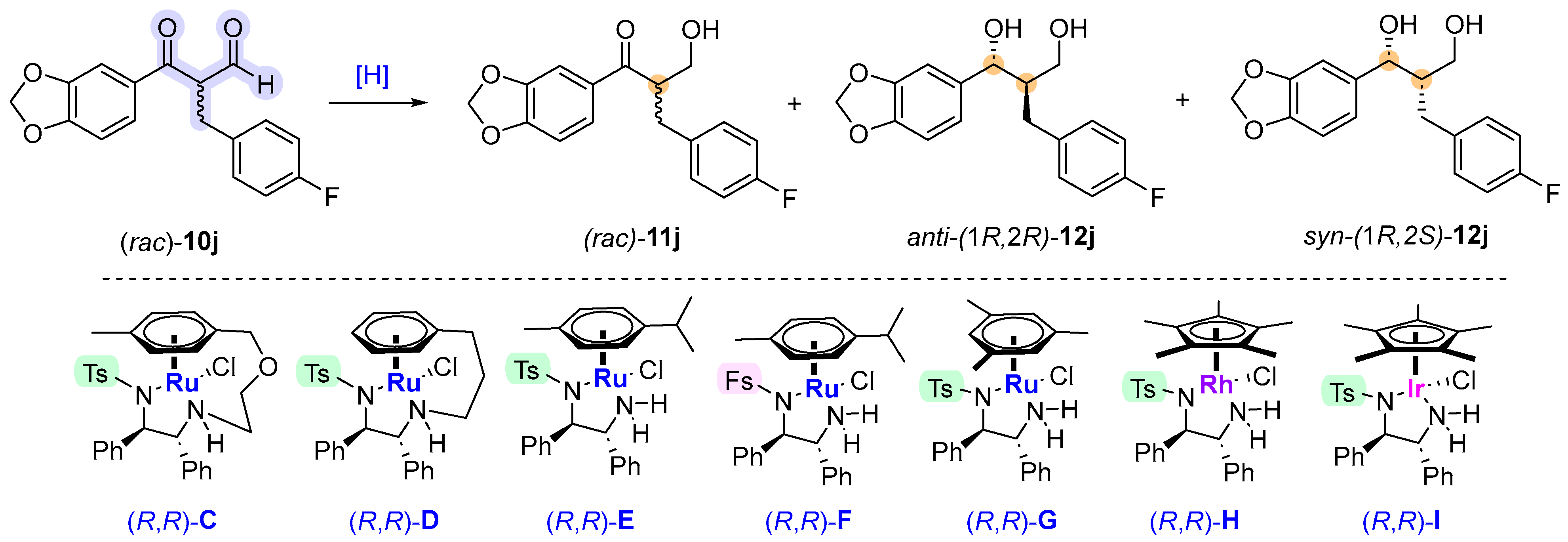
| Entry | [H] Source (Ratio) b | (R,R)-C-I (mol%) | Solvent c | Add. d | T (°C) | t (h) | 11j/12j e | 12j anti/syn e | Yield (%) f | ee (%) g |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | HCO2Na | D (2) | DCE/H2O | CTAB | rt | 16 | >99:1 | nd | nd | nd |
| 2 | HCO2H/Et3N (5:4) | D (2) | DCE | - | rt | 16 | >99:1 | nd | nd | nd |
| 3 | HCO2H/Et3N (5:4) | D (2) | MeCN | - | rt | 16 | 68:32 | 84:16 | nd | nd |
| 4 | HCO2H/Et3N (5:4) | D (2) | THF | - | rt | 16 | 62:38 | 93:7 | 29 | >99 |
| 5 | HCO2H/Et3N (5:4) | D (2) | EtOAc | - | rt | 16 | 54:46 | 92:8 | 36 | 98 |
| 6 | HCO2H/Et3N (5:4) | D (2) | PhMe | - | rt | 16 | 44:56 | 92:8 | 47 | >99 |
| 7 | HCO2H/Et3N (5:4) | D (2) | PhMe | - | 45 | 16 | 7:93 | 87:13 | 92 | >99 |
| 8 | HCO2H/Et3N (5:4) | C (2) | PhMe | - | 45 | 16 | 85:15 | 59:41 | nd | nd |
| 9 | HCO2H/Et3N (5:4) | E (2) | PhMe | - | 45 | 16 | 95:5 | nd | nd | nd |
| 10 | HCO2H/Et3N (5:4) | F (2) | PhMe | - | 45 | 16 | 93:7 | nd | nd | nd |
| 11 | HCO2H/Et3N (5:4) | G (2) | PhMe | - | 45 | 16 | >99:1 | nd | nd | nd |
| 12 | HCO2H/Et3N (5:4) | H (2) | PhMe | - | 45 | 16 | >99:1 | nd | nd | nd |
| 13 | HCO2H/Et3N (5:4) | I (2) | PhMe | - | 45 | 16 | >99:1 | nd | nd | nd |
| 14 | HCO2H/Et3N (5:4) | D (2) | PhMe | Cu(OTf)2 | 45 | 16 | 6:94 | 88:12 | 92 | >99 |
| 15 | HCO2H/Et3N (5:4) | D (2) | PhMe | TfOH | 45 | 16 | 4:96 | 85:15 | 90 | >99 |
| 16 | HCO2H/Et3N (5:4) | D (4) | PhMe | - | rt | 16 | 19:81 | 92:8 | 75 | >99 |
| 17 | HCO2H/Et3N (5:4) | D (4) | PhMe | - | rt | 24 | 20:80 | 90:10 | nd | nd |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lapa, D.P.; Araújo, L.H.S.; Melo, S.R.; Costa, P.R.R.; Caleffi, G.S. Ru(II)-Catalyzed Asymmetric Transfer Hydrogenation of α-Alkyl-β-Ketoaldehydes via Dynamic Kinetic Resolution. Molecules 2024, 29, 3420. https://doi.org/10.3390/molecules29143420
Lapa DP, Araújo LHS, Melo SR, Costa PRR, Caleffi GS. Ru(II)-Catalyzed Asymmetric Transfer Hydrogenation of α-Alkyl-β-Ketoaldehydes via Dynamic Kinetic Resolution. Molecules. 2024; 29(14):3420. https://doi.org/10.3390/molecules29143420
Chicago/Turabian StyleLapa, Daiene P., Leticia H. S. Araújo, Sávio R. Melo, Paulo R. R. Costa, and Guilherme S. Caleffi. 2024. "Ru(II)-Catalyzed Asymmetric Transfer Hydrogenation of α-Alkyl-β-Ketoaldehydes via Dynamic Kinetic Resolution" Molecules 29, no. 14: 3420. https://doi.org/10.3390/molecules29143420
APA StyleLapa, D. P., Araújo, L. H. S., Melo, S. R., Costa, P. R. R., & Caleffi, G. S. (2024). Ru(II)-Catalyzed Asymmetric Transfer Hydrogenation of α-Alkyl-β-Ketoaldehydes via Dynamic Kinetic Resolution. Molecules, 29(14), 3420. https://doi.org/10.3390/molecules29143420