
HOO• as the Chain Carrier for the Autocatalytic Photooxidation of Benzylic Alcohols
 , ,
, ,
Abstract
:1. Introduction
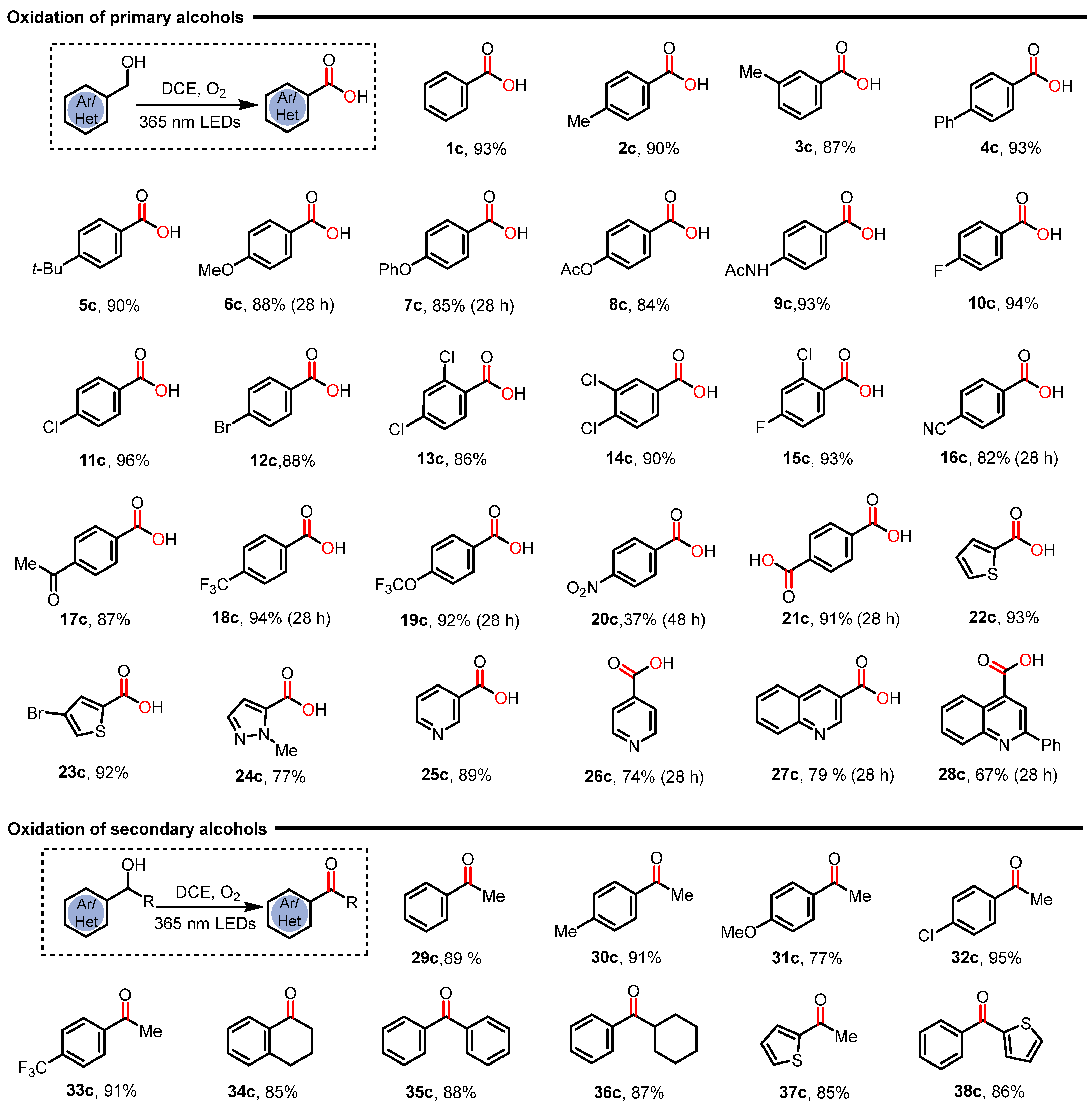
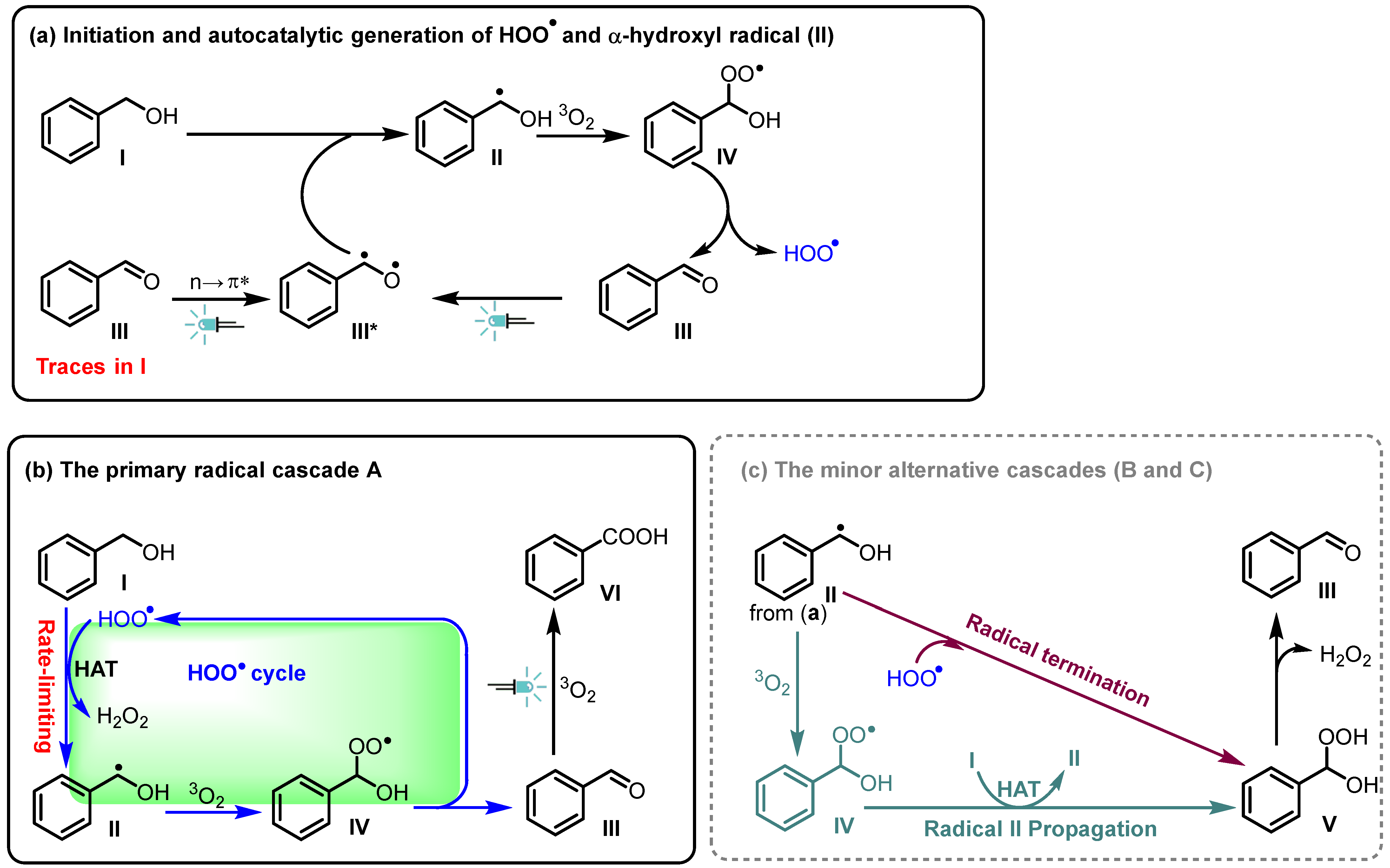
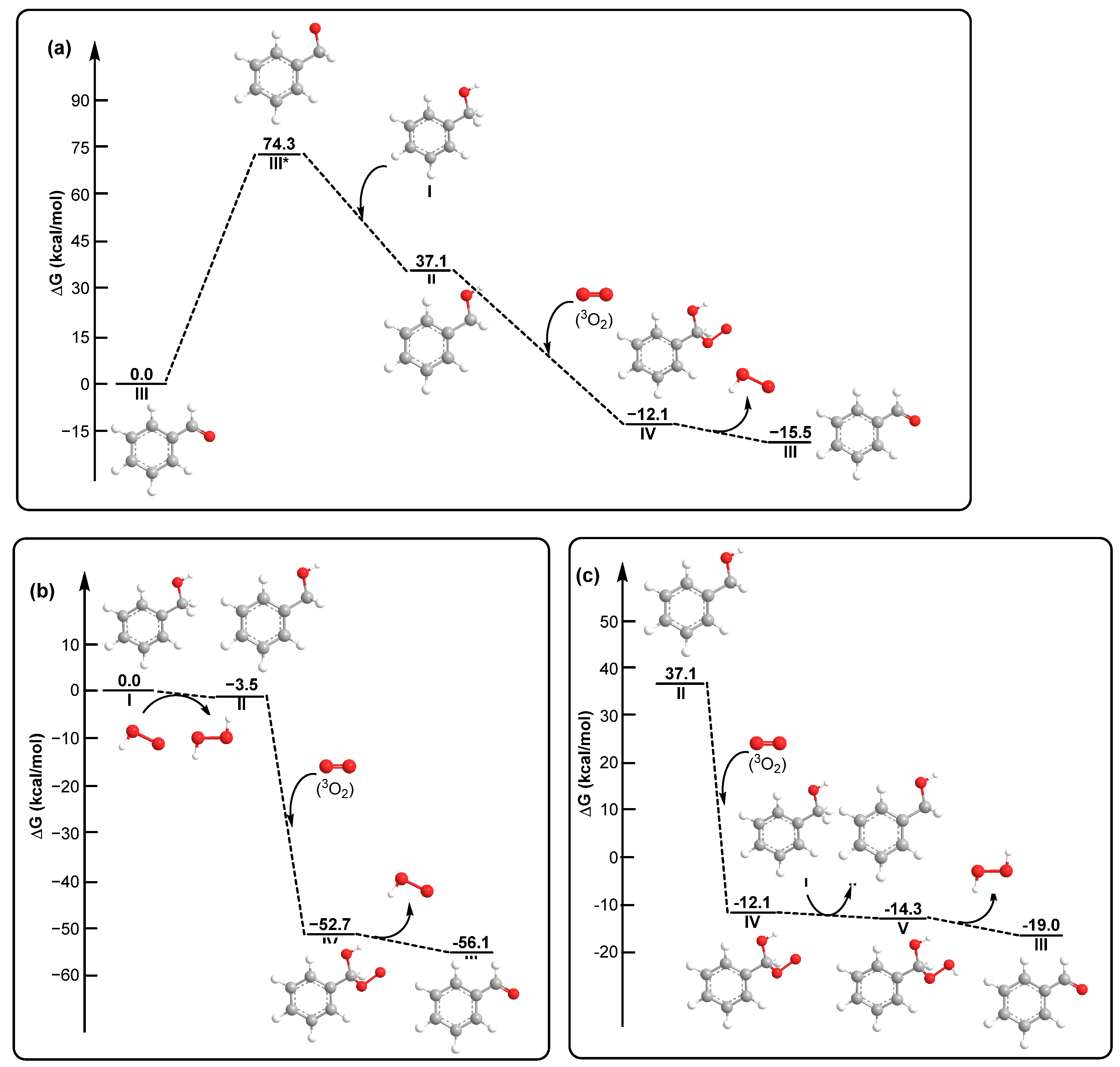
2. Results and Discussions
3. Experimental Section
3.1. General Information
3.2. General Procedure for the Oxidation
3.3. Characterization Data of Products 1–38c
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Backvall, J.E. Modern Oxidation Methods, 2nd ed.; John wiley & Sons: Hoboken, NJ, USA, 2011. [Google Scholar]
- Shen, Z.W.; Hu, Y.Z.; Li, B.F.; Zou, Y.T.; Li, S.J.; Wilma Busser, G.; Wang, X.K.; Zhao, G.X.; Muhler, M. State-of-the-art progress in the selective photo-oxidation of alcohols. J. Energy Chem. 2021, 62, 338–350. [Google Scholar] [CrossRef]
- Nosaka, Y.; Nosaka, A.Y. Generation and detection of reactive oxygen species in photocatalysis. Chem. Rev. 2017, 117, 11302–11336. [Google Scholar] [CrossRef]
- Hajian, R.; Alghour, Z. Selective oxidation of alcohols with H2O2 catalyzed by zinc polyoxometalate immobilized on multi-wall carbon nanotubes modified with ionic liquid. Chin. Chem. Lett. 2017, 28, 971–975. [Google Scholar] [CrossRef]
- Lei, Z.Q.; Wang, J.Q.; Yan, P.H. Highly efficient oxidation of alcohols using Oxone® as oxidant catalyzed by ruthenium complex under mild reaction conditions. Chin. Chem. Lett. 2008, 19, 1031–1034. [Google Scholar] [CrossRef]
- Ji, H.B.; Qian, Y. Highly efficient, green oxidation of alcohols using novel heterogeneous ruthenium catalyst. Chin. Chem. Lett. 2003, 14, 615–618. [Google Scholar]
- Yue, B.; Zhu, S.S.; Xie, G.Y.; Gu, Y.D. Photooxidation of some organic compounds catalyzed by decatungstates. Chin. Chem. Lett. 1995, 6, 215–216. [Google Scholar]
- Corma, A.; García, H. Lewis acids as catalysts in oxidation reactions: From homogeneous to heterogeneous systems. Chem. Rev. 2002, 102, 3837–3892. [Google Scholar] [CrossRef] [PubMed]
- Mallat, T.; Baiker, A. Oxidation of alcohols with molecular oxygen on solid catalysts. Chem. Rev. 2004, 104, 3037–3058. [Google Scholar] [CrossRef] [PubMed]
- Markó, I.E.; Giles, P.R.; Tsukazaki, M.; Chellé-Regnaut, I.; Gautier, A.; Dumeunier, R.; Philippart, F.; Doda, K.; Mutonkole, J.L.; Brown, S.M.; et al. Efficient, ecologically benign, aerobic oxidation of alcohols. Adv. Inorg. Chem. 2004, 56, 211–240. [Google Scholar]
- Schultz, M.J.; Sigman, M.S. Recent advances in homogeneous transition metal-catalyzed aerobic alcohol oxidations. Tetrahedron 2006, 62, 8227–8241. [Google Scholar] [CrossRef]
- Matsumoto, T.; Ueno, M.; Wang, N.; Kobayashi, S. Recent advances in immobilized metal catalysts for environmentally benign oxidation of alcohols. Chem. Asian J. 2008, 3, 196–214. [Google Scholar] [CrossRef] [PubMed]
- Parmeggiani, C.; Cardona, F. Transition metal based catalysts in the aerobic oxidation of alcohols. Green Chem. 2012, 14, 547–564. [Google Scholar] [CrossRef]
- Xiao, C.L.; Zhang, L.; Hao, H.C.; Wang, W.Z. High selective oxidation of benzyl alcohol to benzylaldehyde and benzoic acid with surface oxygen vacancies on W18O49/holey ultrathin g-C3N4 nanosheets. ACS Sustain. Chem. Eng. 2019, 7, 7268–7276. [Google Scholar] [CrossRef]
- Kargar, H.; Fallah-Mehrjardi, M.; Behjatmanesh-Ardakani, R.; Munawar, K.S.; Ashfaq, M.; Tahir, M.N. Selective oxidation of benzyl alcohols to benzaldehydes catalyzed by dioxomolybdenum Schiff base complex: Synthesis, spectral characterization, crystal structure, theoretical and computational studies. Transit. Met. Chem. 2021, 46, 437–455. [Google Scholar] [CrossRef]
- Rana, S.; Jonnalagadda, S.B. Cu doped amine functionalized graphene oxide and its scope as catalyst for selective oxidation. Catal. Commun. 2017, 100, 183–186. [Google Scholar] [CrossRef]
- Hara, T.; Ishikawa, M.; Sawada, J.; Ichikuni, N.; Shimazu, S. Creation of highly stable monomeric Pd(II) species in an anion-exchangeable hydroxy double salt interlayer: Application to aerobic alcohol oxidation under an air atmosphere. Green Chem. 2009, 11, 2034–2040. [Google Scholar] [CrossRef]
- Mori, K.; Hara, T.; Mizugaki, T.; Ebitani, K.; Kaneda, K. Hydroxyapatite-supported palladium nanoclusters: A highly active heterogeneous catalyst for selective oxidation of alcohols by use of molecular oxygen. J. Am. Chem. Soc. 2004, 126, 10657–10666. [Google Scholar] [CrossRef] [PubMed]
- Rana, S.; Maddila, S.; Jonnalagadda, S.B. Synthesis and characterization of Pd(II) dispersed over diamine functionalized graphene oxide and its scope as a catalyst for selective oxidation. Catal. Sci. Technol. 2015, 5, 3235–3241. [Google Scholar] [CrossRef]
- Hao, P.; Dong, X.; Wen, H.G.; Xu, R.R.; Xie, J.F.; Wang, Q.; Cui, G.W.; Tian, J.; Tang, B. In-situ assembly of 2D/3D porous nickel cobalt sulfide solid solution as superior pre-catalysts to boost multi-functional electrocatalytic oxidation. Chin. Chem. Lett. 2023, 34, 107843. [Google Scholar] [CrossRef]
- He, Z.X.; Yin, B.; Li, X.H.; Zhou, X.L.; Song, H.N.; Xu, J.B.; Gao, F. Photochemical selective oxidation of benzyl alcohols to aldehydes or ketones. J. Org. Chem. 2023, 88, 4765–4769. [Google Scholar] [CrossRef]
- Walia, P.K.; Sharma, M.; Kumar, M.; Bhalla, V. UV light promoted ‘Metal’/‘Additive’-free oxidation of alcohols: Investigating the role of alcohols as electron donors. RSC Adv. 2019, 9, 36198–36203. [Google Scholar] [CrossRef]
- Xu, M.; Ou, J.H.; Luo, K.J.; Liang, R.T.; Liu, J.; Li, N.; Hu, B.N.; Liu, K.J. External catalyst- and additive-free photo-oxidation of aromatic alcohols to carboxylic acids or ketones using air/O2. Molecules 2023, 28, 3031. [Google Scholar] [CrossRef] [PubMed]
- Weisheitelová, I.; Varma, N.; Chudoba, J.; Burdzinski, G.; Sikorski, M.; Cibulka, R. Catalyst-free aerobic photooxidation of sensitive benzylic alcohols with chemoselectivity controlled using DMSO as the solvent. Green Chem. 2024, 26, 16628. [Google Scholar] [CrossRef]
- Wu, J.Q.; Chen, J.W.; Wang, L.; Zhu, H.J.; Liu, R.; Song, G.L.; Feng, C.; Li, Y.F. Bronsted acid-catalysed aerobic photo-oxygenation of benzylic C-H bonds. Green Chem. 2023, 25, 940–945. [Google Scholar] [CrossRef]
- Gu, J.F.; Wan, Y.T.; Ma, H.F.; Zhu, H.J.; Bu, H.Z.; Zhou, Y.A.; Zhang, W.J.; Wu, Z.G.; Li, Y.F. Ferric ion concentration-controlled aerobic photo-oxidation of benzylic C-H bond with high selectivity and conversion. Tetrahedron 2021, 93, 132298. [Google Scholar] [CrossRef]
- Huang, Z.L.; Shanmugam, M.; Liu, Z.; Brookfield, A.; Bennett, E.L.; Guan, R.P.; Herrera, D.E.V.; Lopez-Sanchez, J.A.; Slater, A.G.; McInnes, E.J.L.; et al. Chemical recycling of polystyrene to valuable chemicals via selective acid-catalyzed aerobic oxidation under visible light. J. Am. Chem. Soc. 2022, 144, 6532–6542. [Google Scholar] [CrossRef]
- Zhang, Q.H.; An, B.; Lei, Y.; Gao, Z.X.; Zhang, H.A.; Xue, S.; Jin, X.; Xu, W.A.; Wu, Z.H.; Wu, M.B.; et al. Cl2•- mediates direct and selective conversion of inert C(sp3)-H bonds into aldehydes/ketones. Angew. Chem.-Int. Edit. 2023, 62, e202304699. [Google Scholar] [CrossRef]
- Krishnaraj, C.; Jena, H.S.; Bourda, L.; Laemont, A.; Pachfule, P.; Roeser, J.; Chandran, C.V.; Borgmans, S.; Rogge, S.M.J.; Leus, K.; et al. Strongly reducing (diarylamino)benzene-based covalent organic framework for metal-free visible light photocatalytic H2O2 generation. J. Am. Chem. Soc. 2020, 142, 20107–20116. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.Y.; Cao, J.J.; Wang, X.; Liu, Y.; Zhao, Y.J.; Wang, H.; Liu, Y.; Huang, H.; Liao, F.; Shao, M.W.; et al. A metal-free photocatalyst for highly efficient hydrogen peroxide photoproduction in real seawater. Nat. Commun. 2021, 12, 483. [Google Scholar] [CrossRef]
- Santiago-Portillo, A.; Navalón, S.; Cirujano, F.G.; Xamena, F.X.L.I.; Alvaro, M.; Garcia, H. MIL-101 as reusable solid catalyst for autoxidation of benzylic hydrocarbons in the absence of additional oxidizing reagents. ACS Catal. 2015, 5, 3216–3224. [Google Scholar] [CrossRef]
- Mavridi-Printezi, A.; Mollica, F.; Lucernati, R.; Montalti, M.; Amorati, R. Insight into the antioxidant activity of 1,8-dihydroxynaphthalene allomelanin nanoparticles. Antioxidants 2023, 12, 1511. [Google Scholar] [CrossRef] [PubMed]
- Qi, M.Y.; Lin, Q.; Tang, Z.R.; Xu, Y.J. Photoredox coupling of benzyl alcohol oxidation with CO2 reduction over CdS/TiO2 heterostructure under visible light irradiation. Appl. Catal. B-Environ. 2022, 307, 121158. [Google Scholar] [CrossRef]
- Qi, M.Y.; Li, Y.H.; Anpo, M.; Tang, Z.R.; Xu, Y.J. Efficient photoredox-mediated C-C coupling organic synthesis and hydrogen production over engineered semiconductor quantum dots. Acs Catal. 2020, 10, 14327–14335. [Google Scholar] [CrossRef]
- Thiault, G.; Mellouki, A.; Le Bras, G.; Chakir, A.; Sokolowski-Gomez, N.; Daumont, D. UV-absorption cross sections of benzaldehyde, ortho-, meta-, and para-tolualdehyde. J. Photochem. Photobiol. A-Chem. 2004, 162, 273–281. [Google Scholar] [CrossRef]
- Koshino, N.; Funahashi, S.; Takagi, H.D. Oxidations of hydrogen peroxide by bis(1,4,7-triazacyclononane)nickel(III), bis(1,4,7-trithiacyclononane)iron(III) and tris(2,2′-bipyridine)ruthenium(III) ions in acidic aqueous solutions. J. Chem. Soc.-Dalton Trans. 1997, 4175–4180. [Google Scholar] [CrossRef]
- Rao, P.S.; Hayon, E. Redox potentials of free radicals. IV. Superoxide and hydroperoxy radicals •O2− and HO2•. J. Phys. Chem. 2002, 79, 397–402. [Google Scholar] [CrossRef]
- Yang, Q.; Wang, Y.H.; Qiao, Y.; Gau, M.; Carroll, P.J.; Walsh, P.J.; Schelter, E.J. Photocatalytic C–H activation and the subtle role of chlorine radical complexation in reactivity. Science 2021, 372, 847–852. [Google Scholar] [CrossRef]
- Hwang, K.C.; Sagadevan, A.; Kundu, P. The sustainable room temperature conversion of p-xylene to terephthalic acid using ozone and UV irradiation. Green Chem. 2019, 21, 6082–6088. [Google Scholar] [CrossRef]
- Cui, L.Q.; Liu, K.; Zhang, C. Effective oxidation of benzylic and alkane C–H bonds catalyzed by sodium o-iodobenzenesulfonate with Oxone as a terminal oxidant under phase-transfer conditions. Org. Biomol. Chem. 2011, 9, 2258–2265. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Jin, F.L.; Zhou, Q. Ligand-free palladium-catalyzed hydroxycarbonylation of aryl halides under ambient conditions: Synthesis of aromatic carboxylic acids and aromatic esters. Synthesis 2015, 47, 1861–1868. [Google Scholar] [CrossRef]
- Hu, D.Q.; Jiang, X.F. Stepwise benzylic oxygenation via uranyl-photocatalysis. Green Chem. 2022, 24, 124–129. [Google Scholar] [CrossRef]
- Lu, H.T.; Geng, Z.Y.; Li, J.Y.; Zou, D.P.; Wu, Y.S.; Wu, Y.J. Metal-free reduction of aromatic nitro compounds to aromatic amines with B2pin2 in isopropanol. Org. Lett. 2016, 18, 2774–2776. [Google Scholar] [CrossRef]
- Ou, J.H.; Tan, H.; He, S.Y.; Wang, W.; Hu, B.N.; Yu, G.; Liu, K.J. 1,2-dibutoxyethane-promoted oxidative cleavage of olefins into carboxylic acids using O2 under clean conditions. J. Org. Chem. 2021, 86, 14974–14982. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Ota, M.; Kodama, S.; Michimoto, K.; Nomoto, A.; Ogawa, A.; Furuya, M.; Kawakami, K. Au/Ag/Cu-mixed catalysts for the eco-friendly oxidation of 5-hydroxymethylfurfural and related compounds to carboxylic acids under atmospheric oxygen in water. ACS Omega 2021, 6, 2239–2247. [Google Scholar] [CrossRef]
- Joseph, J.T.; Sajith, A.M.; Ningegowda, R.C.; Shashikanth, S. Room temperature carbonylation of (hetero) aryl pentafluoro-benzenesulfonates and triflates using palladium-cobalt bimetallic catalyst: Dual role of cobalt carbonyl. Adv. Synth. Catal. 2017, 359, 419–425. [Google Scholar] [CrossRef]
- Yu, H.; Ru, S.; Zhai, Y.Y.; Dai, G.Y.; Han, S.; Wei, Y.G. An efficient aerobic oxidation protocol of aldehydes to carboxylic acids in water catalyzed by an inorganic-ligand-supported copper catalyst. ChemCatChem 2018, 10, 1253–1257. [Google Scholar] [CrossRef]
- Mahmood, S.; Xu, B.H.; Ren, T.L.; Zhang, Z.B.; Liu, X.M.; Zhang, S.J. Cobalt/N-Hydroxyphthalimide(NHPI)-catalyzed aerobic oxidation of hydrocarbons with ionic liquid additive. Mol. Catal. 2018, 447, 90–96. [Google Scholar] [CrossRef]
- Meng, Q.Y.; Wang, S.; König, B. Carboxylation of aromatic and aliphatic bromides and triflates with CO2 by dual visible-light–nickel catalysis. Angew. Chem. Int. Ed. 2017, 56, 13426–13430. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.K.; Shan, L.; Ma, F.D.; Zhang, Y.L.; Yang, Y.C.; Wang, D.W. The synthesis of methyl triazole-4-carboxylate gold(I) complex and application on allene synthesis and alkyne hydration. Inorg. Chem. Commun. 2019, 109, 107564. [Google Scholar] [CrossRef]
- Oss, G.; Ho, J.M.; Nguyen, T.V. Tropylium ion catalyzes hydration reactions of alkynes. Eur. J. Org. Chem. 2018, 2018, 3974–3981. [Google Scholar] [CrossRef]
- Zhu, X.J.; Liu, C.; Liu, Y.; Yang, H.J.; Fu, H. A sodium trifluoromethanesulfinate-mediated photocatalytic strategy for aerobic oxidation of alcohols. Chem. Commun. 2020, 56, 12443–12446. [Google Scholar] [CrossRef] [PubMed]
- Yue, X.H.; Yan, X.L.; Huo, S.C.; Dong, Q.; Zhang, J.H.; Hao, Z.Q.; Han, Z.G.; Lin, J. Triruthenium carbonyl complexes containing bidentate pyridine–alkoxide ligands for highly efficient oxidation of primary and secondary alcohols. Appl. Organomet. Chem. 2019, 34, e5292. [Google Scholar] [CrossRef]
- Thiruvengetam, P.; Chand, D.K. Controlled and predictably selective oxidation of activated and unactivated C(sp3)–H bonds catalyzed by a molybdenum-based metallomicellar catalyst in water. J. Org. Chem. 2022, 87, 4061–4077. [Google Scholar] [CrossRef] [PubMed]
- Xiong, B.J.; Zeng, X.Q.; Geng, S.S.; Chen, S.; He, Y.; Feng, Z. Thiyl radical promoted chemo- and regioselective oxidation of C=C bonds using molecular oxygenviairon catalysis. Green Chem. 2018, 20, 4521–4527. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Entry | Cat. | Solvent | Wavelength (nm) | Time (h) | 1a/1b/1c (%) b |
| 1 | - | MeCN | 365 | 12 | 1/9/90 |
| 2 | - | Benzene | 365 | 12 | 34/37/29 |
| 3 c | - | DCM | 365 | 8 | 1/2/82 |
| 4 | - | DCE | 365 | 8 | 0/2/98 |
| 5 | - | H2O | 365 | 12 | 99/1/0 |
| 6 | - | DMSO | 365 | 8 | 8/52/40 |
| 7 | - | Acetone | 365 | 8 | 13/0/87 |
| 8 | - | DCE | 400 | 12 | 98/2/0 |
| 9 | - | DCE | 455 | 12 | 100/0/0 |
| 8 d | TsOH | DCE | 365 | 8 | 12/79/9 |
| 9 d | MsOH | DCE | 365 | 8 | 36/60/4 |
| 10 d | TsOH | DCE | 400 | 8 | 96/4/0 |
| 11 d | MsOH | DCE | 400 | 8 | 99/1/0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.-Y.; Lao, H.-E.; Zhang, H.-Y.; Wang, Y.; Zhang, Q.; Wu, J.-Q.; Li, Y.-F.; Zhu, H.-J.; Mao, J.-Y.; Pan, Y. HOO• as the Chain Carrier for the Autocatalytic Photooxidation of Benzylic Alcohols. Molecules 2024, 29, 3429. https://doi.org/10.3390/molecules29143429
Wang X-Y, Lao H-E, Zhang H-Y, Wang Y, Zhang Q, Wu J-Q, Li Y-F, Zhu H-J, Mao J-Y, Pan Y. HOO• as the Chain Carrier for the Autocatalytic Photooxidation of Benzylic Alcohols. Molecules. 2024; 29(14):3429. https://doi.org/10.3390/molecules29143429
Chicago/Turabian StyleWang, Xiao-Yu, Huan-E Lao, Hao-Yue Zhang, Yi Wang, Qing Zhang, Jie-Qing Wu, Yu-Feng Li, Hong-Jun Zhu, Jian-You Mao, and Yi Pan. 2024. "HOO• as the Chain Carrier for the Autocatalytic Photooxidation of Benzylic Alcohols" Molecules 29, no. 14: 3429. https://doi.org/10.3390/molecules29143429