
D-Hexopyranosides with Vicinal Nitrogen-Containing Functionalities


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Nitrogen Functionalities at Positions 2 and 3
2.1. Trans-Configuration
2.1.1. Aziridine Formation
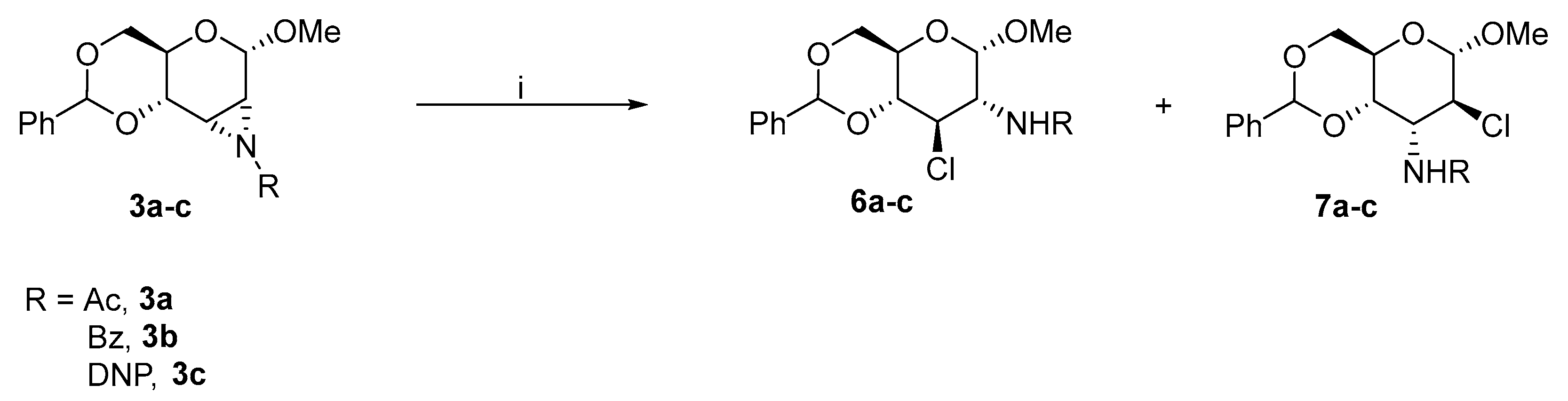
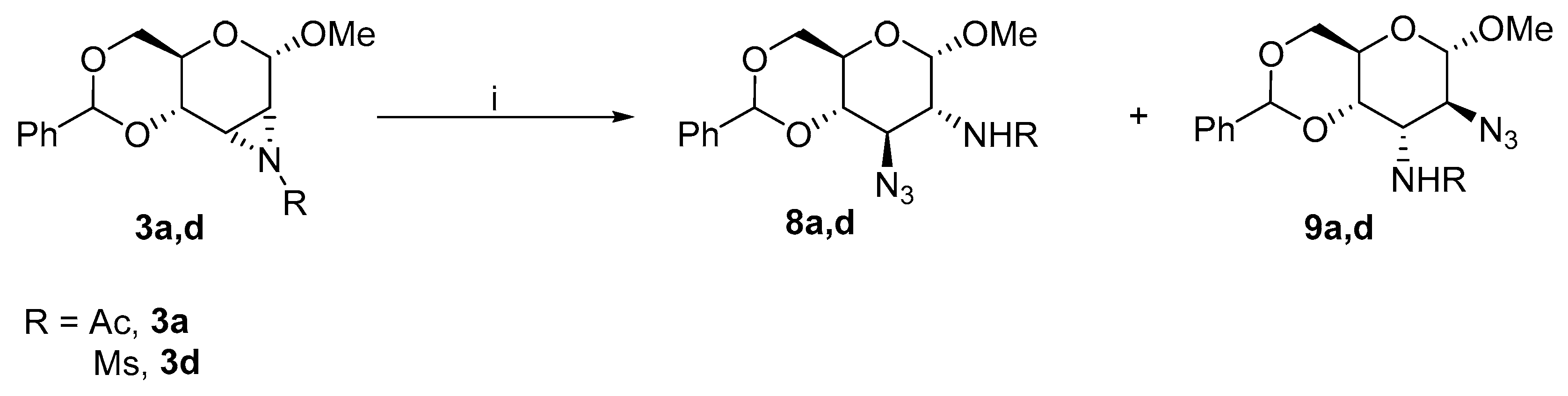
2.1.2. Substitution of the Activated Hydroxyl Group
2.1.3. Michael Addition: Using Addition to Activated Double Bond
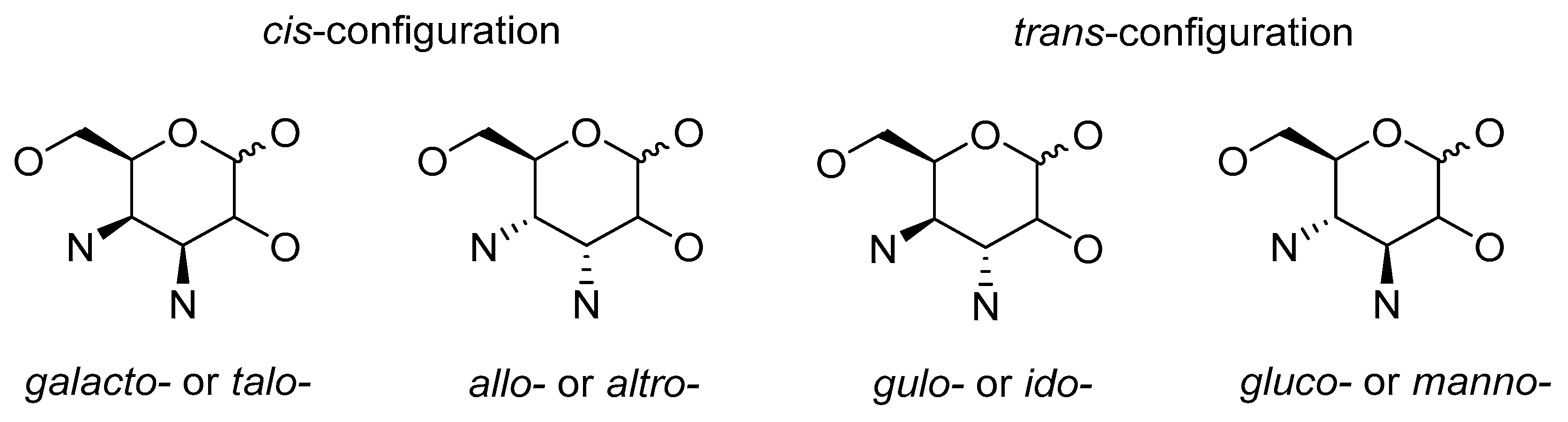
2.2. Cis-Configuration
2.2.1. Substitution of Activated Hydroxyl Group
2.2.2. Miscellaneous Methods
3. Nitrogen Functionalities at Positions 3 and 4
3.1. Trans-Configuration
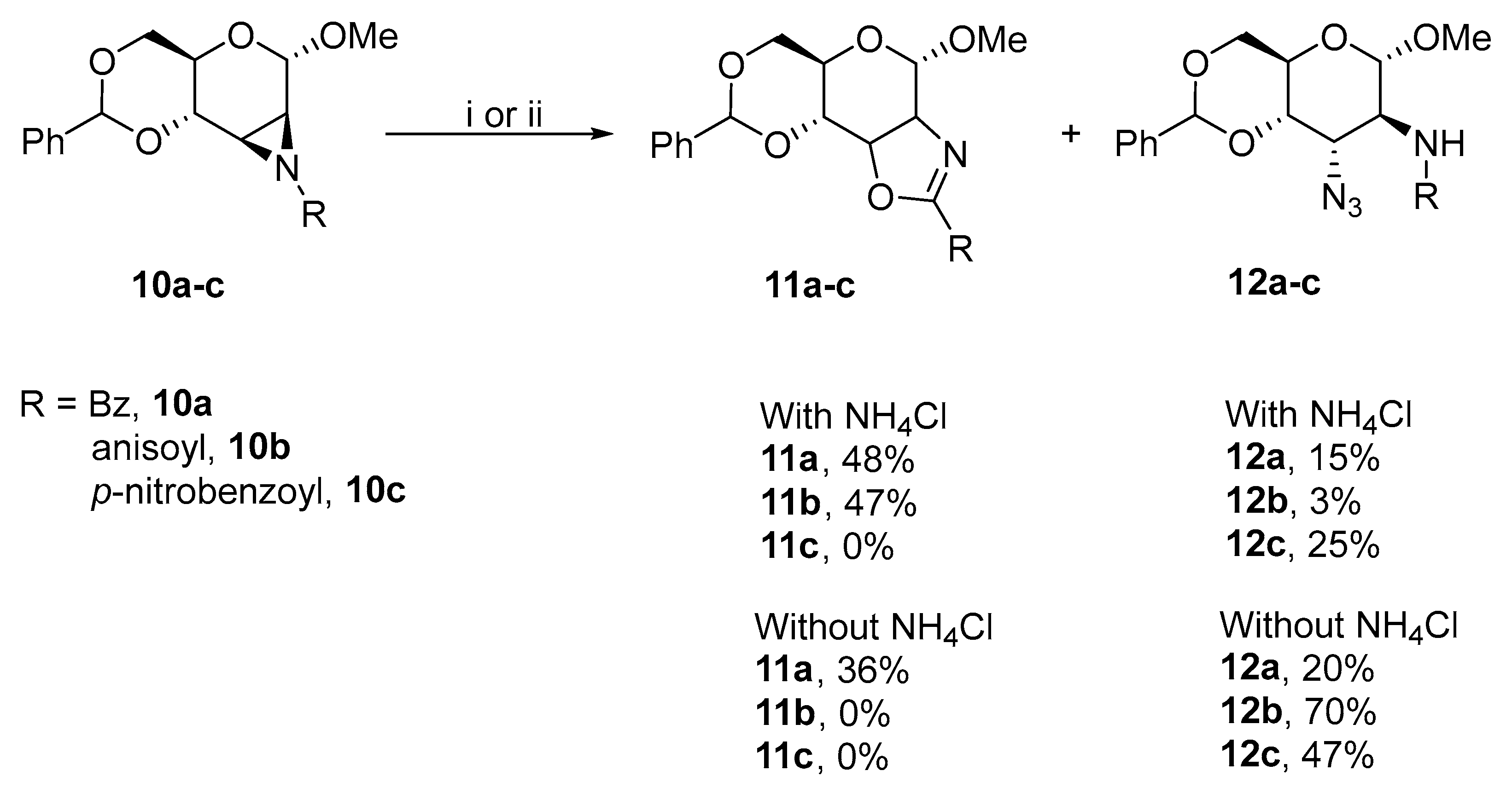
3.1.1. Oxazoline Ring Formation
3.1.2. Cyclization of Acyclic Intermediates with Vicinal Dinitrogen-Containing Functionalities
3.1.3. Michael Addition
3.1.4. Aziridine Ring Formation
3.2. Cis-Configuration
3.3. Methods Resulting in Cis- and Trans-Configurations
4. Nitrogen Functionalities at Positions 2, 3, and 4
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Cahoreau, C.; Klett, D.; Combarnous, Y. Structure-Function Relationships of Glycoprotein Hormones and Their Subunits’ Ancestors. Front. Endocrinol. 2015, 6, 26. [Google Scholar] [CrossRef]
- Wang, B.; Brand-Miller, J. The Role and Potential of Sialic Acid in Human Nutrition. Eur. J. Clin. Nutr. 2003, 57, 1351–1369. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Zhu, Y.; Wang, H.; Zhang, W.; Mu, W. Recent Advances on N-Acetylneuraminic Acid: Physiological Roles, Applications, and Biosynthesis. Synth. Syst. Biotechnol. 2023, 8, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Thuy-Boun, P.S.; Pfeiffer, W.; Vartabedian, V.F.; Torkamani, A.; Teijaro, J.R.; Wolan, D.W. Identification of an N-Acetylneuraminic Acid-Presenting Bacteria Isolated from a Human Microbiome. Sci. Rep. 2021, 11, 4763. [Google Scholar] [CrossRef] [PubMed]
- Hadházi, Á.; Pascolutti, M.; Bailly, B.; Dyason, J.C.; Borbás, A.; Thomson, R.J.; von Itzstein, M. A Sialosyl Sulfonate as a Potent Inhibitor of Influenza Virus Replication. Org. Biomol. Chem. 2017, 15, 5249–5253. [Google Scholar] [CrossRef] [PubMed]
- Hale, K.J.; Domostoj, M.M.; Tocher, D.A.; Irving, E.; Scheinmann, F. Enantiospecific Formal Total Synthesis of the Tumor and GSK-3β Inhibiting Alkaloid, (−)-Agelastatin A. Org. Lett. 2003, 5, 2927–2930. [Google Scholar] [CrossRef]
- Charon, D.; Mondange, M.; Pons, J.-F.; Le Blay, K.; Chaby, R. Synthesis and in Vitro Activities of a Spacer-Containing Glycophospholipid Ligand of a Lipopolysaccharide Receptor Involved in Endotoxin Tolerance. Bioorg. Med. Chem. 1998, 6, 755–765. [Google Scholar] [CrossRef]
- Del Litto, R.; Benessere, V.; Ruffo, F.; Moberg, C. Carbohydrate-Based Pyridine-2-Carboxamides for Mo-Catalyzed Asymmetric Allylic Alkylations. Eur. J. Org. Chem. 2009, 2009, 1352–1356. [Google Scholar] [CrossRef]
- Böge, M.; Fowelin, C.; Bednarski, P.; Heck, J. Diaminohexopyranosides as Ligands in Half-Sandwich Ruthenium(II), Rhodium(III), and Iridium(III) Complexes. Organometallics 2015, 34, 1507–1521. [Google Scholar] [CrossRef]
- Abrous, L.; Hynes, J., Jr.; Friedrich, S.R.; Smith, A.B., III; Hirschmann, R. Design and Synthesis of Novel Scaffolds for Drug Discovery: Hybrids of β-D-Glucose with 1,2,3,4-Tetrahydrobenzo[e][1,4]Diazepin-5-One, the Corresponding 1-Oxazepine, and 2- and 4-Pyridyldiazepines. Org. Lett. 2001, 3, 1089–1092. [Google Scholar] [CrossRef]
- Gibbs, C.F.; Hough, L.; Richardson, A.C. A New Synthesis of a 2,3-Epimino-α-D-Allopyranoside. Carbohydr. Res. 1965, 1, 290–296. [Google Scholar] [CrossRef]
- Ali, Y.; Richardson, A.C.; Gibbs, C.F.; Hough, L. Ring-Opening Reactions of Methyl 4,6-O-Benzylidene-2,3-Dideoxy-2,3-Epimino-α-D-Allopyranoside and Its Derivatives. Carbohydr. Res. 1968, 7, 255–271. [Google Scholar] [CrossRef]
- Guthrie, R.D.; Williams, G.J. Nitrogen-Containing Carbohydrate Derivatives. Part XXXII. Further Studies on the Ring-Opening of Epimino-Sugars. J. Chem. Soc. Perkin Trans. 1 1976, 801–804. [Google Scholar] [CrossRef]
- Guthrie, R.D.; Murphy, D. Nitrogen-Containing Carbohydrate Derivatives. VII. Ring-Opening Reactions of Epimino Sugars. J. Chem. Soc. 1965, 3828–3834. [Google Scholar] [CrossRef]
- Meyer zu Reckendorf, W.; Lenzen, H.J. Neighboring Group Participation in the Substitution of 2-Amino-2-Deoxy-α-D-Glucopyranosides in Position 3. Tetrahedron Lett. 1979, 11, 3657–3658. [Google Scholar] [CrossRef]
- Meyer zu Reckendorf, W.; Lenzen, H.J. Di- and Polyamino Sugars. XXVIII. Substitution of 2-Amino-2-Deoxy-α-D-Glucopyranosides at the 3-Position. Liebigs Ann. Chemie 1982, 13, 265–274. [Google Scholar] [CrossRef]
- Meyer zu Reckendorf, W.; Lenzen, H.J. Di- and Polyamino Sugars, XXXI. Substitution of 2-Amino-2-Deoxy-β-D-Glucopyranosides at Position 3. Liebigs Ann. Chemie 1985, 16, 477–484. [Google Scholar] [CrossRef]
- Rucil, T.; Travnicek, Z.; Cankar, P. Ring-Opening Reactions of the N-4-Nosyl Hough-Richardson Aziridine with Nitrogen Nucleophiles. J. Org. Chem. 2017, 82, 723–730. [Google Scholar] [CrossRef]
- Gibbs, C.F.; Hough, L. Further Reactions of Carbohydrate Epimines. Carbohydr. Res. 1971, 18, 363–371. [Google Scholar] [CrossRef]
- Rejzek, M.; Kannathasan, V.S.; Wing, C.; Preston, A.; Westman, E.L.; Lam, J.S.; Naismith, J.H.; Maskell, D.J.; Field, R.A. Chemical Synthesis of UDP-Glc-2,3-DiNAcA, a Key Intermediate in Cell Surface Polysaccharide Biosynthesis in the Human Respiratory Pathogens B. Pertussis and P. Aeruginosa. Org. Biomol. Chem. 2009, 7, 1203–1210. [Google Scholar] [CrossRef]
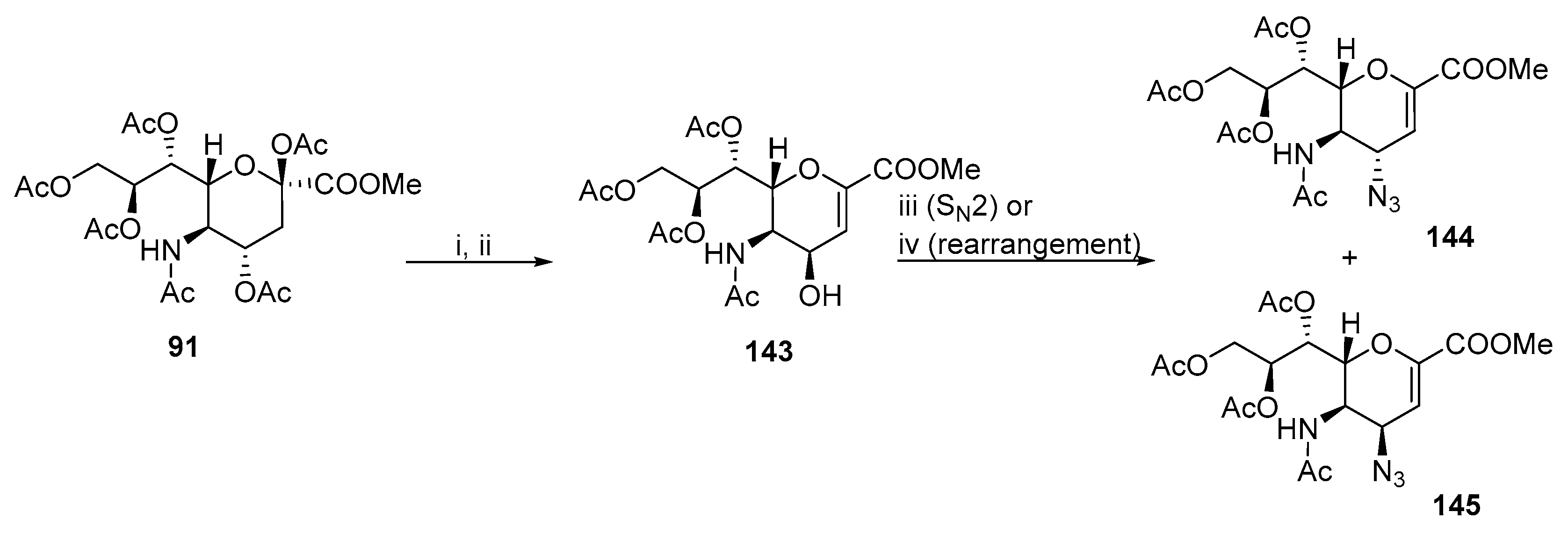
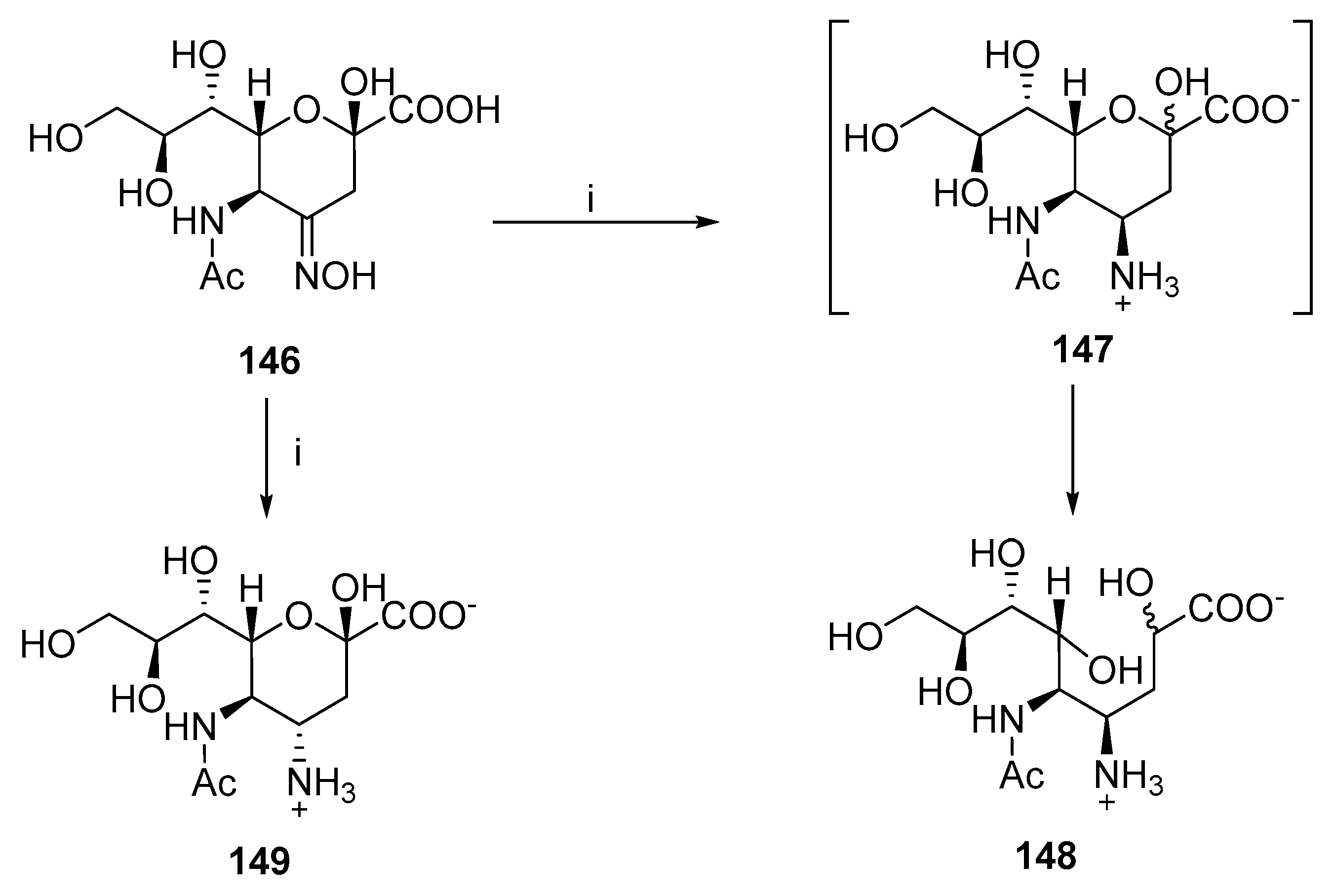
- Qin, C.; Schumann, B.; Zou, X.; Pereira, C.L.; Tian, G.; Hu, J.; Seeberger, P.H.; Yin, J. Total Synthesis of a Densely Functionalized Plesiomonas Shigelloides Serotype 51 Aminoglycoside Trisaccharide Antigen. J. Am. Chem. Soc. 2018, 140, 3120–3127. [Google Scholar] [CrossRef] [PubMed]
- Vega-Perez, J.M.; Vega, M.; Blanco, E.; Iglesias-Guerra, F. Stereoselective Epoxidation of Alkenylidene Acetals Derived from Carbohydrates with D-Allo, D-Altro, D-Galacto, D-Gluco and D-Xylo Configurations. Tetrahedron Asymmetry 2007, 18, 1850–1867. [Google Scholar] [CrossRef]
- Berger, I.; Nazarov, A.A.; Hartinger, C.G.; Groessl, M.; Valiahdi, S.M.; Jakupec, M.A.; Keppler, B.K. A Glucose Derivative as Natural Alternative to the Cyclohexane-1,2-Diamine Ligand in the Anticancer Drug Oxaliplatin? ChemMedChem 2007, 2, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Böge, M.; Heck, J. One-Step Preparation and Crystallization of Almost Insoluble Palladium(II) and Platinum(II/IV) Complexes from a Biphasic Solvent System. Cryst. Growth Des. 2015, 15, 5280–5287. [Google Scholar] [CrossRef]
- Böge, M.; Heck, J. Catalytic Diamino-Sugar-Assisted Enantioselective Hydrogenation. Eur. J. Inorg. Chem. 2015, 2015, 2858–2864. [Google Scholar] [CrossRef]
- Fowelin, C.; Matyja, A.; Schmidt, M.; Heck, J. Diamino Monosaccharide Ligands in Group 6 Carbonyl Complexes. Z. Für Anorg. Und Allg. Chem. 2007, 633, 2395–2399. [Google Scholar] [CrossRef]
- Borriello, C.; Del Litto, R.; Panunzi, A.; Ruffo, F. A Supported Mn(III) Catalyst Based on D-Glucose in the Asymmetric Epoxidation of Styrenes. Inorg. Chem. Commun. 2005, 8, 717–721. [Google Scholar] [CrossRef]
- Borriello, C.; Cucciolito, M.E.; Panunzi, A.; Ruffo, F.; Saporito, A. A Hydrophilic Chiral Diamine from D-Glucose in the Rh(I) Catalysed Asymmetric Hydrogenation of Acetophenone. Inorg. Chem. Commun. 2003, 6, 1081–1085. [Google Scholar] [CrossRef]
- Borriello, C.; Cucciolito, M.E.; Panunzi, A.; Ruffo, F. Novel Chiral Diimines and Diamines Derived from Sugars in Copper-Catalyzed Asymmetric Cyclopropanation. Tetrahedron Asymmetry 2001, 12, 2467–2471. [Google Scholar] [CrossRef]
- Borriello, C.; De Renzi, A.; Fusto, M.; Molinaro, A.; Ruffo, F. Transition Metals and Carbohydrates: The Methyl-4,6-O-Benzylidene-2,3-Diazo-2,3-Dideoxy-α-D-Mannopyranoside Skeleton as Building Block for New Chiral Nitrogen Chelates. Carbohydr. Res. 2001, 331, 209–212. [Google Scholar] [CrossRef]
- Tsubomura, T.; Yano, S.; Kobayashi, K.; Sakurai, T.; Yoshikawa, S. First Synthesis and Characterization of Platinum(II) Complexes of Amino Sugars Having Antitumor Activity; Crystal Structure of Dichloro(Methyl 2,3-Diamino-2,3-Dideoxy-α-D-Mannopyranoside)Platinum Monohydrate. J. Chem. Soc. Chem. Commun. 1986, 6, 459–460. [Google Scholar] [CrossRef]
- Calvo-Mateo, A.; De las Heras, F.G. Synthesis of N-(2-Acetamido-2,3-Dideoxy-D-Glucopyranos-3-Yl)Glycyl-L-Alanyl-D-Isoglutamine Analogs of Muramyl Dipeptide. Carbohydr. Res. 1986, 155, 266–271. [Google Scholar] [CrossRef] [PubMed]
- Baer, H.H.; Neilson, T. A New Synthesis of 2,3-Diamino-2,3-Dideoxy-D-Glucose. J. Org. Chem. 1967, 32, 1068–1072. [Google Scholar] [CrossRef]
- Baer, H.H.; Kienzle, F. Reactions of Nitro Sugars. XIV. Anomalous Stereochemical Course and a Color Phenomenon in the Addition of Anthranilic Acid to a Sugar Nitro Olefin. Synthesis of 2,3-Diamino-2,3-Dideoxy-D-Mannose. J. Org. Chem. 1969, 34, 3848–3853. [Google Scholar] [CrossRef]
- Baer, H.H.; Rajabalee, F. Reactions of Nitro Sugars. XIII. Bisglycosidylamines, a New Class of Amino Sugars. Can. J. Chem. 1969, 47, 4086–4089. [Google Scholar] [CrossRef]
- Nakagawa, T.; Sakakibara, T.; Kumazawa, S. Polyamino Sugars. IV. Synthesis of a New Type of Nucleoside: Methyl 4,6-O-Benzylidene-2,3-Dideoxy-3-Nitro-2-(7′-Theophyllinyl)-β-D-Glucopyranoside and Its 2-(2′,6′-Dichloro-9′-Purinyl) Homolog. Tetrahedron Lett. 1970, 11, 1645–1648. [Google Scholar] [CrossRef]
- Rajabalee, F.J.M. Carbohydrate Derivatives of Amino Acids. I. Novel Route to Carbohydrate Derivatives of Amino Acids. Synthesis 1972, 1972, 318–319. [Google Scholar] [CrossRef]
- Sakakibara, T.; Sudoh, R. Preparation of Methyl 4,6-O-Benzylidene-2-Deoxy-2-Nitro-β-D-Glucopyranoside from the Corresponding 3-Deoxy-3-Nitro Derivatives with Sodium Nitrite. Carbohydr. Res. 1976, 50, 197–202. [Google Scholar] [CrossRef]
- Sakakibara, T.; Sudoh, R. Stereochemistry of Nucleophilic Addition-Reactions. Part I. Stereoselective Synthesis of the Thermodynamically Less Stable Manno Isomers from a Nitro Sugar. Carbohydr. Res. 1976, 50, 191–196. [Google Scholar] [CrossRef]
- Vega-Perez, J.M.; Candela, J.I.; Blanco, E.; Iglesias-Guerra, F. Potential Anticancer Drugs. Part 3. Alkylating Agents from Sugars. Stereoselective Synthesis of 2,3-Diaminoglucoses from 2-Nitroalkenes, as Intermediates in the Synthesis of Carriers of Chlorambucil. Tetrahedron 1999, 55, 9641–9650. [Google Scholar] [CrossRef]
- Walvoort, M.T.C.; Moggre, G.-J.; Lodder, G.; Overkleeft, H.S.; Codee, J.D.C.; van der Marel, G.A. Stereoselective Synthesis of 2,3-Diamino-2,3-Dideoxy-β-D-Mannopyranosyl Uronates. J. Org. Chem. 2011, 76, 7301–7315. [Google Scholar] [CrossRef]
- Baer, H.H.; Radatus, B. Further Applications of Selective Displacements in an Unsymmetrical Ditriflate. Synthesis of New Triamino and Tetraamino Disaccharides of the Trehalose Type. Carbohydr. Res. 1986, 146, 73–88. [Google Scholar] [CrossRef]
- Kok, G.B.; Campbell, M.; Mackey, B.L.; von Itzstein, M. Synthesis of C-3 Nitrogen-Containing Derivatives of N-Acetyl-α,β-D-Mannosamine as Substrates for N-Acetylneuraminic Acid Aldolase. Carbohydr. Res. 2001, 332, 133–139. [Google Scholar] [CrossRef]
- Posakony, J.J.; Ferré-D’Amaré, A.R. Glucosamine and Glucosamine-6-Phosphate Derivatives: Catalytic Cofactor Analogues for the GlmS Ribozyme. J. Org. Chem. 2013, 78, 4730–4743. [Google Scholar] [CrossRef] [PubMed]
- Ágoston, K.; Fügedi, P. Preparation of New Type of Organocatalysts Having a Carbohydrate Scaffold. Carbohydr. Res. 2014, 389, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Baer, H.H.; Rank, W. The Addition of N -Bromoacetamide to 2,3-Unsaturated Nitro Sugars, a New Approach to 2,3-Diamino Sugars. Synthesis of Derivatives of 2,3-Diamino-2,3-Dideoxy-D-Mannose and-D-Talose. Can. J. Chem. 1974, 52, 2257–2267. [Google Scholar] [CrossRef]
- Zhang, J.; Eisink, N.N.H.M.; Witte, M.D.; Minnaard, A.J. Regioselective Manipulation of GlcNAc Provides Allosamine, Lividosamine, and Related Compounds. J. Org. Chem. 2019, 84, 516–525. [Google Scholar] [CrossRef] [PubMed]
- Baker, B.R.; Neilson, T. Synthetic Nucleosides. LXI. Studies on the Synthesis of Cis-2,3-Diamino Sugars. 4. The Guanidine Neighboring Group. J. Org. Chem. 1964, 29, 1063–1067. [Google Scholar] [CrossRef]
- Baker, B.R.; Neilson, T. Synthetic Nucleosides. LVIII. Studies on the Synthesis of Cis-2,3-Diamino Sugars. I. The Nitroguanidine Neighboring Group. J. Org. Chem. 1964, 29, 1047–1050. [Google Scholar] [CrossRef]
- Baker, B.R.; Neilson, T. Synthetic Nucleosides. LX. Studies on the Synthesis of Cis-2,3-Diamino Sugars. 3. The Urea Neighboring Group. J. Org. Chem. 1964, 29, 1057–1062. [Google Scholar] [CrossRef]
- Baker, B.R.; Hullar, T.L. Synthetic Nucleosides. LXV. Studies on the Synthesis of Cis-2,3-Diamino Sugars. 5. Neighboring Group Reactions with Derivatives of Methyl 2-Amino-4,6-O-Benzylidene-2-Deoxy-α-D-Altropyranoside. J. Org. Chem. 1965, 30, 4038–4044. [Google Scholar] [CrossRef] [PubMed]
- von Itzstein, M.; Jin, B.; Wu, W.Y.; Chandler, M. A Convenient Method for the Introduction of Nitrogen and Sulfur at C-4 on a Sialic Acid Analog. Carbohydr. Res. 1993, 244, 181–185. [Google Scholar] [CrossRef]
- Prescher, H.; Schweizer, A.; Kuhfeldt, E.; Nitschke, L.; Brossmer, R. Discovery of Multifold Modified Sialosides as Human CD22/Siglec-2 Ligands with Nanomolar Activity on B-Cells. ACS Chem. Biol. 2014, 9, 1444–1450. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Gervay-Hague, J. Synthesis of C-4 and C-7 Triazole Analogs of Zanamivir as Multivalent Sialic Acid Containing Scaffolds. Carbohydr. Res. 2007, 342, 1636–1650. [Google Scholar] [CrossRef] [PubMed]
- El-Deeb, I.M.; Guillon, P.; Winger, M.; Eveno, T.; Haselhorst, T.; Dyason, J.C.; Von Itzstein, M. Exploring Human Parainfluenza Virus Type-1 Hemagglutinin-Neuraminidase as a Target for Inhibitor Discovery. J. Med. Chem. 2014, 57, 7613–7623. [Google Scholar] [CrossRef] [PubMed]
- Shidmoossavee, F.S.; Watson, J.N.; Bennet, A.J. Chemical Insight into the Emergence of Influenza Virus Strains That Are Resistant to Relenza. J. Am. Chem. Soc. 2013, 135, 13254–13257. [Google Scholar] [CrossRef] [PubMed]
- Ciccotosto, S.; von Itzstein, M. Synthesis of Methyl 5-Acetamido-3,4,5-Trideoxy-4-Guanidinyl-D-Glycero-D-Galacto-2-Nonulopyranosidonic Acid (4-Deoxy-4-Guanidino-Neu5Acα2Me). Tetrahedron Lett. 1995, 36, 5405–5408. [Google Scholar] [CrossRef]
- Sabesan, S. Synthesis and Neuraminidase Inhibition Studies of 4-Azido, Amino, and Acetamido Substituted Sialosides. Bioorg. Med. Chem. Lett. 1994, 4, 2457–2460. [Google Scholar] [CrossRef]
- Dirr, L.; El-Deeb, I.M.; Guillon, P.; Carroux, C.J.; Chavas, L.M.G.; Von Itzstein, M. The Catalytic Mechanism of Human Parainfluenza Virus Type 3 Haemagglutinin-Neuraminidase Revealed. Angew. Chem. Int. Ed. 2015, 54, 2936–2940. [Google Scholar] [CrossRef]
- Hadhazi, A.; Li, L.; Bailly, B.; Maggioni, A.; Martin, G.; Dirr, L.; Dyason, J.C.; Thomson, R.J.; Gao, G.F.; Borbas, A.; et al. A Sulfonozanamivir Analogue Has Potent Anti-Influenza Virus Activity. ChemMedChem 2018, 13, 785–789. [Google Scholar] [CrossRef]
- Ye, D.; Shin, W.J.; Li, N.; Tang, W.; Feng, E.; Li, J.; He, P.L.; Zuo, J.P.; Kim, H.; Nam, K.Y.; et al. Synthesis of C-4-Modified Zanamivir Analogs as Neuraminidase Inhibitors and Their Anti-AIV Activities. Eur. J. Med. Chem. 2012, 54, 764–770. [Google Scholar] [CrossRef] [PubMed]
- Ye, D.; Li, J.; Zhang, J.; Liu, H.; Jiang, H. Simultaneous Stereoselective 4-Amination with Cyclic Secondary Amines and 2-O-Deacetylation of Peracetylated Sialic Acid Derivatives. Tetrahedron Lett. 2007, 48, 4023–4027. [Google Scholar] [CrossRef]
- Ye, D.; Deng, G.; Liu, W.; Zhou, Y.; Feng, E.; Jiang, H.; Liu, H. Simultaneous 2-O-Deacetylation and 4-Amination of Peracetylated Neu5Ac: Application to the Synthesis of (4→4)-Piperazine Derivatives Linked Sialic Acid Dimers. Tetrahedron 2008, 64, 6544–6550. [Google Scholar] [CrossRef]
- Bozzola, T.; Johnsson, R.E.; Nilsson, U.J.; Ellervik, U. Sialic Acid 4-N-Piperazine and Piperidine Derivatives Bind with High Affinity to the P. Mirabilis Sialic Acid Sodium Solute Symporter. ChemMedChem 2022, 17, e202200351. [Google Scholar] [CrossRef] [PubMed]
- Rota, P.; Allevi, P.; Agnolin, I.S.; Mattina, R.; Papini, N.; Anastasia, M. A Simple Synthesis of N-Perfluoroacylated and N-Acylated Glycals of Neuraminic Acid with a Cyclic Aminic Substituent at the 4α Position as Possible Inhibitors of Sialidases. Org. Biomol. Chem. 2012, 10, 2885–2894. [Google Scholar] [CrossRef] [PubMed]
- Ye, D.; Liu, W.; Zhang, D.; Feng, E.; Jiang, H.; Liu, H. Efficient Dehydrative Sialylation of C-4-Aminated Sialyl-Hemiketal Donors with Ph2SO/Tf2O. J. Org. Chem. 2009, 74, 1733–1735. [Google Scholar] [CrossRef]
- Liu, K.-G.; Zhou, H.-B.; Wu, Y.-L.; Yao, Z.-J. Synthesis of a New Stable Conformationally Constrained 2,7-Anhydrosialic Acid Derivative. J. Org. Chem. 2003, 68, 9528–9531. [Google Scholar] [CrossRef]
- Liu, K.G.; Yan, S.; Wu, Y.L.; Yao, Z.J. Synthesis of 4-Azido-4-Deoxy-Neu5,7,8,9ac42en1me. A Key Intermediate for the Synthesis of GG167 from D-Glucono-δ-Lactone. Org. Lett. 2004, 6, 2269–2272. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.X.; Wang, M.; Wang, S.; Yao, Z.J. Efficient Synthesis of 4-Amido-N5-Acetyl-4-Deoxyneuraminic Acid and Its Application to the C-4 Modification of Sialic Acids. Org. Lett. 2009, 11, 3678–3681. [Google Scholar] [CrossRef]
- Zhu, X.B.; Wang, M.; Wang, S.; Yao, Z.J. Concise Synthesis of Zanamivir and Its C4-Thiocarbamido Derivatives Utilizing a [3+2]-Cycloadduct Derived from d-Glucono-δ-Lactone. Tetrahedron 2012, 68, 2041–2044. [Google Scholar] [CrossRef]
- Tian, J.; Zhong, J.; Li, Y.; Ma, D. Organocatalytic and Scalable Synthesis of the Anti-Influenza Drugs Zanamivir, Laninamivir, and CS-8958. Angew. Chem. Int. Ed. 2014, 53, 13885–13888. [Google Scholar] [CrossRef] [PubMed]
- Hemeon, I.; Bennet, A.J. Synthesis of 4-Deoxy-4-Nitrosialic Acid. Org. Biomol. Chem. 2006, 4, 2986–2992. [Google Scholar] [CrossRef] [PubMed]
- Takamoto, T.; Sakakibara, T.; Matsumoto, M.; Sudoh, R. Studies on Nitro Sugars. Part VIII. Synthesis of Phenyl 4-O-Acetyl-2-O-Benzyl-3-Deoxy-3-Nitro-β-D-Glucopyranoside Derivatives and Amination at C-4. Carbohydr. Res. 1977, 53, 129–133. [Google Scholar] [CrossRef]
- Chen, X.; Xu, Y.; Zhao, Z.; Lei, P. Synthesis of Several Novel 14-Membered Ketolides Bearing Modified 5-O-4′-[1,2,3] Triazol Desosamine Side Chain. Tetrahedron Lett. 2014, 55, 6128–6130. [Google Scholar] [CrossRef]
- Zhao, Z.; Jin, L.; Wang, A.; Yang, S.; Lei, P. Two Novel N-Glycoside 17,18-Unsaturated Acid Quinolin-3-Ylmethyl Ester Josamycin Derivatives. Tetrahedron Lett. 2018, 59, 1208–1211. [Google Scholar] [CrossRef]
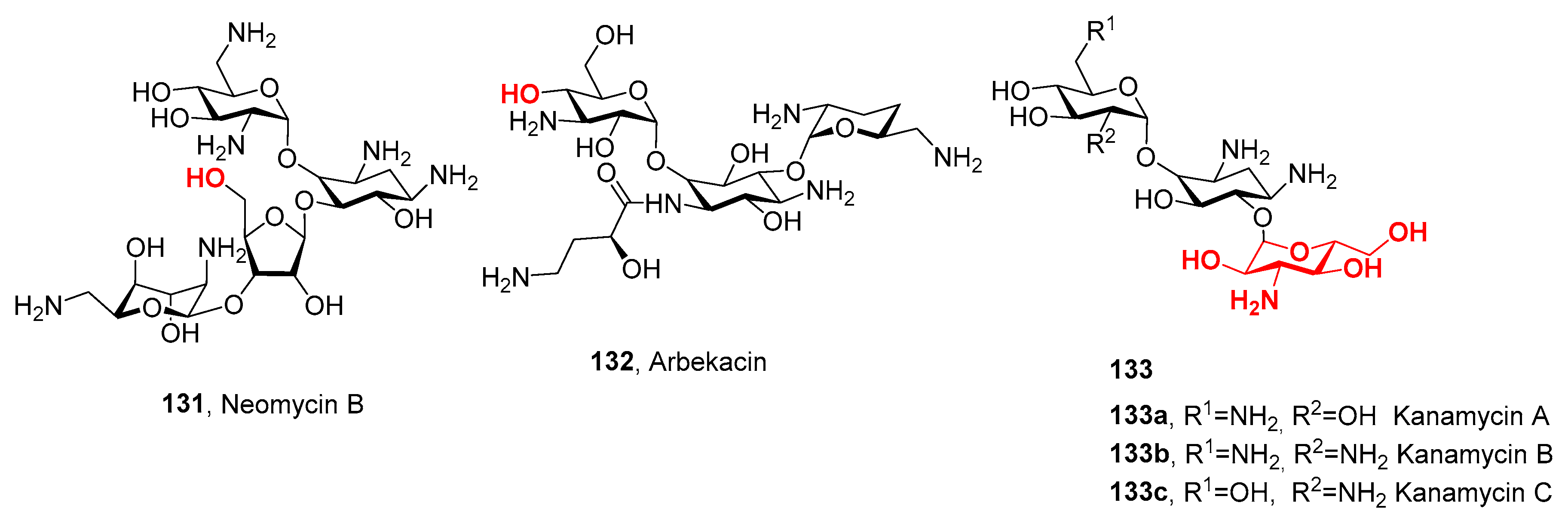
- Inoue, M.; Nonoyama, M.; Okamoto, R.; Ida, T. Antimicrobial Activity of Arbekacin, a New Aminoglycoside Antibiotic, against Methicillin-Resistant Staphylococcus Aureus. Drugs Exp. Clin. Res. 1994, 20, 233–240. [Google Scholar]
- Sasaki, K.; Kobayashi, Y.; Kurihara, T.; Yamashita, Y.; Takahashi, Y.; Miyake, T.; Akamatsu, Y. Synthesis and Antibacterial Activity of 4″ or 6″-Alkanoylamino Derivatives of Arbekacin. J. Antibiot. 2015, 68, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Heidary, N.; Cohen, D.E. Hypersensitivity Reactions to Vaccine Components. Dermatitis 2005, 16, 115–120. [Google Scholar]
- Hiraiwa, Y.; Minowa, N.; Usui, T.; Akiyama, Y.; Maebashi, K.; Ikeda, D. Effect of Varying the 4″-Position of Arbekacin Derivatives on Antibacterial Activity against MRSA and Pseudomonas Aeruginosa. Bioorg. Med. Chem. Lett. 2007, 17, 6369–6372. [Google Scholar] [CrossRef]
- Li, J.; Wang, J.; Czyryca, P.G.; Chang, H.; Orsak, T.W.; Evanson, R.; Chang, C.-W.T. Application of Glyco-Diversification: Expedient Synthesis and Antibacterial Evaluation of a Library of Kanamycin B Analogs. Org. Lett. 2004, 6, 1381–1384. [Google Scholar] [CrossRef]
- Fridman, M.; Belakhov, V.; Yaron, S.; Baasov, T. A New Class of Branched Aminoglycosides: Pseudo-Pentasaccharide Derivatives of Neomycin B. Org. Lett. 2003, 5, 3575–3578. [Google Scholar] [CrossRef]
- Fridman, M.; Belakhov, V.; Lee, L.V.; Liang, F.-S.; Wong, C.-H.; Baasov, T. Dual Effect of Synthetic Amino-Glycosides: Antibacterial Activity against Bacillus Anthracis and Inhibition of Anthrax Lethal Factor. Angew. Chem. Int. Ed. 2005, 44, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Zbiral, E.; Schreiner, E.; Christian, R. Synthesis of the 4-Acetamido-4-Deoxy Analogue of N-Acetylneuraminic Acid and Its Behaviour towards CMP-Sialate Synthase. Carbohydr. Res. 1989, 194, c15–c18. [Google Scholar] [CrossRef] [PubMed]
- Schreiner, E.; Zbiral, E.; Kleineidam, R.G.; Schauer, R. Structural Variations on N-acetylneuraminic Acid, 20. Synthesis of Some 2,3-didehydro-2-deoxysialic Acids Structurally Varied at C-4 and Their Behavior towards Sialidase from Vibrio Cholerae. Liebigs Ann. Chem. 1991, 1991, 129–134. [Google Scholar] [CrossRef]
- Bandgar, B.P.; Patil, S.V.; Zbiral, E. Synthesis of Methyl 4-Acetamido-N-Acetyl-4-Deoxy-a- and Fl-4-Epi-Neuraminic Acids. Carbohydr. Res. 1995, 276, 337–345. [Google Scholar] [CrossRef]
- Mack, H.; Brossmer, R. Chain Extension of 1-Deoxy-1-Nitroalditols in Nitrile Oxide Cycloaddition. Synthesis of 4-N-Substituted 3,4-Dideoxy-2-Ulosonic Acids. Tetrahedron 1998, 54, 4539–4560. [Google Scholar] [CrossRef]
- Bailliez, V.; Olesker, A.; Cleophax, J. Synthesis of Polynitrogenated Analogues of Glucopyranoses from Levoglucosan. Tetrahedron 2004, 60, 1079–1085. [Google Scholar] [CrossRef]
- Nakagawa, T.; Sato, Y.; Takamoto, T.; Lichtenthaler, F.W.; Majer, N. Polyamino Sugars. VI. Synthesis of Derivatives of 2,3,4-Triamino-2,3,4-Trideoxy-β-D-Glucose. Bull. Chem. Soc. Jpn. 1970, 43, 3866–3869. [Google Scholar] [CrossRef]
- Baer, H.H.; Rajabalee, F. Reactions of Nitro Sugars. XVI. Synthesis of Derivatives of 2,3-Diamino-2,3-Dideoxy-α-D-Glucose and 2,3,4-Triamino-2,3,4-Trideoxy-α-D-Glucose. Carbohydr. Res. 1970, 12, 241–251. [Google Scholar] [CrossRef]
- Lichtenthaler, F.W.; Voss, P.; Majer, N. Route to Inosatriamines and Triamino Sugars. Angew. Chem. Int. Ed. Engl. 1969, 8, 211–212. [Google Scholar] [CrossRef]
- Baer, H.H.; Bayer, M. Synthesis of Methyl 2,3,4,6-Tetraamino-2,3,4,6-Tetradeoxy-α-D-Glucopyranoside Tetrahydrochloride. Carbohydr. Res. 1970, 14, 114–118. [Google Scholar] [CrossRef]
- Meyer zu Reckendorf, W. Di- and Polyamino Sugars. XIV. Synthesis of 2,3,4,6-Tetraamino-2,3,4,6-Tetradeoxy-D-Glucose. Tetrahedron Lett. 1970, 4, 287–288. [Google Scholar] [CrossRef] [PubMed]
- Ali, Y.; Richardson, A.C. Nucleophilic Replacement Reactions of Sulfonate Esters. V. Synthesis of Derivatives of 2,3,4,6-Tetraamino-2,3,4,6-Tetradeoxy-D-Glucose. J. Chem. Soc. C 1969, 320–329. [Google Scholar] [CrossRef]
- Ali, Y.; Richardson, A.C. Nucleophilic Replacement Reactions of Sulfonates. III. The Synthesis of Derivatives of 2,3,4,6-Tetraamino-2,3,4,6-Tetradeoxy-D-Galactose and -D-Idose. J. Chem. Soc. C 1968, 1764–1769. [Google Scholar] [CrossRef]
- Ali, Y.; Richardson, A.C. Synthesis of Derivatives of 2,3,4,6-Tetraamino-2,3,4,6-Tetradeoxy-D-Galactose and -D-Idose. Chem. Commun. 1967, 554–556. [Google Scholar] [CrossRef]






























Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pospíšilová, J.; Toman, D.; Ručil, T.; Cankař, P. D-Hexopyranosides with Vicinal Nitrogen-Containing Functionalities. Molecules 2024, 29, 3465. https://doi.org/10.3390/molecules29153465
Pospíšilová J, Toman D, Ručil T, Cankař P. D-Hexopyranosides with Vicinal Nitrogen-Containing Functionalities. Molecules. 2024; 29(15):3465. https://doi.org/10.3390/molecules29153465
Chicago/Turabian StylePospíšilová, Jana, Daniel Toman, Tomáš Ručil, and Petr Cankař. 2024. "D-Hexopyranosides with Vicinal Nitrogen-Containing Functionalities" Molecules 29, no. 15: 3465. https://doi.org/10.3390/molecules29153465