
Structural Unfolding of G-Quadruplexes: From Small Molecules to Antisense Strategies


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. G4-Unfolding Small Molecules
2.1. TMPyP4: A Controversial Story
2.2. Effective Metal Complexes with G4-Disrupting Activity
2.3. Other G4-Destabilizing Organic Compounds
2.4. Small Molecules with Tunable G4-Unfolding Properties
3. Antisense Strategies
3.1. 2′-OMe Antisense Oligonucleotides in mRNA-G4 Unfolding
3.2. Effective Peptide Nucleic Acid Sequences
3.3. Locked Nucleic Acids as One of the Latest Advancements in Nucleotide Analogs
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Watson, J.D.; Crick, F.H.C. Molecular Structure of Nucleic Acids: A Structure for Deoxyribose Nucleic Acid. Nature 1953, 171, 737–738. [Google Scholar] [CrossRef] [PubMed]
- Makova, K.D.; Weissensteiner, M.H. Noncanonical DNA structures are drivers of genome evolution. Trends Genet. 2023, 39, 109–124. [Google Scholar] [CrossRef] [PubMed]
- Zuffo, M.; Pirota, V.; Doria, F. Photoresponsive molecular devices targeting nucleic acid secondary structures. In Photochemistry: Volume 46; The Royal Society of Chemistry: London, UK, 2019; Volume 46, pp. 281–318. [Google Scholar] [CrossRef]
- Yang, D. G-Quadruplex DNA and RNA. Methods Mol. Biol. 2019, 2035, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Farag, M.; Mouawad, L. Comprehensive analysis of intramolecular G-quadruplex structures: Furthering the understanding of their formalism. Nucleic Acids Res. 2024, 52, 3522–3546. [Google Scholar] [CrossRef] [PubMed]
- Kharel, P.; Becker, G.; Tsvetkov, V.; Ivanov, P. Properties and biological impact of RNA G-quadruplexes: From order to turmoil and back. Nucleic Acids Res. 2020, 48, 12534–12555. [Google Scholar] [CrossRef] [PubMed]
- Biver, T. Discriminating between Parallel, Anti-Parallel and Hybrid G-Quadruplexes: Mechanistic Details on Their Binding to Small Molecules. Molecules 2022, 27, 4165. [Google Scholar] [CrossRef]
- Spiegel, J.; Adhikari, S.; Balasubramanian, S. The Structure and Function of DNA G-Quadruplexes. Trends Chem. 2020, 2, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.; Raguseo, F.; Nuccio, S.P.; Liano, D.; Di Antonio, M. DNA G-quadruplex structures: More than simple roadblocks to transcription? Nucleic Acids Res. 2021, 49, 8419–8431. [Google Scholar] [CrossRef] [PubMed]
- Varshney, D.; Spiegel, J.; Zyner, K.; Tannahill, D.; Balasubramanian, S. The regulation and functions of DNA and RNA G-quadruplexes. Nat. Rev. Mol. Cell Biol. 2020, 21, 459–474. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Chen, Y.-Q.; Wang, S.-R.; Zhou, X. G-Quadruplex: A Regulator of Gene Expression and Its Chemical Targeting. Chem 2018, 4, 1314–1344. [Google Scholar] [CrossRef]
- Rigo, R.; Palumbo, M.; Sissi, C. G-quadruplexes in human promoters: A challenge for therapeutic applications. Biochim. Biophys. Acta (BBA) Gen. Subj. 2017, 1861, 1399–1413. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-Y.; Joshi, M.; Wang, A.; Myong, S. 5′UTR G-quadruplex structure enhances translation in size dependent manner. Nat. Commun. 2024, 15, 3963. [Google Scholar] [CrossRef]
- Lyu, K.; Chen, S.-B.; Chow, E.Y.-C.; Zhao, H.; Yuan, J.-H.; Cai, M.; Shi, J.; Chan, T.-F.; Tan, J.-H.; Kwok, C.K. An RNA G-Quadruplex Structure within the ADAR 5′UTR Interacts with DHX36 Helicase to Regulate Translation. Angew. Chem. Int. Ed. 2022, 61, e202203553. [Google Scholar] [CrossRef]
- Sharma, S.; Mukherjee, A.K.; Roy, S.S.; Bagri, S.; Lier, S.; Verma, M.; Sengupta, A.; Kumar, M.; Nesse, G.; Pandey, D.P.; et al. Human telomerase is directly regulated by non-telomeric TRF2-G-quadruplex interaction. Cell Rep. 2021, 35, 109154. [Google Scholar] [CrossRef]
- Lee, H.-T.; Sanford, S.; Paul, T.; Choe, J.; Bose, A.; Opresko, P.L.; Myong, S. Position-Dependent Effect of Guanine Base Damage and Mutations on Telomeric G-Quadruplex and Telomerase Extension. Biochemistry 2020, 59, 2627–2639. [Google Scholar] [CrossRef]
- Reina, C.; Cavalieri, V. Epigenetic Modulation of Chromatin States and Gene Expression by G-Quadruplex Structures. Int. J. Mol. Sci. 2020, 21, 4172. [Google Scholar] [CrossRef] [PubMed]
- Lawler, N.B.; Ou, A.; King, J.J.; Evans, C.W.; Iyer, K.S.; Smith, N.M. G4-DNA formation and chromatin remodelling are interdependent in human cells. Chem. Sci. 2023, 14, 7681–7687. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Martin-Pintado, N.; Post, H.; Altelaar, M.; Knipscheer, P. Multistep mechanism of G-quadruplex resolution during DNA replication. Sci. Adv. 2021, 7, eabf8653. [Google Scholar] [CrossRef]
- Prorok, P.; Artufel, M.; Aze, A.; Coulombe, P.; Peiffer, I.; Lacroix, L.; Guédin, A.; Mergny, J.-L.; Damaschke, J.; Schepers, A.; et al. Involvement of G-quadruplex regions in mammalian replication origin activity. Nat. Commun. 2019, 10, 3274. [Google Scholar] [CrossRef]
- Pavlova, A.V.; Kubareva, E.A.; Monakhova, M.V.; Zvereva, M.I.; Dolinnaya, N.G. Impact of G-Quadruplexes on the Regulation of Genome Integrity, DNA Damage and Repair. Biomolecules 2021, 11, 1284. [Google Scholar] [CrossRef]
- Rider, S.D., Jr.; Gadgil, R.Y.; Hitch, D.C.; Damewood, F.J.t.; Zavada, N.; Shanahan, M.; Alhawach, V.; Shrestha, R.; Shin-Ya, K.; Leffak, M. Stable G-quadruplex DNA structures promote replication-dependent genome instability. J. Biol. Chem. 2022, 298, 101947. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, J.; Mergny, J.L.; Cruz, C. G-quadruplex ligands in cancer therapy: Progress, challenges, and clinical perspectives. Life Sci. 2024, 340, 122481. [Google Scholar] [CrossRef]
- Doria, F.; Salvati, E.; Pompili, L.; Pirota, V.; D’Angelo, C.; Manoli, F.; Nadai, M.; Richter, S.N.; Biroccio, A.; Manet, I.; et al. Dyads of G-Quadruplex Ligands Triggering DNA Damage Response and Tumour Cell Growth Inhibition at Subnanomolar Concentration. Chemistry 2019, 25, 11085–11097. [Google Scholar] [CrossRef] [PubMed]
- Ruggiero, E.; Zanin, I.; Terreri, M.; Richter, S.N. G-Quadruplex Targeting in the Fight against Viruses: An Update. Int. J. Mol. Sci. 2021, 22, 10984. [Google Scholar] [CrossRef]
- Tassinari, M.; Zuffo, M.; Nadai, M.; Pirota, V.; Sevilla Montalvo, A.C.; Doria, F.; Freccero, M.; Richter, S.N. Selective targeting of mutually exclusive DNA G-quadruplexes: HIV-1 LTR as paradigmatic model. Nucleic Acids Res. 2020, 48, 4627–4642. [Google Scholar] [CrossRef] [PubMed]
- Belmonte-Reche, E.; Benassi, A.; Peñalver, P.; Cucchiarini, A.; Guédin, A.; Mergny, J.L.; Rosu, F.; Gabelica, V.; Freccero, M.; Doria, F.; et al. Thiosugar naphthalene diimide conjugates: G-quadruplex ligands with antiparasitic and anticancer activity. Eur. J. Med. Chem. 2022, 232, 114183. [Google Scholar] [CrossRef] [PubMed]
- Monti, L.; Di Antonio, M. G-Quadruplexes as Key Transcriptional Regulators in Neglected Trypanosomatid Parasites. ChemBioChem 2023, 24, e202300265. [Google Scholar] [CrossRef] [PubMed]
- Benassi, A.; Peñalver, P.; Pérez-Soto, M.; Pirota, V.; Freccero, M.; Morales, J.C.; Doria, F. Structure–Activity Study on Substituted, Core-Extended, and Dyad Naphthalene Diimide G-Quadruplex Ligands Leading to Potent Antitrypanosomal Agents. J. Med. Chem. 2024, 67, 10643–10654. [Google Scholar] [CrossRef] [PubMed]
- Cebrián, R.; Belmonte-Reche, E.; Pirota, V.; de Jong, A.; Morales, J.C.; Freccero, M.; Doria, F.; Kuipers, O.P. G-Quadruplex DNA as a Target in Pathogenic Bacteria: Efficacy of an Extended Naphthalene Diimide Ligand and Its Mode of Action. J. Med. Chem. 2021, 65, 4752–4766. [Google Scholar] [CrossRef]
- Yadav, P.; Kim, N.; Kumari, M.; Verma, S.; Sharma Tarun, K.; Yadav, V.; Kumar, A. G-Quadruplex Structures in Bacteria: Biological Relevance and Potential as an Antimicrobial Target. J. Bacteriol. 2021, 203, e00577-20. [Google Scholar] [CrossRef]
- Ciaco, S.; Aronne, R.; Fiabane, M.; Mori, M. The Rise of Bacterial G-Quadruplexes in Current Antimicrobial Discovery. ACS Omega 2024, 9, 24163–24180. [Google Scholar] [CrossRef]
- Wang, E.; Thombre, R.; Shah, Y.; Latanich, R.; Wang, J. G-Quadruplexes as pathogenic drivers in neurodegenerative disorders. Nucleic Acids Res. 2021, 49, 4816–4830. [Google Scholar] [CrossRef]
- Pirota, V.R.; Fantini, V.; Esposito, L.; Pandini, C.; Di Geraldo, R.; Doria, F.; Mella, M.; Pansarasa, O.; Gandellini, P.; Freccero, M.; et al. Effective lowering of α-synuclein expression by targeting G-quadruplex structures within the SNCA genome. bioRxiv 2024. [Google Scholar] [CrossRef]
- Xu, H.; Hurley, L.H. A first-in-class clinical G-quadruplex-targeting drug. The bench-to-bedside translation of the fluoroquinolone QQ58 to CX-5461 (Pidnarulex). Bioorg. Med. Chem. Lett. 2022, 77, 129016. [Google Scholar] [CrossRef]
- Alexandrou, E.; Guneri, D.; Neidle, S.; Waller, Z.A.E. QN-302 demonstrates opposing effects between i-motif and G-quadruplex DNA structures in the promoter of the S100P gene. Org. Biomol. Chem. 2024, 22, 55–58. [Google Scholar] [CrossRef]
- Pirota, V.; Stasi, M.; Benassi, A.; Doria, F. Chapter Six—An overview of quadruplex ligands: Their common features and chemotype diversity. In Annual Reports in Medicinal Chemistry; Neidle, S., Ed.; Academic Press: Cambridge, MA, USA, 2020; Volume 54, pp. 163–196. [Google Scholar] [CrossRef]
- Iachettini, S.; Biroccio, A.; Zizza, P. Therapeutic Use of G4-Ligands in Cancer: State-of-the-Art and Future Perspectives. Pharmaceuticals 2024, 17, 771. [Google Scholar] [CrossRef]
- Santos, T.; Salgado, G.F.; Cabrita, E.J.; Cruz, C. G-Quadruplexes and Their Ligands: Biophysical Methods to Unravel G-Quadruplex/Ligand Interactions. Pharmaceuticals 2021, 14, 769. [Google Scholar] [CrossRef]
- Berner, A.; Das, R.N.; Bhuma, N.; Golebiewska, J.; Abrahamsson, A.; Andréasson, M.; Chaudhari, N.; Doimo, M.; Bose, P.P.; Chand, K.; et al. G4-Ligand-Conjugated Oligonucleotides Mediate Selective Binding and Stabilization of Individual G4 DNA Structures. J. Am. Chem. Soc. 2024, 146, 6926–6935. [Google Scholar] [CrossRef]
- Yadav, K.; Meka, P.N.R.; Sadhu, S.; Guggilapu, S.D.; Kovvuri, J.; Kamal, A.; Srinivas, R.; Devayani, P.; Babu, B.N.; Nagesh, N. Telomerase Inhibition and Human Telomeric G-Quadruplex DNA Stabilization by a β-Carboline-Benzimidazole Derivative at Low Concentrations. Biochemistry 2017, 56, 4392–4404. [Google Scholar] [CrossRef]
- Asamitsu, S.; Shioda, N.; Sugiyama, H. Chapter Three—Telomeric quadruplexes as therapeutic targets. In Annual Reports in Medicinal Chemistry; Neidle, S., Ed.; Academic Press: Cambridge, MA, USA, 2020; Volume 54, pp. 77–99. [Google Scholar] [CrossRef]
- Frasson, I.; Pirota, V.; Richter, S.N.; Doria, F. Multimeric G-quadruplexes: A review on their biological roles and targeting. Int. J. Biol. Macromol. 2022, 204, 89–102. [Google Scholar] [CrossRef]
- Biffi, G.; Tannahill, D.; McCafferty, J.; Balasubramanian, S. Quantitative visualization of DNA G-quadruplex structures in human cells. Nat. Chem. 2013, 5, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Hänsel-Hertsch, R.; Spiegel, J.; Marsico, G.; Tannahill, D.; Balasubramanian, S. Genome-wide mapping of endogenous G-quadruplex DNA structures by chromatin immunoprecipitation and high-throughput sequencing. Nat. Protoc. 2018, 13, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Di Antonio, M.; Ponjavic, A.; Radzevičius, A.; Ranasinghe, R.T.; Catalano, M.; Zhang, X.; Shen, J.; Needham, L.-M.; Lee, S.F.; Klenerman, D.; et al. Single-molecule visualization of DNA G-quadruplex formation in live cells. Nat. Chem. 2020, 12, 832–837. [Google Scholar] [CrossRef] [PubMed]
- Zheng, K.W.; Zhang, J.Y.; He, Y.D.; Gong, J.Y.; Wen, C.J.; Chen, J.N.; Hao, Y.H.; Zhao, Y.; Tan, Z. Detection of genomic G-quadruplexes in living cells using a small artificial protein. Nucleic Acids Res. 2020, 48, 11706–11720. [Google Scholar] [CrossRef]
- Abdel-Monem, M.; Hoffmann-Berling, H. Enzymic unwinding of DNA. 1. Purification and characterization of a DNA-dependent ATPase from Escherichia coli. Eur. J. Biochem. 1976, 65, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Monem, M.; Dürwald, H.; Hoffmann-Berling, H. Enzymic unwinding of DNA. 2. Chain separation by an ATP-dependent DNA unwinding enzyme. Eur. J. Biochem. 1976, 65, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Brosh, R.M., Jr. DNA helicases involved in DNA repair and their roles in cancer. Nat. Rev. Cancer 2013, 13, 542–558. [Google Scholar] [CrossRef] [PubMed]
- Brosh, R.M., Jr.; Matson, S.W. History of DNA Helicases. Genes 2020, 11, 255. [Google Scholar] [CrossRef] [PubMed]
- Khreiss, A.; Bohnsack, K.E.; Bohnsack, M.T. Molecular functions of RNA helicases during ribosomal subunit assembly. Biol. Chem. 2023, 404, 781–789. [Google Scholar] [CrossRef]
- Suhasini, A.N.; Brosh, R.M. DNA Helicases Associated with Genetic Instability, Cancer, and Aging. In DNA Helicases and DNA Motor Proteins; Spies, M., Ed.; Springer: New York, NY, USA, 2013; pp. 123–144. [Google Scholar] [CrossRef]
- Lejault, P.; Mitteaux, J.; Sperti, F.R.; Monchaud, D. How to untie G-quadruplex knots and why? Cell Chem. Biol. 2021, 28, 436–455. [Google Scholar] [CrossRef]
- Childs-Disney, J.L.; Yang, X.; Gibaut, Q.M.R.; Tong, Y.; Batey, R.T.; Disney, M.D. Targeting RNA structures with small molecules. Nat. Rev. Drug Discov. 2022, 21, 736–762. [Google Scholar] [CrossRef]
- Maurer, T.S.; Edwards, M.; Hepworth, D.; Verhoest, P.; Allerton, C.M.N. Designing small molecules for therapeutic success: A contemporary perspective. Drug Discov. Today 2022, 27, 538–546. [Google Scholar] [CrossRef]
- Mitteaux, J.; Lejault, P.; Wojciechowski, F.; Joubert, A.; Boudon, J.; Desbois, N.; Gros, C.P.; Hudson, R.H.E.; Boulé, J.-B.; Granzhan, A.; et al. Identifying G-Quadruplex-DNA-Disrupting Small Molecules. J. Am. Chem. Soc. 2021, 143, 12567–12577. [Google Scholar] [CrossRef]
- Fujimori, J.; Matsuo, T.; Shimose, S.; Kubo, T.; Ishikawa, M.; Yasunaga, Y.; Ochi, M. Antitumor effects of telomerase inhibitor TMPyP4 in osteosarcoma cell lines. J. Orthop. Res. 2011, 29, 1707–1711. [Google Scholar] [CrossRef]
- Shalaby, T.; Fiaschetti, G.; Nagasawa, K.; Shin-ya, K.; Baumgartner, M.; Grotzer, M. G-Quadruplexes as Potential Therapeutic Targets for Embryonal Tumors. Molecules 2013, 18, 12500–12537. [Google Scholar] [CrossRef]
- Mikami-Terao, Y.; Akiyama, M.; Yuza, Y.; Yanagisawa, T.; Yamada, O.; Kawano, T.; Agawa, M.; Ida, H.; Yamada, H. Antitumor activity of TMPyP4 interacting G-quadruplex in retinoblastoma cell lines. Exp. Eye Res. 2009, 89, 200–208. [Google Scholar] [CrossRef]
- Weisman-Shomer, P.; Cohen, E.; Hershco, I.; Khateb, S.; Wolfovitz-Barchad, O.; Hurley, L.H.; Fry, M. The cationic porphyrin TMPyP4 destabilizes the tetraplex form of the fragile X syndrome expanded sequence d(CGG)n. Nucleic Acids Res. 2003, 31, 3963–3970. [Google Scholar] [CrossRef]
- Nolin, S.L.; Glicksman, A.; Tortora, N.; Allen, E.; Macpherson, J.; Mila, M.; Vianna-Morgante, A.M.; Sherman, S.L.; Dobkin, C.; Latham, G.J.; et al. Expansions and contractions of the FMR1 CGG repeat in 5,508 transmissions of normal, intermediate, and premutation alleles. Am. J. Med. Genet. A 2019, 179, 1148–1156. [Google Scholar] [CrossRef]
- Ofer, N.; Weisman-Shomer, P.; Shklover, J.; Fry, M. The quadruplex r(CGG)n destabilizing cationic porphyrin TMPyP4 cooperates with hnRNPs to increase the translation efficiency of fragile X premutation mRNA. Nucleic Acids Res. 2009, 37, 2712–2722. [Google Scholar] [CrossRef] [PubMed]
- Khateb, S.; Weisman-Shomer, P.; Hershco-Shani, I.; Ludwig, A.L.; Fry, M. The tetraplex (CGG)n destabilizing proteins hnRNP A2 and CBF-A enhance the in vivo translation of fragile X premutation mRNA. Nucleic Acids Res. 2007, 35, 5775–5788. [Google Scholar] [CrossRef]
- Dutikova, Y.V.; Borisova, O.F.; Shchyolkina, A.K.; Lin, J.; Huang, S.; Shtil, A.A.; Kaluzhny, D.N. 5,10,15,20-Tetra-(N-methyl-3-pyridyl)porphyrin destabilizes the antiparallel telomeric quadruplex d(TTAGGG)4. Mol. Biol. 2010, 44, 823–831. [Google Scholar] [CrossRef]
- Singh, A.; Joshi, S.; Kukreti, S. Cationic porphyrins as destabilizer of a G-quadruplex located at the promoter of human MYH7 β gene. J. Biomol. Struct. Dyn. 2020, 38, 4801–4816. [Google Scholar] [CrossRef]
- Joshi, S.; Singh, A.; Kukreti, S. Porphyrin induced structural destabilization of a parallel DNA G-quadruplex in human MRP1 gene promoter. J. Mol. Recognit. 2022, 35, e2950. [Google Scholar] [CrossRef]
- Joachimi, A.; Mayer, G.; Hartig, J.S. A New Anticoagulant−Antidote Pair: Control of Thrombin Activity by Aptamers and Porphyrins. J. Am. Chem. Soc. 2007, 129, 3036–3037. [Google Scholar] [CrossRef]
- Morris, M.J.; Wingate, K.L.; Silwal, J.; Leeper, T.C.; Basu, S. The porphyrin TmPyP4 unfolds the extremely stable G-quadruplex in MT3-MMP mRNA and alleviates its repressive effect to enhance translation in eukaryotic cells. Nucleic Acids Res. 2012, 40, 4137–4145. [Google Scholar] [CrossRef]
- Bhattacharjee, A.J.; Ahluwalia, K.; Taylor, S.; Jin, O.; Nicoludis, J.M.; Buscaglia, R.; Brad Chaires, J.; Kornfilt, D.J.P.; Marquardt, D.G.S.; Yatsunyk, L.A. Induction of G-quadruplex DNA structure by Zn(II) 5,10,15,20-tetrakis(N-methyl-4-pyridyl)porphyrin. Biochimie 2011, 93, 1297–1309. [Google Scholar] [CrossRef]
- D’Urso, A.; Randazzo, R.; Rizzo, V.; Gangemi, C.M.A.; Romanucci, V.; Zarrelli, A.; Tomaselli, G.; Milardi, D.; Borbone, N.; Purrello, R.; et al. Stabilization vs. destabilization of G-quadruplex superstructures: The role of the porphyrin derivative having spermine arms. Phys. Chem. Chem. Phys. 2017, 19, 17404–17410. [Google Scholar] [CrossRef]
- Raza, A.; Archer, S.A.; Fairbanks, S.D.; Smitten, K.L.; Botchway, S.W.; Thomas, J.A.; MacNeil, S.; Haycock, J.W. A Dinuclear Ruthenium(II) Complex Excited by Near-Infrared Light through Two-Photon Absorption Induces Phototoxicity Deep within Hypoxic Regions of Melanoma Cancer Spheroids. J. Am. Chem. Soc. 2020, 142, 4639–4647. [Google Scholar] [CrossRef]
- Holden, L.; Burke, C.S.; Cullinane, D.; Keyes, T.E. Strategies to promote permeation and vectorization, and reduce cytotoxicity of metal complex luminophores for bioimaging and intracellular sensing. RSC Chem. Biol. 2021, 2, 1021–1049. [Google Scholar] [CrossRef] [PubMed]
- Weynand, J.; Episkopou, H.; Le Berre, G.; Gillard, M.; Dejeu, J.; Decottignies, A.; Defrancq, E.; Elias, B. Photo-induced telomeric DNA damage in human cancer cells. RSC Chem. Biol. 2022, 3, 1375–1379. [Google Scholar] [CrossRef]
- Holden, L.; Gkika, K.S.; Burke, C.S.; Long, C.; Keyes, T.E. Selective, Disruptive Luminescent Ru(II) Polypyridyl Probes of G-Quadruplex. Inorg. Chem. 2023, 62, 2213–2227. [Google Scholar] [CrossRef]
- Ghosh, A.; Trajkovski, M.; Teulade-Fichou, M.-P.; Gabelica, V.; Plavec, J. Phen-DC3 Induces Refolding of Human Telomeric DNA into a Chair-Type Antiparallel G-Quadruplex through Ligand Intercalation. Angew. Chem. Int. Ed. 2022, 61, e202207384. [Google Scholar] [CrossRef]
- Sathyaseelan, C.; Veerapathiran, S.; Das, U.; Ravichandran, G.; Ajjugal, Y.; Singh, J.; Rengan, A.K.; Rathinavelan, T.; Prabusankar, G. Destabilizing Effect of Organo Ru(II) Salts on the Intermolecular Parallel CGG Repeat DNA Quadruplex Associated with Neurodegenerative/Neuromuscular Diseases. ACS Chem. Neurosci. 2023, 14, 3646–3654. [Google Scholar] [CrossRef]
- Yang, H.; Cui, H.; Wang, L.; Yan, L.; Qian, Y.; Zheng, X.E.; Wei, W.; Zhao, J. A label-free G-quadruplex DNA-based fluorescence method for highly sensitive, direct detection of cisplatin. Sens. Actuators B Chem. 2014, 202, 714–720. [Google Scholar] [CrossRef]
- Yett, A.; Lin, L.Y.; Beseiso, D.; Miao, J.; Yatsunyk, L.A. N-methyl mesoporphyrin IX as a highly selective light-up probe for G-quadruplex DNA. J. Porphyr. Phthalocyanines 2019, 23, 1195–1215. [Google Scholar] [CrossRef]
- Rajczak, E.; Gluszynska, A.; Juskowiak, B. Interaction of metallacrown complexes with G-quadruplex DNA. J. Inorg. Biochem. 2016, 155, 105–114. [Google Scholar] [CrossRef]
- Waller, Z.A.E.; Sewitz, S.A.; Hsu, S.-T.D.; Balasubramanian, S. A Small Molecule That Disrupts G-Quadruplex DNA Structure and Enhances Gene Expression. J. Am. Chem. Soc. 2009, 131, 12628–12633. [Google Scholar] [CrossRef]
- Adrian, M.; Heddi, B.; Phan, A.T. NMR spectroscopy of G-quadruplexes. Methods 2012, 57, 11–24. [Google Scholar] [CrossRef]
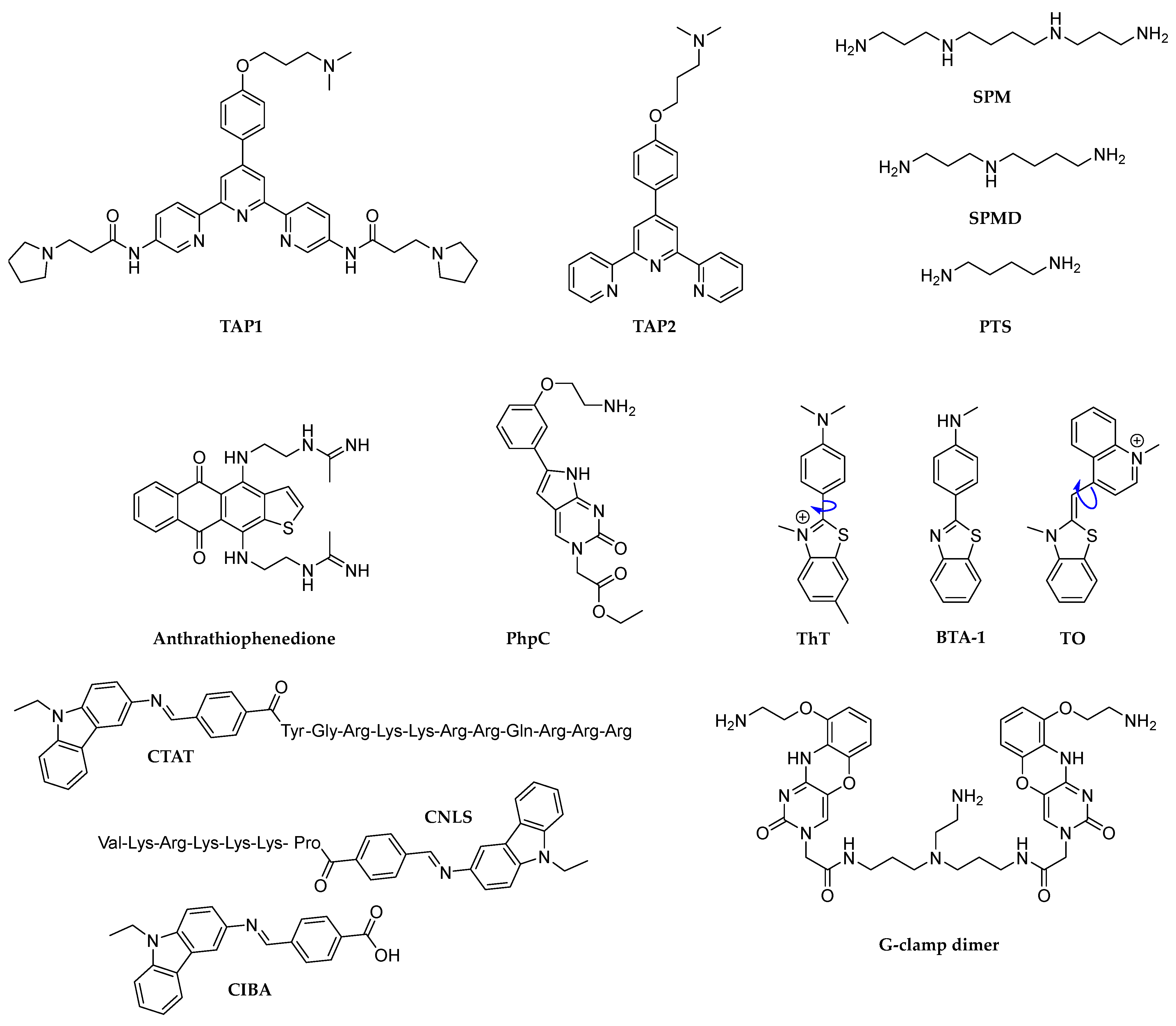
- Sun, H.; Xiang, J.; Liu, Y.; Li, L.; Li, Q.; Xu, G.; Tang, Y. A stabilizing and denaturing dual-effect for natural polyamines interacting with G-quadruplexes depending on concentration. Biochimie 2011, 93, 1351–1356. [Google Scholar] [CrossRef]
- Fornier, M.N. Approved agents for metastatic breast cancer. Semin. Oncol. 2011, 38 (Suppl. 2), S3–S10. [Google Scholar] [CrossRef]
- Kaluzhny, D.; Ilyinsky, N.; Shchekotikhin, A.; Sinkevich, Y.; Tsvetkov, P.O.; Tsvetkov, V.; Veselovsky, A.; Livshits, M.; Borisova, O.; Shtil, A.; et al. Disordering of human telomeric G-quadruplex with novel antiproliferative anthrathiophenedione. PLoS ONE 2011, 6, e27151. [Google Scholar] [CrossRef]
- Mitteaux, J.; Raevens, S.; Wang, Z.; Pirrotta, M.; Valverde, I.E.; Hudson, R.H.E.; Monchaud, D. PhpC modulates G-quadruplex-RNA landscapes in human cells. Chem. Commun. 2024, 60, 424–427. [Google Scholar] [CrossRef]
- Xu, Q.; Yang, M.; Chang, Y.; Peng, S.; Wang, D.; Zhou, X.; Shao, Y. Switching G-quadruplex to parallel duplex by molecular rotor clustering. Nucleic Acids Res. 2022, 50, 10249–10263. [Google Scholar] [CrossRef]
- Xu, P.; Yuan, L.; Wang, K.; Pan, B.; Ye, Y.; Lu, K. Interaction of bifunctional peptide-carbazole complexes with DNA and antimicrobial activity. Int. J. Biol. Macromol. 2023, 237, 124070. [Google Scholar] [CrossRef]
- Murase, H.; Nagatsugi, F.; Sasaki, S. Development of a selective ligand for G–G mismatches of CGG repeat RNA inducing the RNA structural conversion from the G-quadruplex into a hairpin-like structure. Org. Biomol. Chem. 2022, 20, 3375–3381. [Google Scholar] [CrossRef]
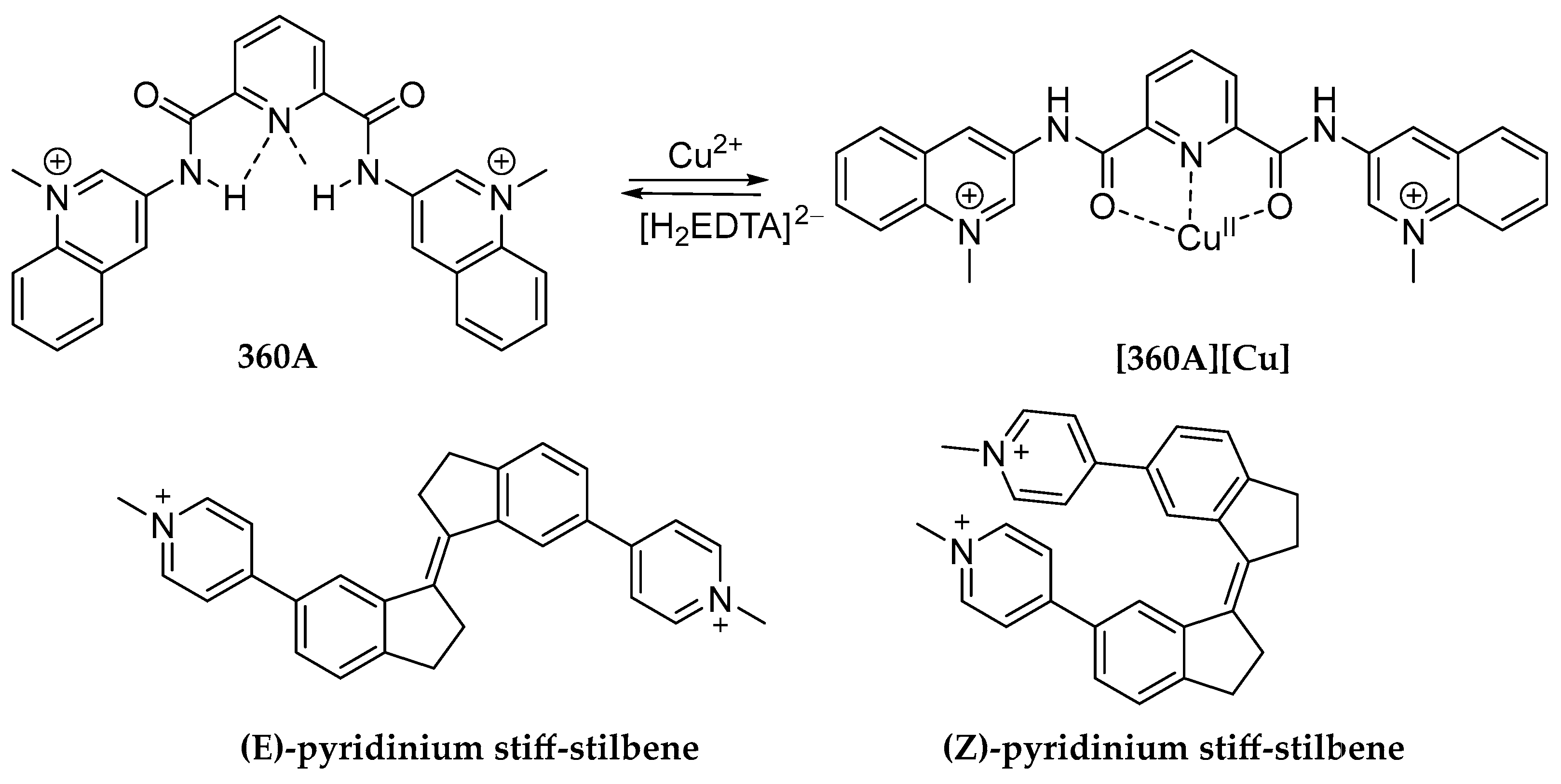
- Monchaud, D.; Yang, P.; Lacroix, L.; Teulade-Fichou, M.-P.; Mergny, J.-L. A Metal-Mediated Conformational Switch Controls G-Quadruplex Binding Affinity. Angew. Chem. Int. Ed. 2008, 47, 4858–4861. [Google Scholar] [CrossRef]
- O’Hagan, M.P.; Haldar, S.; Duchi, M.; Oliver, T.A.A.; Mulholland, A.J.; Morales, J.C.; Galan, M.C. A Photoresponsive Stiff-Stilbene Ligand Fuels the Reversible Unfolding of G-Quadruplex DNA. Angew. Chem. Int. Ed. 2019, 58, 4334–4338. [Google Scholar] [CrossRef]
- Disney, M.D.; Dwyer, B.G.; Childs-Disney, J.L. Drugging the RNA World. Cold Spring Harb. Perspect. Biol. 2018, 10, a034769. [Google Scholar] [CrossRef]
- Dias, N.; Stein, C.A. Antisense oligonucleotides: Basic concepts and mechanisms. Mol. Cancer Ther. 2002, 1, 347–355. [Google Scholar]
- Quemener, A.M.; Bachelot, L.; Forestier, A.; Donnou-Fournet, E.; Gilot, D.; Galibert, M.-D. The powerful world of antisense oligonucleotides: From bench to bedside. WIREs RNA 2020, 11, e1594. [Google Scholar] [CrossRef] [PubMed]
- Mansoor, M.; Melendez, A.J. Advances in antisense oligonucleotide development for target identification, validation, and as novel therapeutics. Gene Regul. Syst. Biol. 2008, 2, 275–295. [Google Scholar] [CrossRef]
- Le, B.T.; Agarwal, S.; Veedu, R.N. Evaluation of DNA segments in 2′-modified RNA sequences in designing efficient splice switching antisense oligonucleotides. RSC Adv. 2021, 11, 14029–14035. [Google Scholar] [CrossRef]
- Khvorova, A.; Watts, J.K. The chemical evolution of oligonucleotide therapies of clinical utility. Nat. Biotechnol. 2017, 35, 238–248. [Google Scholar] [CrossRef]
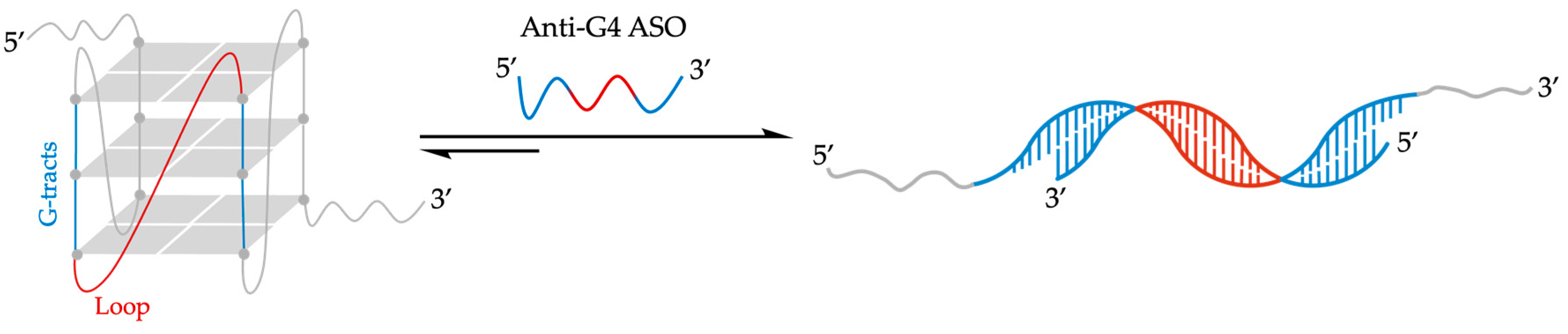
- Rouleau, S.G.; Beaudoin, J.D.; Bisaillon, M.; Perreault, J.P. Small antisense oligonucleotides against G-quadruplexes: Specific mRNA translational switches. Nucleic Acids Res. 2015, 43, 595–606. [Google Scholar] [CrossRef]
- Murat, P.; Zhong, J.; Lekieffre, L.; Cowieson, N.P.; Clancy, J.L.; Preiss, T.; Balasubramanian, S.; Khanna, R.; Tellam, J. G-quadruplexes regulate Epstein-Barr virus–encoded nuclear antigen 1 mRNA translation. Nat. Chem. Biol. 2014, 10, 358–364. [Google Scholar] [CrossRef]
- Nielsen, P.E.; Egholm, M.; Berg, R.H.; Buchardt, O. Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science 1991, 254, 1497–1500. [Google Scholar] [CrossRef]
- Pellestor, F.; Paulasova, P. The peptide nucleic acids (PNAs), powerful tools for molecular genetics and cytogenetics. Eur. J. Hum. Genet. 2004, 12, 694–700. [Google Scholar] [CrossRef]
- Gupta, A.; Mishra, A.; Puri, N. Peptide nucleic acids: Advanced tools for biomedical applications. J. Biotechnol. 2017, 259, 148–159. [Google Scholar] [CrossRef]
- Komiyama, M.; Ye, S.; Liang, X.; Yamamoto, Y.; Tomita, T.; Zhou, J.-M.; Aburatani, H. PNA for One-Base Differentiating Protection of DNA from Nuclease and Its Use for SNPs Detection. J. Am. Chem. Soc. 2003, 125, 3758–3762. [Google Scholar] [CrossRef]
- Jasiński, M.; Miszkiewicz, J.; Feig, M.; Trylska, J. Thermal Stability of Peptide Nucleic Acid Complexes. J. Phys. Chem. B 2019, 123, 8168–8177. [Google Scholar] [CrossRef]
- Porcheddu, A.; Giacomelli, G. Peptide nucleic acids (PNAs), a chemical overview. Curr. Med. Chem. 2005, 12, 2561–2599. [Google Scholar] [CrossRef]
- Datta, B.; Armitage, B.A. Hybridization of PNA to Structured DNA Targets: Quadruplex Invasion and the Overhang Effect. J. Am. Chem. Soc. 2001, 123, 9612–9619. [Google Scholar] [CrossRef]
- Schultze, P.; Macaya, R.F.; Feigon, J. Three-dimensional solution structure of the thrombin-binding DNA aptamer d(GGTTGGTGTGGTTGG). J. Mol. Biol. 1994, 235, 1532–1547. [Google Scholar] [CrossRef]
- Zacchia, M.; Abategiovanni, M.L.; Stratigis, S.; Capasso, G. Potassium: From Physiology to Clinical Implications. Kidney Dis. 2016, 2, 72–79. [Google Scholar] [CrossRef]
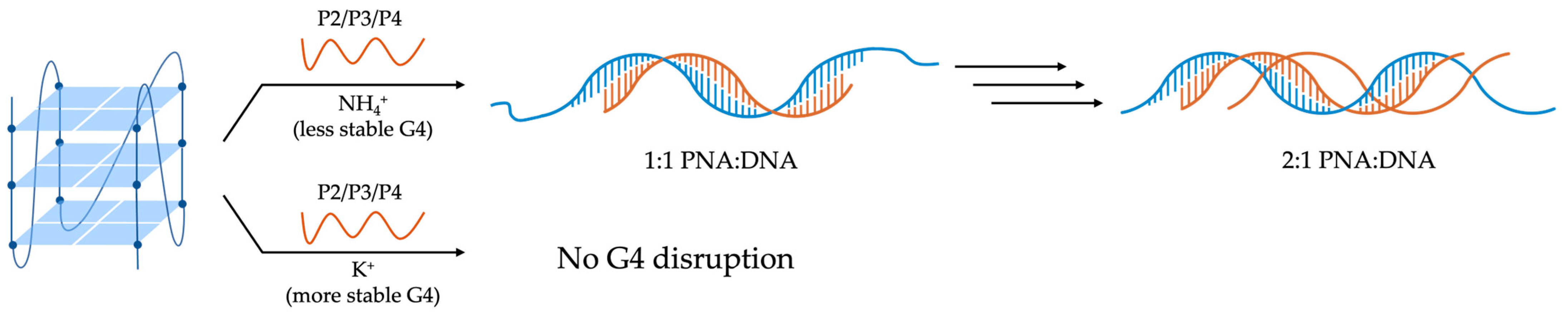
- Gupta, A.; Lee, L.-L.; Roy, S.; Tanious, F.A.; Wilson, W.D.; Ly, D.H.; Armitage, B.A. Strand Invasion of DNA Quadruplexes by PNA: Comparison of Homologous and Complementary Hybridization. ChemBioChem 2013, 14, 1476–1484. [Google Scholar] [CrossRef]
- Roy, S.; Zanotti, K.J.; Murphy, C.T.; Tanious, F.A.; Wilson, W.D.; Ly, D.H.; Armitage, B.A. Kinetic discrimination in recognition of DNA quadruplex targets by guanine-rich heteroquadruplex-forming PNA probes. Chem. Commun. 2011, 47, 8524–8526. [Google Scholar] [CrossRef] [PubMed]
- Green, J.J.; Ying, L.; Klenerman, D.; Balasubramanian, S. Kinetics of Unfolding the Human Telomeric DNA Quadruplex Using a PNA Trap. J. Am. Chem. Soc. 2003, 125, 3763–3767. [Google Scholar] [CrossRef]
- Amato, J.; Pagano, B.; Borbone, N.; Oliviero, G.; Gabelica, V.; Pauw, E.D.; D’Errico, S.; Piccialli, V.; Varra, M.; Giancola, C.; et al. Targeting G-Quadruplex Structure in the Human c-Kit Promoter with Short PNA Sequences. Bioconjug. Chem. 2011, 22, 654–663. [Google Scholar] [CrossRef]
- Oyaghire, S.N.; Cherubim, C.J.; Telmer, C.A.; Martinez, J.A.; Bruchez, M.P.; Armitage, B.A. RNA G-Quadruplex Invasion and Translation Inhibition by Antisense γ-Peptide Nucleic Acid Oligomers. Biochemistry 2016, 55, 1977–1988. [Google Scholar] [CrossRef]
- Campbell, M.A.; Wengel, J. Locked vs. unlocked nucleic acids (LNA vs. UNA): Contrasting structures work towards common therapeutic goals. Chem. Soc. Rev. 2011, 40, 5680–5689. [Google Scholar] [CrossRef]
- Koshkin, A.A.; Singh, S.K.; Nielsen, P.; Rajwanshi, V.K.; Kumar, R.; Meldgaard, M.; Olsen, C.E.; Wengel, J. LNA (Locked Nucleic Acids): Synthesis of the adenine, cytosine, guanine, 5-methylcytosine, thymine and uracil bicyclonucleoside monomers, oligomerisation, and unprecedented nucleic acid recognition. Tetrahedron 1998, 54, 3607–3630. [Google Scholar] [CrossRef]
- Nielsen, K.E.; Rasmussen, J.; Kumar, R.; Wengel, J.; Jacobsen, J.P.; Petersen, M. NMR Studies of Fully Modified Locked Nucleic Acid (LNA) Hybrids: Solution Structure of an LNA:RNA Hybrid and Characterization of an LNA:DNA Hybrid. Bioconjug. Chem. 2004, 15, 449–457. [Google Scholar] [CrossRef]
- Hagedorn, P.H.; Persson, R.; Funder, E.D.; Albæk, N.; Diemer, S.L.; Hansen, D.J.; Møller, M.R.; Papargyri, N.; Christiansen, H.; Hansen, B.R.; et al. Locked nucleic acid: Modality, diversity, and drug discovery. Drug Discov. Today 2018, 23, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Papargyri, N.; Pontoppidan, M.; Andersen, M.R.; Koch, T.; Hagedorn, P.H. Chemical Diversity of Locked Nucleic Acid-Modified Antisense Oligonucleotides Allows Optimization of Pharmaceutical Properties. Mol. Ther. Nucleic Acids 2020, 19, 706–717. [Google Scholar] [CrossRef]
- Petersen, M.; Wengel, J. LNA: A versatile tool for therapeutics and genomics. Trends Biotechnol. 2003, 21, 74–81. [Google Scholar] [CrossRef]
- Battey, J.; Moulding, C.; Taub, R.; Murphy, W.; Stewart, T.; Potter, H.; Lenoir, G.; Leder, P. The human c-myc oncogene: Structural consequences of translocation into the IgH locus in Burkitt lymphoma. Cell 1983, 34, 779–787. [Google Scholar] [CrossRef]
- Kumar, N.; Patowary, A.; Sivasubbu, S.; Petersen, M.; Maiti, S. Silencing c-MYC Expression by Targeting Quadruplex in P1 Promoter Using Locked Nucleic Acid Trap. Biochemistry 2008, 47, 13179–13188. [Google Scholar] [CrossRef]
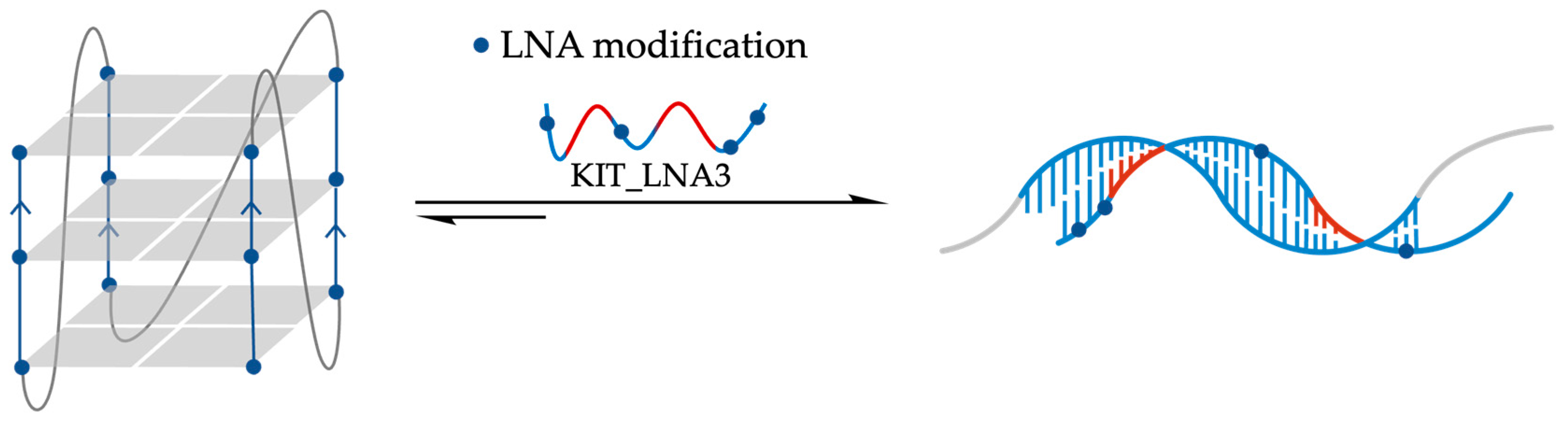
- Chowdhury, S.; Wang, J.; Nuccio, S.P.; Mao, H.; Di Antonio, M. Short LNA-modified oligonucleotide probes as efficient disruptors of DNA G-quadruplexes. Nucleic Acids Res. 2022, 50, 7247–7259. [Google Scholar] [CrossRef]
- Xu, H.; Di Antonio, M.; McKinney, S.; Mathew, V.; Ho, B.; O’Neil, N.J.; Santos, N.D.; Silvester, J.; Wei, V.; Garcia, J.; et al. CX-5461 is a DNA G-quadruplex stabilizer with selective lethality in BRCA1/2 deficient tumours. Nat. Commun. 2017, 8, 14432. [Google Scholar] [CrossRef]
- Ducani, C.; Bernardinelli, G.; Högberg, B.; Keppler, B.K.; Terenzi, A. Interplay of Three G-Quadruplex Units in the KIT Promoter. J. Am. Chem. Soc. 2019, 141, 10205–10213. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fracchioni, G.; Vailati, S.; Grazioli, M.; Pirota, V. Structural Unfolding of G-Quadruplexes: From Small Molecules to Antisense Strategies. Molecules 2024, 29, 3488. https://doi.org/10.3390/molecules29153488
Fracchioni G, Vailati S, Grazioli M, Pirota V. Structural Unfolding of G-Quadruplexes: From Small Molecules to Antisense Strategies. Molecules. 2024; 29(15):3488. https://doi.org/10.3390/molecules29153488
Chicago/Turabian StyleFracchioni, Giorgia, Sabrina Vailati, Marta Grazioli, and Valentina Pirota. 2024. "Structural Unfolding of G-Quadruplexes: From Small Molecules to Antisense Strategies" Molecules 29, no. 15: 3488. https://doi.org/10.3390/molecules29153488
APA StyleFracchioni, G., Vailati, S., Grazioli, M., & Pirota, V. (2024). Structural Unfolding of G-Quadruplexes: From Small Molecules to Antisense Strategies. Molecules, 29(15), 3488. https://doi.org/10.3390/molecules29153488