

Phytochemical Investigation of Carex praecox Schreb. and ACE-Inhibitory Activity of Oligomer Stilbenes of the Plant

 , , ,
, , ,  ,
,  , and
, and
Abstract

1. Introduction
2. Results
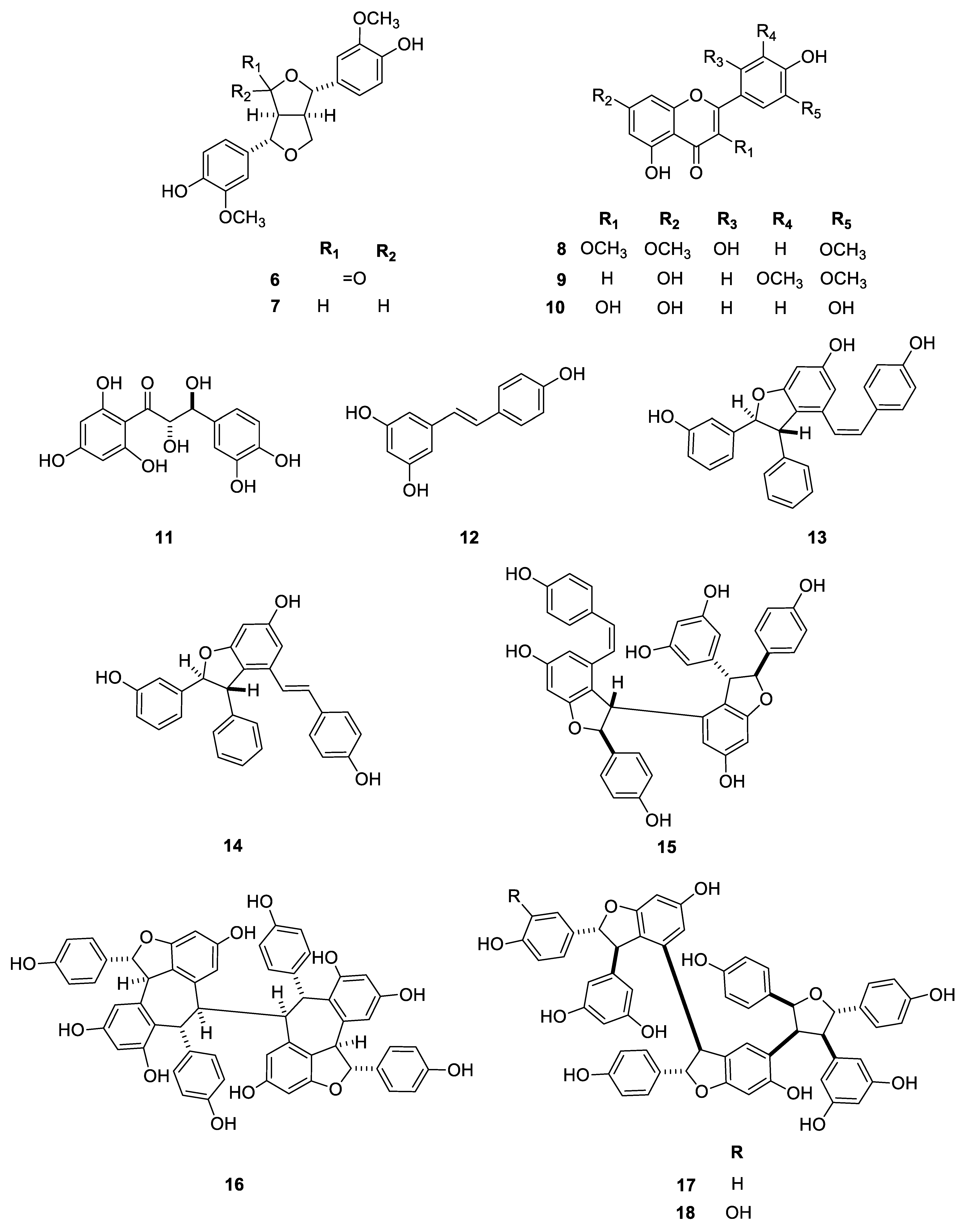
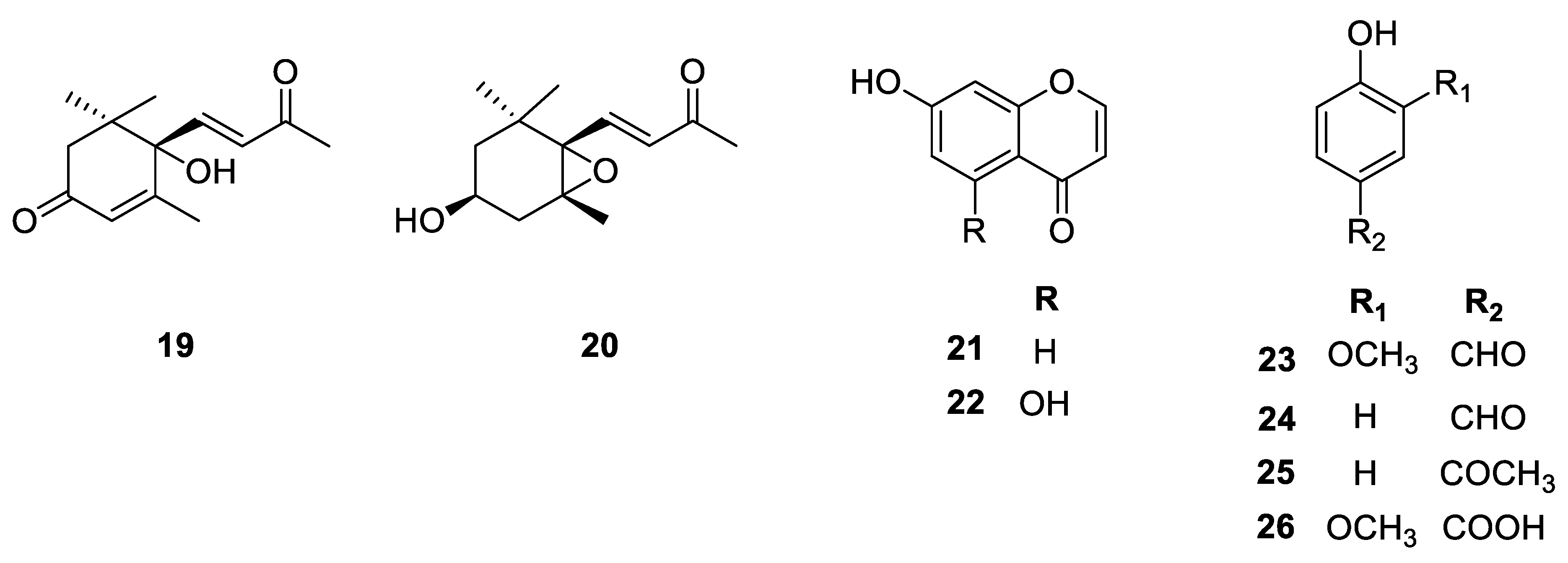
2.1. Isolation and Structure Determination of the Compounds
2.2. Pharmacological Assays
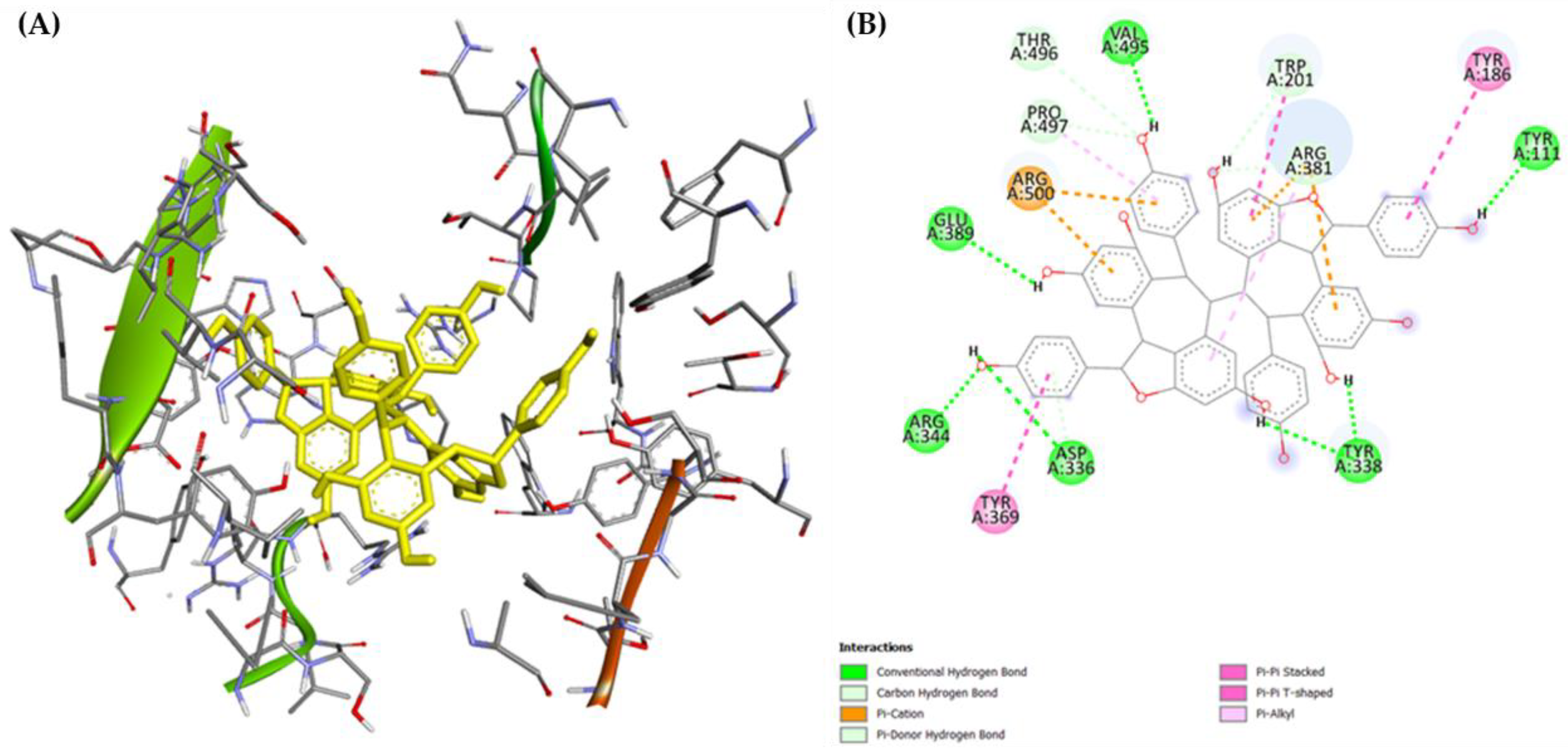
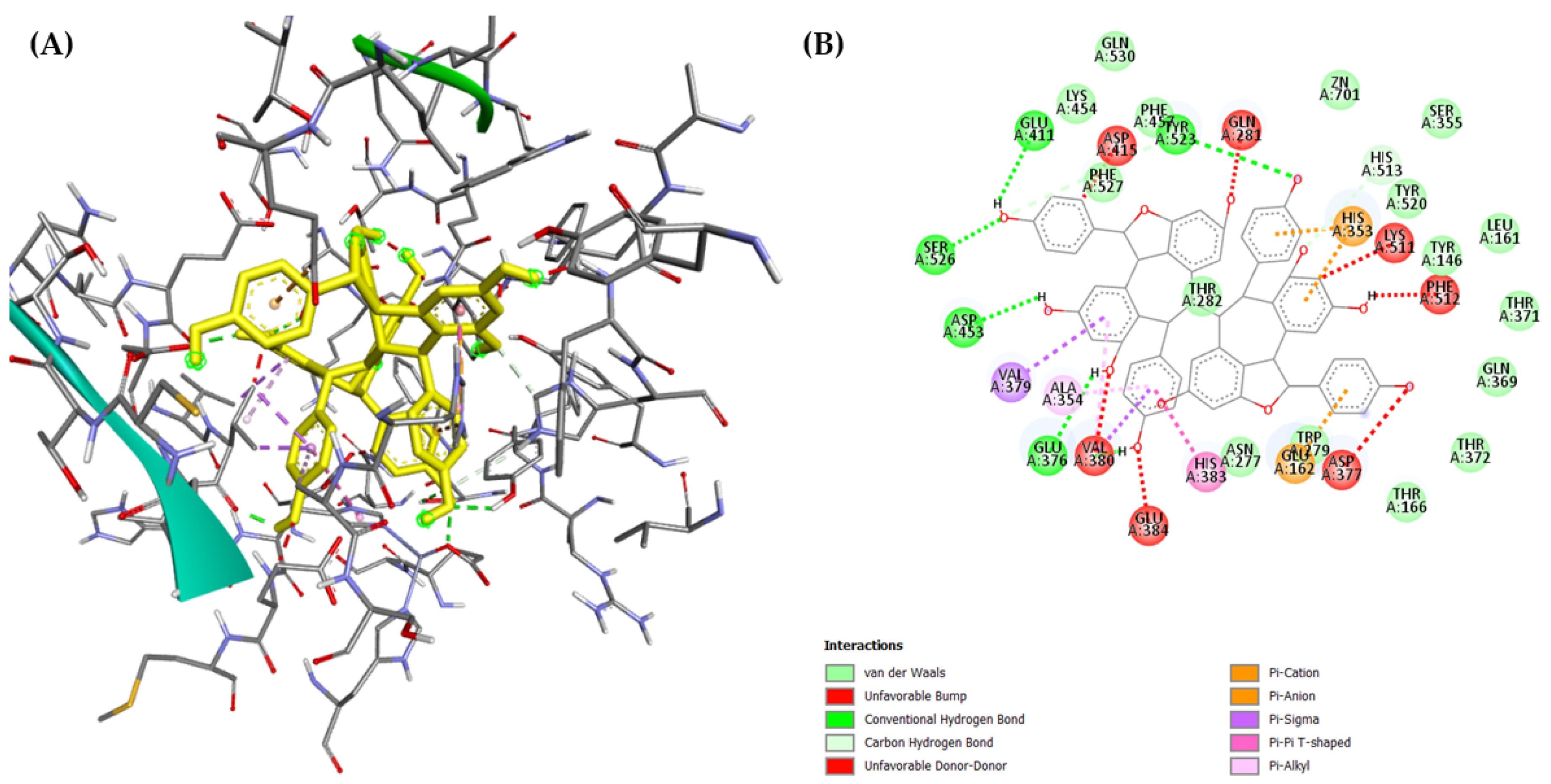
2.3. Molecular Docking
2.4. Other Pharmacological Assays
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Plant Material
3.3. Extraction and Isolation
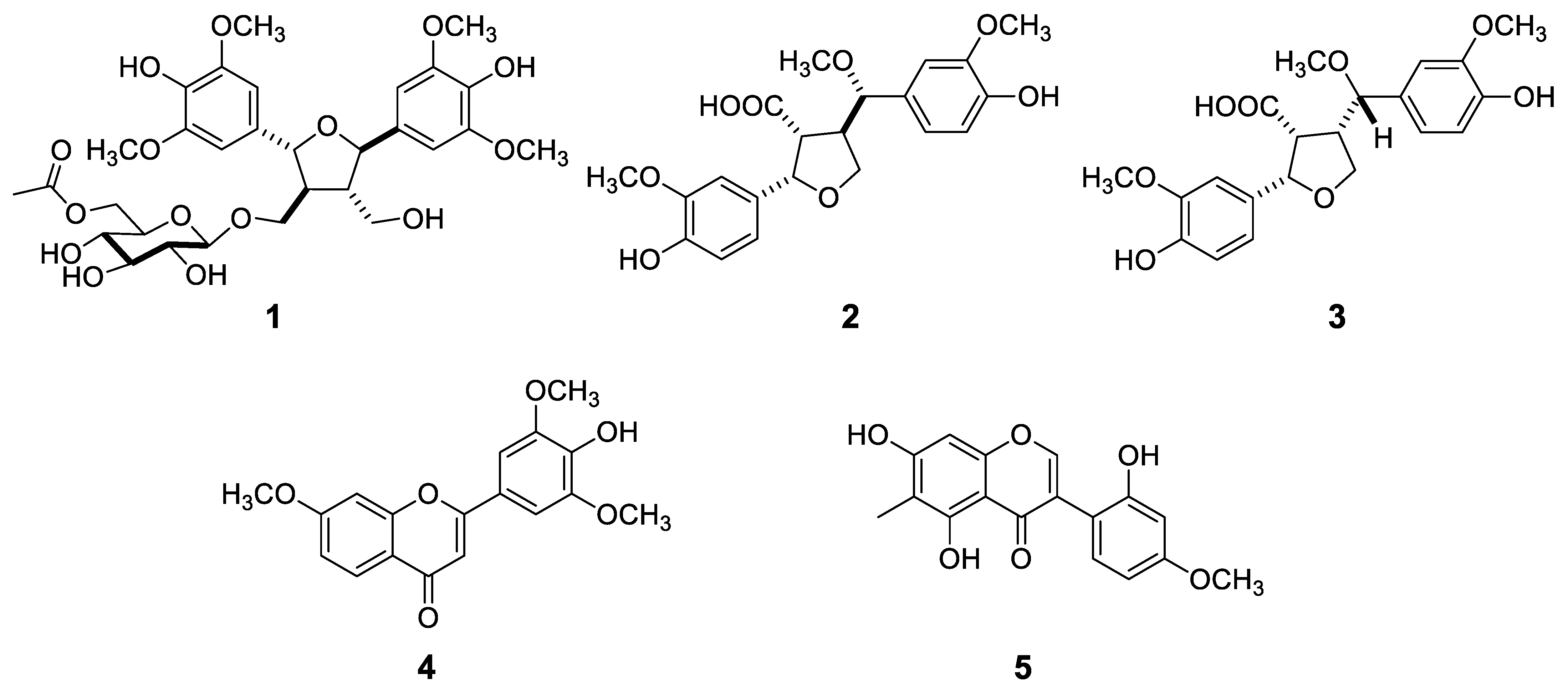
- Carexine A (1): yellowish oil; [α −6.0 (c 0.20, MeOH); 1H and 13C NMR data (CD3OD, see Table 1); HRESIMS m/z 658.2711 [M + NH4]+ (calcd for C30H44NO15 658.2711), 663.2258 [M + Na]+ (calcd for C30H40O15Na 663.2264).
- Carexine B (2): white amorphous solid; [α−14.4 (c 0.05, MeOH); 1H and 13C NMR data (CD3OD, see Table 2); HRESIMS m/z 403.1443 [M − H]− (calcd for C21H23 O8 403.1441).
- Carexine C (3): pale yellow amorphous solid; [α +7.6 (c 0.05, MeOH); 1H and 13C NMR data (CD3OD, see Table 2); HRESIMS m/z 403.1443 [M −H]− (calcd for C21H23 O8 403.1441).
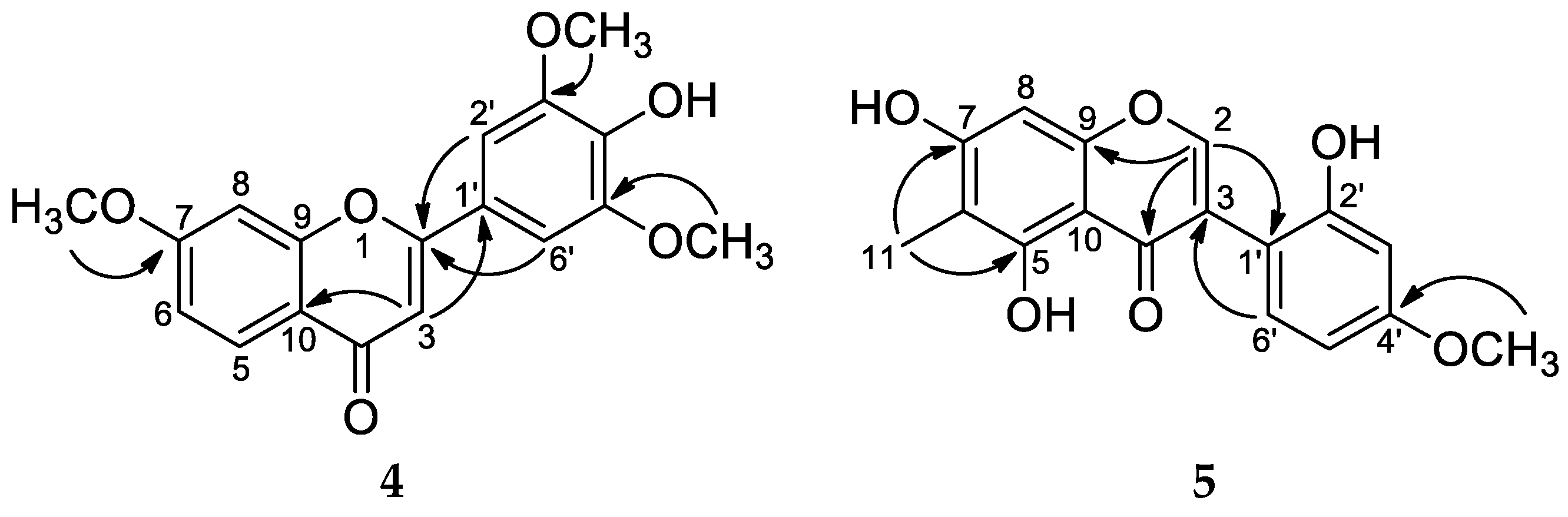
- Carexine D (4): pale yellow amorphous solid; 1H and 13C NMR data (CD3OD, see Table 3); HRESIMS m/z 329.1018 [M + H]+ (calcd for C18H17O6 329.1020).
- Carexine E (5): light brown amorphous solid; 1H and 13C NMR data (CD3OD, see Table 3); HRESIMS m/z 315.0865 [M + H]+ (calcd for C17H14O6 315.0869).
- Compound 8 (chrysosplenol F): 1H NMR (CD3OD) δH 7.02 (1H, s, H-6′), 6.55 (1H, d, J = 2.2 Hz, H-8), 6.48 (1H, s, H-3′), 6.34 (1H, d, J = 2.2 Hz, H-6), 3.87 (3H, s, 7-OMe), 3.83 (3H, s, 5′-OMe), 3.74 (3H, s, 3-OMe); δC 180.0 (C-4), 167.3 (C-7), 162.9 (C-5), 159.2* (C-2), 159.0* (C-9), 152.3# (C-2′), 152.1# (C-4′), 142.8 (C-5′), 140.4 (C-3), 114.6 (C-6′), 109.1 (C-1′), 107.1 (C-10), 93.3 (C-8), 105.3 (C-3′), 99.0 (C-6), 56.5 (OMe-7), 57.3 (OMe-5′), 61.2 (OMe-3); *,# are interchangeable.
- Compound 9 (tricin): 1H NMR (CD3OD) δH 7.25 (2H, s, H-2′,6′), 6.63 (1H, s, H-3), 6.45 (1H, d, J = 2.0 Hz, H-8), 6.2 (1H, d, J = 2.0 Hz, H-6), 3.95 (6H, s, 2 × OMe, 3′,5′).
3.4. Pharmacological Assays
3.4.1. ACE-Inhibitory Assay
3.4.2. Domain-Specific Studies
3.4.3. Molecular Docking
3.4.4. Cell Line Cultures
3.4.5. Antiproliferative Assay
3.4.6. Bacterial Strains and Culture Conditions for Antimicrobial Assays
3.4.7. Determination of Antibacterial Activity Using the Disk Diffusion Method
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO: The Top 10 Causes of Death. Available online: https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death (accessed on 29 February 2024).
- Curtin, S.C.; Tejada-Vera, B.; Bastian, B.A. Deaths: Leading causes for 2020. In National Vital Statistics Reports; National Center for Health Statistics: Hyattsville, MD, USA, 2023; Volume 72, No 13. [Google Scholar]
- OECD: Main Causes of Mortality. Available online: https://www.oecd-ilibrary.org/sites/a72a34af-en/index.html?itemId=/content/component/a72a34af-en (accessed on 29 February 2024).
- Bernstein, K.E.; Shen, X.Z.; Gonzalez-Villalobos, R.A.; Billet, S.; Okwan-Duodu, D.; Ong, F.S.; Fuchs, S. Different in vivo functions of the two catalytic domains of angiotensin-converting enzyme (ACE). Curr. Opin. Pharmacol. 2011, 11, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Masuyer, G.; Schwager, S.; Sturrock, E.; Isaac, R.E.; Acharya, K.R. Molecular recognition and regulation of human angiotensin-I converting enzyme (ACE) activity by natural inhibitory peptides. Sci. Rep. 2012, 2, 717. [Google Scholar] [CrossRef] [PubMed]
- Alves-Lopes, R.; Montezano, A.C.; Neves, K.B.; Harvey, A.; Rios, F.J.; Skiba, D.S.; Arendse, L.B.; Guzik, T.J.; Graham, D.; Poglitsch, M.; et al. Selective inhibition of the C-domain of ACE (Angiotensin-Converting Enzyme) combined with inhibition of NEP (Neprilysin)—A potential new therapy for hypertension. Hypertension 2021, 78, 604–616. [Google Scholar] [CrossRef] [PubMed]
- Fiorentino, A.; D’Abrosca, B.; Pacifico, S.; Izzo, A.; Letizia, M.; Esposito, A.; Monaco, P. Potential allelopatic effects of stilbenoids and flavonoids from leaves of Carex distachya Desf. Biochem. Syst. Ecol. 2008, 36, 691–698. [Google Scholar] [CrossRef]
- Dhar, P.; Dhar, D.G.; Rawat, A.K.S.; Srivastava, S. Medicinal chemistry and biological potential of Cyperus rotundus Linn.: An overview to discover elite chemotype(s) for industrial use. Ind. Crops Prod. 2017, 108, 232–247. [Google Scholar] [CrossRef]
- Cho, N.; Valenciano, A.L.; Du, Y.; Clement, J.; Cassera, M.B.; Goetz, M.; Kingston, D.G. Antiplasmodial flavanones and a stilbene from Carpha glomerata. Bioorg. Med. Chem. Lett. 2018, 28, 3368–3371. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, K.; Taguchi, H.; Endo, T.; Yosioka, I. The constituents of Scirpus fluviatilis (Torr). A. Gray. I. The structures of two new hydroxystilbene dimers, scirpusin A and B. Chem. Pharm. Bull. 1978, 26, 3050–3057. [Google Scholar] [CrossRef]
- Gamal, M.A.; Hani, K.M.K.; Sameh, E.S.; Sabrin, I.R.M. A Review: Compounds isolated from Cyperus species (Part I): Phenolics and nitrogenous. Int. J. Pharm. Phytochem. Res. 2015, 7, 51–67. [Google Scholar]
- Dávid, C.Z.; Hohmann, J.; Vasas, A. Chemistry and pharmacology of Cyperaceae stilbenoids: A review. Molecules 2021, 26, 2794. [Google Scholar] [CrossRef]
- Fiorentino, A.; D’Abrosca, B.; Pacifico, S.; Natale, A.; Monaco, P. Structures of bioactive carexanes from the roots of Carex distachya Desf. Phytochemistry 2006, 67, 971–977. [Google Scholar] [CrossRef]
- Kurihara, H.; Kawabata, J.; Ichikawa, S.; Mizutani, J. (–)-ε-Viniferin and related oligostilbenes from Carex pumila Thunb. (Cyperaceae). Agric. Biol. Chem. 1990, 54, 1097–1099. [Google Scholar] [CrossRef]
- Arraki, K.; Richard, T.; Badoc, A.; Pédrot, E.; Bisson, J.; Waffo-Téguo, P.; Mahjoub, A.; Mérillon, J.M.; Decendit, A. Isolation, characterization and quantification of stilbenes from some Carex species. Rec. Nat. Prod. 2013, 7, 281–291. [Google Scholar]
- Sotheeswaran, S.; Pasupathy, V. Distribution of resveratrol oligomers in plants. Phytochemistry 1993, 32, 1083–1092. [Google Scholar] [CrossRef]
- Teka, T.; Zhang, L.; Ge, X.; Li, Y.; Han, L.; Yan, X. Stilbenes: Source plants, chemistry, biosynthesis, pharmacology, application and problems related to their clinical application—A comprehensive review. Phytochemistry 2022, 197, 113128. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, H.; Miyase, T.; Ueno, A. Lignan and terpene glycosides from Epimedium sagittatum. Phytochemistry 1991, 30, 2025–2027. [Google Scholar] [CrossRef]
- Yamauchi, H.; Kakuda, R.; Yaoita, Y.; Machida, K.; Kikuchi, M. Two new glycosides from the whole plant of Gleochoma hederacea L. Chem. Pharm. Bull. 2007, 55, 346–347. [Google Scholar] [CrossRef] [PubMed]
- In, S.J.; Seo, K.H.; Song, N.Y.; Lee, D.S.; Kim, Y.C.; Baek, N.I. Lignans and neolignans from the stems of Viburnum erosum and their neuroprotective and anti-inflammatory activity. Arch. Pharm. Res. 2015, 38, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.S.; Rahman, A.A.; Kim, J.Y.; Kee, S.H. Hanultarin, a cytotoxic lignan as an inhibitor of actin cytoskeleton polymerization from the seeds of Trichosanthes kirilowii. Bioorg. Med. Chem. 2008, 16, 7264–7269. [Google Scholar] [CrossRef]
- Xie, L.H.; Akao, T.; Hamasaki, K.; Deyama, T.; Hattori, M. Biotransformation of pinoresinol diglucoside to mammalian lignans by human intestinal microflora, and isolation of Enterococcus faecalis strain PDG-1 responsible for the transformation of (+)-pinoresinol to (+)-lariciresinol. Chem. Pharm. Bull. 2003, 51, 508–515. [Google Scholar] [CrossRef]
- Arisawa, M.; Hayashi, T.; Shimizu, M.; Morita, N. Isolation and cytotoxicity of two new flavonoids from Chrysosplenium grayanum and related flavonols. J. Nat. Prod. 1991, 54, 898–901. [Google Scholar] [CrossRef]
- Wang, Y.; Shen, J.Z.; Chan, Y.W.; Ho, W.S. Identification and growth inhibitory activity of the chemical constituents from Imperata cylindrica aerial part ethyl acetate extract. Molecules 2018, 23, 1807. [Google Scholar] [CrossRef] [PubMed]
- Sinha, R.; Gadhwal, M.K.; Joshi, U.J.; Srivastava, S.; Govil, G. Modifying effect of quercetin on model biomembranes: Studied by molecular dynamic simulation, DSC and NMR. Int. J. Curr. Pharm. Res. 2012, 4, 70–79. [Google Scholar]
- Ahmed, B.; Al-Howiriny, T.A. Two new hydroxy chalcone derivatives from Thymus cilicicus. Z. Naturforsch B 2007, 62, 121–124. [Google Scholar] [CrossRef]
- Guiso, M.; Marra, C.; Farina, A. A new efficient resveratrol synthesis. Tetrahedron Lett. 2002, 43, 597–598. [Google Scholar] [CrossRef]
- Kim, H.J.; Chang, E.J.; Bae, S.J.; Shim, S.M.; Park, H.D.; Rhee, C.H.; Park, J.H.; Choi, S.W. Cytotoxic and antimutagenic stilbenes from seeds of Paeonia lactiflora. Arch. Pharm. Res. 2002, 25, 293–299. [Google Scholar] [CrossRef]
- Kurihara, H.; Kawabata, J.; Ichikawa, S.; Mishima, M.; Mizutani, J. Oligostilbenes from Carex kobomugi. Phytochemistry 1991, 30, 649–653. [Google Scholar] [CrossRef]
- Aisha, F.; Din, L.B.; Yaacob, W.A. Resveratrol tetramer of hopeaphenol isolated from Shorea johorensis (Dipterocarpaceae). In Proceedings of the 2014 UKM FST Postgraduate Colloquium, Selangor, Malaysia, 9–11 April 2014; Volume 1614, pp. 302–308. [Google Scholar] [CrossRef]
- Häusler, M.; Montag, A. Isolation, identification and quantitative determination of the norisoprenoid (S)-(+)-dehydrovomifoliol in honey. Z. Lebensm. Unters. Forsch. 1989, 189, 113–115. [Google Scholar] [CrossRef]
- Yan, Z.H.; Han, Z.Z.; Hu, X.Q.; Liu, Q.X.; Zhang, W.D.; Liu, R.H.; Li, H.L. Chemical constituents of Euonymus alatus. Chem. Nat. Compd. 2013, 49, 340–342. [Google Scholar] [CrossRef]
- Kataoka, K.I.; Shiota, T.; Takeyasu, T.; Mochizuki, T.; Taneda, K.; Ota, M.; Tanabe, H.; Yamaguchi, H. Potent inhibitors of acyl-CoA:cholesterol acyltransferase. Structure-activity relationships of novel N-(4-oxo-8-chromanyl) amides. J. Med. Chem. 1995, 38, 3174–3186. [Google Scholar] [CrossRef]
- Pendse, R.; Rao, A.V.R.; Venkataraman, K. 5,7-Dihydroxychromone from Arachis hypogoea shells. Phytochemistry 1973, 12, 2033–2034. [Google Scholar] [CrossRef]
- Suresh, D.; Gurudutt, K.N.; Srinivasan, K. Degradation of bioactive spice compound: Curcumin during domestic cooking. Eur. Food Res. Technol. 2008, 228, 807–812. [Google Scholar] [CrossRef]
- Fishman, A.; Tao, Y.; Wood, T.K. Toluene 3-monooxygenase of Ralstonia pickettii PKO1 is a para-hydroxylating enzyme. J. Bacteriol. 2004, 186, 3117–3123. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Polyphenols and health: Update and perspectives. Arch. Biochem. Biophys. 2010, 501, 2–5. [Google Scholar] [CrossRef]
- Manach, C.; Scalbert, A.; Morand, C.; Rémésy, C.; Jiménez, L. Polyphenols: Food sources and bioavailability. Am. J. Clin. Nutr. 2004, 79, 727–747. [Google Scholar] [CrossRef] [PubMed]
- Rudrapal, M.; Khairnar, S.J.; Khan, J.; Dukhyil, A.B.; Ansari, M.A.; Alomary, M.N.; Alshabrmi, F.M.; Palai, S.; Kumar, P.; Devi, R. Dietary polyphenols and their role in oxidative stress-induced human diseases: Insights into protective effects, antioxidant potentials and mechanism(s) of action. Front. Pharmacol. 2022, 13, 806470. [Google Scholar] [CrossRef] [PubMed]
- Loupit, G.; Fonayet, J.V.; Lorensen, M.D.B.B.; Franc, C.; De Revel, G.; Janfelt, C.; Cookson, S.J. Tissue-specific stilbene accumulation is an early response to wounding/grafting as revealed by using spatial and temporal metabolomics. Plant Cell Environ. 2023, 46, 3871–3886. [Google Scholar] [CrossRef] [PubMed]
- DellaGreca, M.; Cutillo, F.; D’Abrosca, B.; Fiorentino, A.; Zarrelli, A. Isolation of seed germination and plant growth inhibitors from mediterranean plants: Their potential use as herbicides. In Natural Products for Pest Management; Rimando, A.M., Duke, S.O., Eds.; American Chemical Society: New York, NY, USA, 2006; Volume 927, pp. 24–36. [Google Scholar] [CrossRef]
- Spencer, G.F.; Tjarks, L.W. Germination inhibition by 5,7-dihydroxychromone, a flavonoid decomposition product. J. Plant Growth Regul. 1985, 4, 177–180. [Google Scholar] [CrossRef]
- Tena, C.; Santiago, A.R.; Osuna, D.; Sosa, T. Phytotoxic activity of p-cresol, 2-phenylethanol and 3-phenyl-1-propanol, phenolic compounds present in Cistus ladanifer L. Plants 2021, 10, 1136. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Hwang, B.; Cho, S.; Kim, W.J.; Myung, S.C.; Choi, Y.H.; Kim, W.J.; Lee, S.; Moon, S.K. The ethanol extract of Cyperus exaltatus var. iwasakii exhibits cell cycle dysregulation, ERK1/2/p38 MAPK/AKT phosphorylation, and reduced MMP-9-mediated metastatic capacity in prostate cancer models in vitro and in vivo. Phytomedicine 2023, 114, 154794. [Google Scholar] [CrossRef]
- Pagning, A.; Tamokou, J.D.D.; Lateef, M.; Tapondjou, L.; Kuiate, J.R.; Ngnokam, D.; Ali, M. New triterpene and new flavone glucoside from Rhynchospora corymbosa (Cyperaceae) with their antimicrobial, tyrosinase and butyrylcholinesterase inhibitory activities. Phytochem. Lett. 2016, 16, 121–128. [Google Scholar] [CrossRef]
- Sayed, H.M.; Mohamed, M.H.; Farag, S.F.; Mohamed, G.A.; Proksch, P. A new steroid glycoside and furochromones from Cyperus rotundus L. Nat. Prod. Res. 2007, 21, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Santos, A.L.; Yamamoto, E.S.; Passero, L.F.D.; Laurenti, M.D.; Martins, L.F.; Lima, M.L.; Uemi, M.; Soares, M.G.; Lago, J.H.G.; Tempone, A.G.; et al. Antileishmanial activity and immunomodulatory effects of tricin isolated from leaves of Casearia arborea (Salicaceae). Chem. Biodivers. 2017, 14, e1600458. [Google Scholar] [CrossRef] [PubMed]
- Kuwabara, H.; Mouri, K.; Otsuka, H.; Kasai, R.; Yamasaki, K. Tricin from a malagasy connaraceous plant with potent antihistaminic activity. J. Nat. Prod. 2003, 66, 1273–1275. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.; Park, S.; Jhee, K.; Yang, S. Protection against UVB-induced wrinkle formation in SKH-1 hairless mice: Efficacy of tricin isolated from enzyme-treated Zizania latifolia extract. Molecules 2018, 23, 2254. [Google Scholar] [CrossRef] [PubMed]
- Coggon, P.; Janes, N.F.; King, F.E.; King, T.J.; Molyneux, R.J.; Morgan, J.W.W.; Sellars, K. Hopeaphenol, an extractive of the heartwood of Hopea odorata and Balanocarpus heimii. J. Chem. Soc. 1965, 406–409. [Google Scholar] [CrossRef]
- Tietjen, I.; Schonhofer, C.; Sciorillo, A.; Naidu, M.E.; Haq, Z.; Kannan, T.; Kossenkov, A.V.; Rivera-Ortiz, J.; Mounzer, K.; Hart, C.; et al. The natural stilbenoid (–)-hopeaphenol inhibits HIV transcription by targeting both PKC and NF-κB signaling and cyclin-dependent kinase 9. Antimicrob. Agents Chemother. 2023, 67, e01600-22. [Google Scholar] [CrossRef] [PubMed]
- Tietjen, I.; Cassel, J.; Register, E.T.; Zhou, X.Y.; Messick, T.E.; Keeney, F.; Lu, L.D.; Beattie, K.D.; Rali, T.; Tebas, P.; et al. The natural stilbenoid (–)-hopeaphenol inhibits cellular entry of SARS-CoV-2 USA-WA1/2020, B.1.1.7, and B.1.351 variants. Antimicrob. Agents Chemother. 2021, 65, e0077221. [Google Scholar] [CrossRef]
- Sharifi, N.; Khajeh, K.; Mahernia, S.; Balalaie, S.; Ataie, G.; Jahanbani, R.; Amanlou, M. Probing angiotensin converting enzyme (ACE) domain-dependent inhibition of onopordia, isolated from Onopordon acanthium L., using a continuous fluorescent assay. Pharm. Sci. 2018, 24, 31–37. [Google Scholar] [CrossRef]
- Dive, V.; Cotton, J.; Yiotakis, A.; Michaud, A.; Vassiliou, S.; Jiracek, J.; Vazeux, G.; Chauvet, M.T.; Cuniasse, P.; Corvol, P. RXP 407, a phosphinic peptide, is a potent inhibitor of angiotensin I converting enzyme able to differentiate between its two active sites. Proc. Natl. Acad. Sci. USA 1999, 96, 4330–4335. [Google Scholar] [CrossRef]
- Forli, S.; Huey, R.; Pique, M.E.; Sanner, M.F.; Goodsell, D.S.; Olson, A.J. Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 2016, 11, 905–919. [Google Scholar] [CrossRef]
- Corradi, H.R.; Schwager, S.L.; Nchinda, A.T.; Sturrock, E.D.; Acharya, K.R. Crystal structure of the N domain of human somatic angiotensin I-converting enzyme provides a structural basis for domain-specific inhibitor design. J. Mol. Biol. 2006, 357, 964–974. [Google Scholar] [CrossRef] [PubMed]
- Carmona, A.K.; Schwager, S.L.; Juliano, M.A.; Juliano, L.; Sturrock, E.D. A continuous fluorescence resonance energy transfer angiotensin I-converting enzyme assay. Nat. Protoc. 2006, 1, 1971–1976. [Google Scholar] [CrossRef]
- Lunow, D.; Kaiser, S.; Rückriemen, J.; Pohl, C.; Henle, T. Tryptophan-containing dipeptides are C-domain selective inhibitors of angiotensin converting enzyme. Food Chem. 2015, 166, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Natesh, R.; Schwager, S.L.; Sturrock, E.D.; Acharya, K.R. Crystal structure of the human angiotensin-converting enzyme–lisinopril complex. Nature 2003, 421, 551–554. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Xie, D.; Du, L.; Lin, H.; Su, E.; Shen, Y.; Xie, J.; Wei, D. In vitro-in silico screening strategy and mechanism of angiotensin I-converting enzyme inhibitory peptides from α-lactalbumin. LWT 2022, 156, 112984. [Google Scholar] [CrossRef]
- Douglas, R.G.; Sharma, R.K.; Masuyer, G.; Lubbe, L.; Zamora, I.; Acharya, K.R.; Chibale, K.; Sturrock, E.D. Fragment-based design for the development of N-domain-selective angiotensin-1-converting enzyme inhibitors. Clin. Sci. 2014, 126, 305–313. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeerch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef]
- Ghazal, T.S.A.; Schelz, Z.; Vidács, L.; Szemerédi, N.; Veres, K.; Spengler, G.; Hohmann, J. Antimicrobial, multidrug resistance reversal and biofilm formation inhibitory effect of Origanum majorana extracts, essential oil and monoterpenes. Plants 2022, 11, 1432. [Google Scholar] [CrossRef]
 ) and HMBCs (H → C) of compound 1.
) and HMBCs (H → C) of compound 1.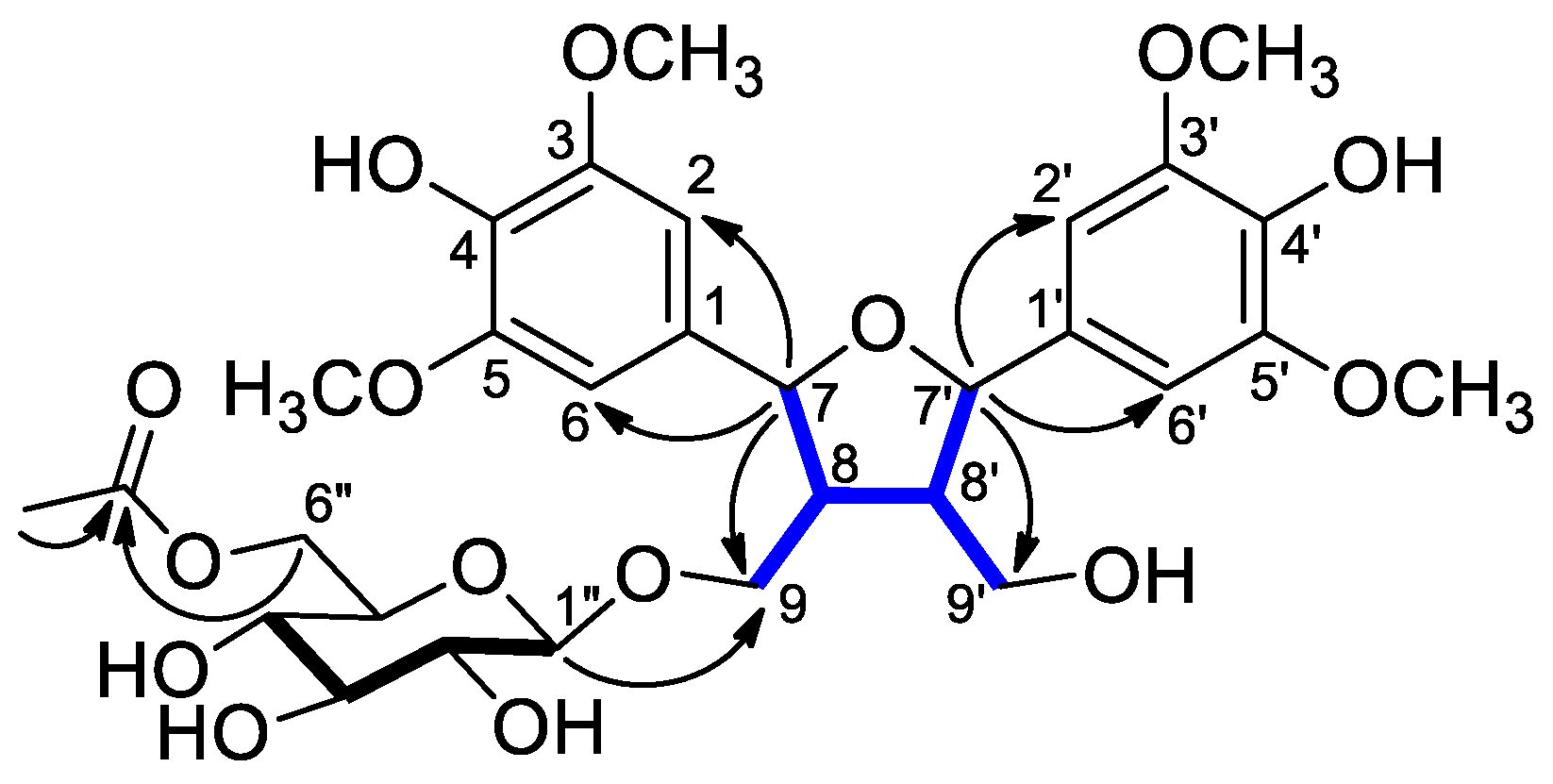 ) and HMBCs (H → C) of compound 2.
) and HMBCs (H → C) of compound 2.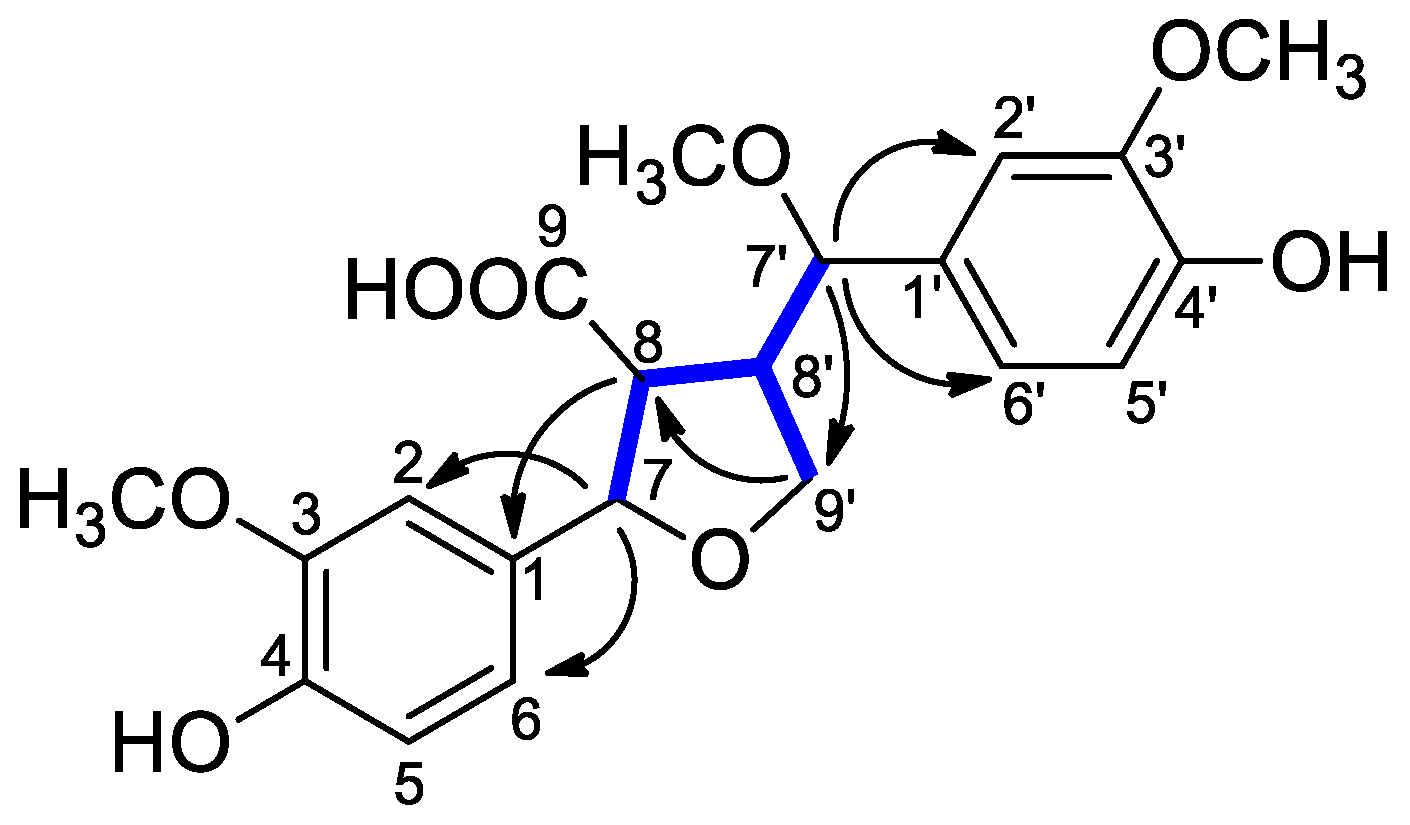
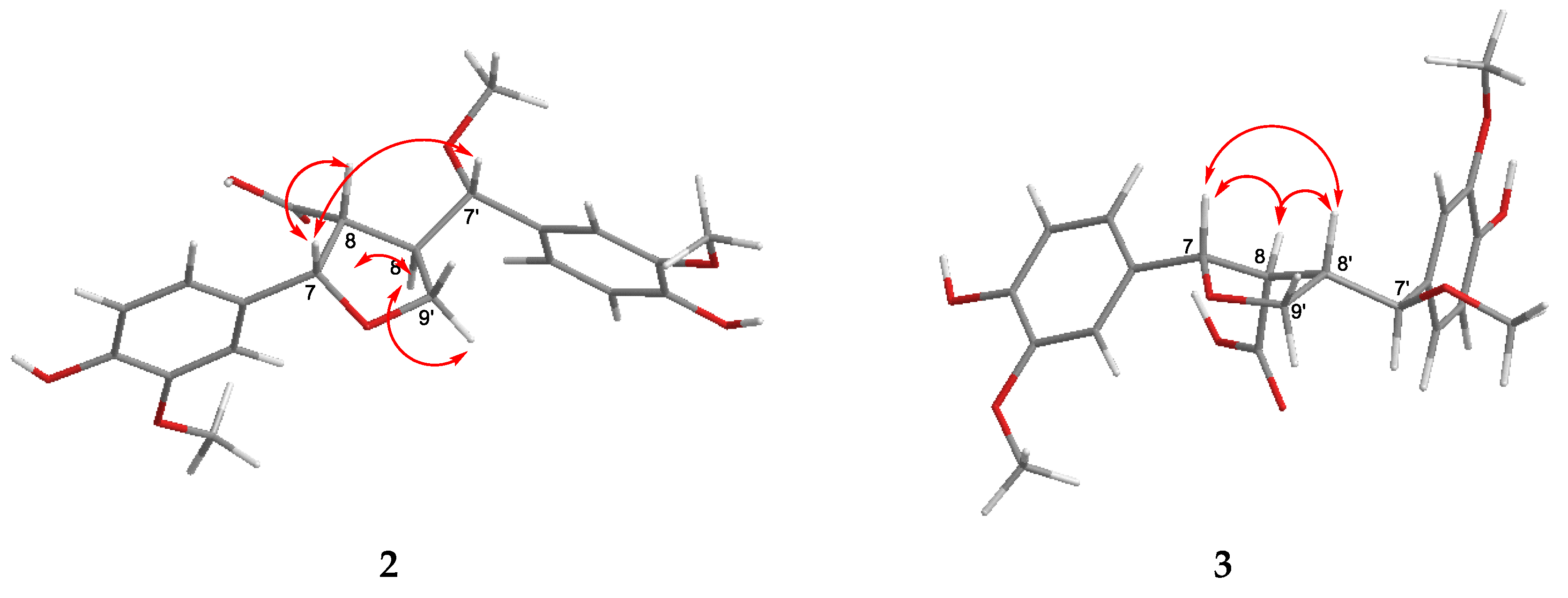

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1H | 13C | Position | 1H | 13C |
|---|---|---|---|---|---|
| δH (ppm), J (Hz) | δC, Type | δH (ppm), J (Hz) | δC, Type | ||
| 1 | 134.3, C | 1′ | 134.3, C | ||
| 2,6 | 6.74 s | 104.9, CH | 2′,6′ | 6.71 s | 104.9, CH |
| 3 | 149.3, C | 3′ | 149.3, C | ||
| 4 | 136.1, * C | 4′ | 136.2, * C | ||
| 5 | 149.3, C | 5′ | 149.3, C | ||
| 7 | 5.05 d (8.4) | 84.6, CH | 7′ | 4.98 d (8.5) | 84.3, CH |
| 8 | 2.44 m | 51.9, CH | 8′ | 2.32 m | 54.5, CH |
| 9 | 3.96 dd (10.1, 5.0), 3.65 dd (10.1, 5.0) | 69.9, CH2 | 9′ | 3.71 dd (11.6, 4.4), 3.59 dd (11.6, 4.4) | 61.0, CH2 |
| 1″ | 4.25 d (7.9) | 104.7, CH | |||
| 2″ | 3.16 br t (7.9) | 75.1, CH | |||
| 3″ | 3.30 m | 78.0, CH | |||
| 4″ | 3.25 m | 71.6, CH | |||
| 5″ | 3.41 m | 75.3, CH | |||
| 6″ | 4.33 dd (11.9, 2.1), 4.15 dd (11.9, 5.8) | 64.6, CH2 | |||
| 3/3′-OCH3 | 3.83 s | 56.9, CH3 | |||
| 5/5′-OCH3 | 3.83 s | 56.9, CH3 | |||
| Ac-CO | 172.7, C | ||||
| Ac-Me | 1.94 s | 20.6, CH3 |
| 2 | 3 | |||
|---|---|---|---|---|
| Position | 1H | 13C | 1H | 13C |
| δH (ppm), J (Hz) | δC, Type | δH (ppm), J (Hz) | δC, Type | |
| 1 | - | 133.9, C | - | 134.9, C |
| 2 | 6.91 d (1.8) | 110.6, CH | 6.90 d (1.6) | 110.6, CH |
| 3 | - | 149.1, C | - | 148.9, C |
| 4 | - | 147.9, * C | - | 147.1, C |
| 5 | 6.78 d (8.1) | 116.1, CH | 6.77 d (8.1) | 116.2, CH |
| 6 | 6.77 dd (8.1, 1.8) | 119.7, CH | 6.78 dd (8.1, 1.6) | 119.6, CH |
| 7 | 5.07 d (7.2) | 86.0, CH | 5.12 d (5.9) | 85.5, CH |
| 8 | 3.07 m | 56.4, CH | 2.72 dd (8.4, 5.9) | 56.4, CH |
| 9 | - | 176.6, C | - | 176.8, C |
| 1′ | - | 132.8, C | - | 132.9, C |
| 2′ | 6.92 d (1.8) | 111.4, CH | 6.85 d (1.6) | 111.7, CH |
| 3′ | - | 149.4, C | - | 149.1, C |
| 4′ | - | 147.4, * C | - | 147.3, C |
| 5′ | 6.80 d (8.0) | 116.1, CH | 6.73 d (8.1) | 116.0, CH |
| 6′ | 6.78 dd (8.0, 1.8) | 121.5, CH | 6.76 dd (8.1, 1.6) | 121.0, CH |
| 7′ | 4.26 d (10.4) | 83.6, CH | 4.51 d (7.1) | 83.3, CH |
| 8′ | 3.00 m | 51.2, CH | 2.90 m | 50.3, CH |
| 9′a (α) | 3.63 t (8.6, 7.6) | 71.4, CH2 | 4.15 d (7.9) 2H | 71.6, CH2 |
| 9′b (β) | 3.56 t (8.6) | |||
| 3-OCH3 | 3.84 s | 56.4, CH3 | 3.80 s | 56.4, CH3 |
| 3′-OCH3 | 3.87 s | 56.4, CH3 | 3.84 s | 56.4, CH3 |
| 7′-OCH3 | 3.06 s | 56.0, CH3 | 3.15 s | 56.6, CH3 |
| 4 | 5 | |||
|---|---|---|---|---|
| Position | 1H | 13C | 1H | 13C |
| δH (ppm), J (Hz) | δC, Type | δH (ppm), J (Hz) | δC, Type | |
| 2 | 166.1, C | 8.00 s | 156.5, C | |
| 3 | 6.78 s | 106.1, CH | 122.3, C | |
| 4 | 180.2, C | 182.6, C | ||
| 5 | 8.04 d (8.9) | 127.5, C | 160.5, C | |
| 6 | 7.07 dd (8.9, 2.4) | 116.1, CH | 109.1, C | |
| 7 | 166.4, C | 164.0, C | ||
| 8 | 7.28 d (2.4) | 101.7, CH | 6.41 s | 93.8, CH |
| 9 | 159.7, C | 157.5, C | ||
| 10 | 118.2 | 105.9, C | ||
| 11 | - | - | 2.07 s | 7.4, CH3 |
| 1′ | 123.0, C | 112.3, C | ||
| 2′ | 7.33 s | 105.5, CH | 157.8, C | |
| 3′ | 149.8, C | 6.48 br s | 103.1, CH | |
| 4′ | 141.2, CH | 162.7, C | ||
| 5′ | 149.8, C | 6.49 dd (8.3, 2.5) | 106.6, CH | |
| 6′ | 7.33 s | 105.5, CH2 | 7.13 d (8.3) | 133.2, CH |
| 7-OCH3 | 3.98 s | 56.7, CH3 | ||
| 3′-OCH3 | 3.97 s | 57.2, CH3 | ||
| 4′-OCH3 | 3.78 s | 55.7, CH3 | ||
| 5′-OCH3 | 3.97 s | 57.2, CH3 | ||
| Compound | Inhibition at 90 μM (%) ± SD | IC50 (μM) ± SD |
|---|---|---|
| 12 | 35.4 ± 3.9 | 185.8 ± 12.8 |
| 13 | 95.5 ± 2.8 | 18.0 ± 1.2 |
| 14 | 106.7 ± 1.1 | 14.0 ± 0.7 |
| 15 | 96.7 ± 6.3 | 15.2 ± 0.4 |
| 16 | 102.8 ± 1.5 | 7.7 ± 0.9 |
| 17 | 98.5 ± 1.2 | 14.8 ± 0.8 |
| 18 | 98.7 ± 3.8 | 22.3 ± 0.9 |
| Captopril | 80.8 ± 1.2 | 0.2 ± 0.1 |
| Inhibition (%) ± SD | ||
|---|---|---|
| N-Domain | C-Domain | |
| BPPb (200 nM) | 5.2 ± 0.2 | 83.20 ± 3.6 |
| 16 (10 μM) | 42.41 ± 0.8 | 18.17 ± 0.5 |
| 16 (50 μM) | 55.74 ± 3.2 | 23.39 ± 0.6 |
| Colo 205 | Colo 320 | |
|---|---|---|
| Compound | IC50 (μM) ± SD | IC50 (μM) ± SD |
| 12 | 48.33 ± 1.54 | 40.4 ± 1.84 |
| 13 | >100 | >100 |
| 14 | >100 | >100 |
| 15 | >100 | >100 |
| 16 | 1.59 ± 0.11 | 6.08 ± 0.24 |
| 17 | >100 | >100 |
| 18 | >100 | >100 |
| DMSO | >1% | >1% |
| Cisplatin | 53.93 ± 3.92 | 64.68 ± 3.56 |
| Doxorubicin | 0.33 ± 0.03 | 0.67 ± 0.06 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dávid, C.Z.; Kúsz, N.; Agbadua, O.G.; Berkecz, R.; Kincses, A.; Spengler, G.; Hunyadi, A.; Hohmann, J.; Vasas, A. Phytochemical Investigation of Carex praecox Schreb. and ACE-Inhibitory Activity of Oligomer Stilbenes of the Plant. Molecules 2024, 29, 3427. https://doi.org/10.3390/molecules29143427
Dávid CZ, Kúsz N, Agbadua OG, Berkecz R, Kincses A, Spengler G, Hunyadi A, Hohmann J, Vasas A. Phytochemical Investigation of Carex praecox Schreb. and ACE-Inhibitory Activity of Oligomer Stilbenes of the Plant. Molecules. 2024; 29(14):3427. https://doi.org/10.3390/molecules29143427
Chicago/Turabian StyleDávid, Csilla Zsuzsanna, Norbert Kúsz, Orinamhe Godwin Agbadua, Róbert Berkecz, Annamária Kincses, Gabriella Spengler, Attila Hunyadi, Judit Hohmann, and Andrea Vasas. 2024. "Phytochemical Investigation of Carex praecox Schreb. and ACE-Inhibitory Activity of Oligomer Stilbenes of the Plant" Molecules 29, no. 14: 3427. https://doi.org/10.3390/molecules29143427
APA StyleDávid, C. Z., Kúsz, N., Agbadua, O. G., Berkecz, R., Kincses, A., Spengler, G., Hunyadi, A., Hohmann, J., & Vasas, A. (2024). Phytochemical Investigation of Carex praecox Schreb. and ACE-Inhibitory Activity of Oligomer Stilbenes of the Plant. Molecules, 29(14), 3427. https://doi.org/10.3390/molecules29143427