
Synthesis, X-ray Crystallography, Spectroscopic Characterizations, Density Functional Theory, and Hirshfeld Surface Analyses of a Novel (Carbonato) Picket Fence Iron(III) Complex

 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Experimental
2.1.1. Materials and Methods
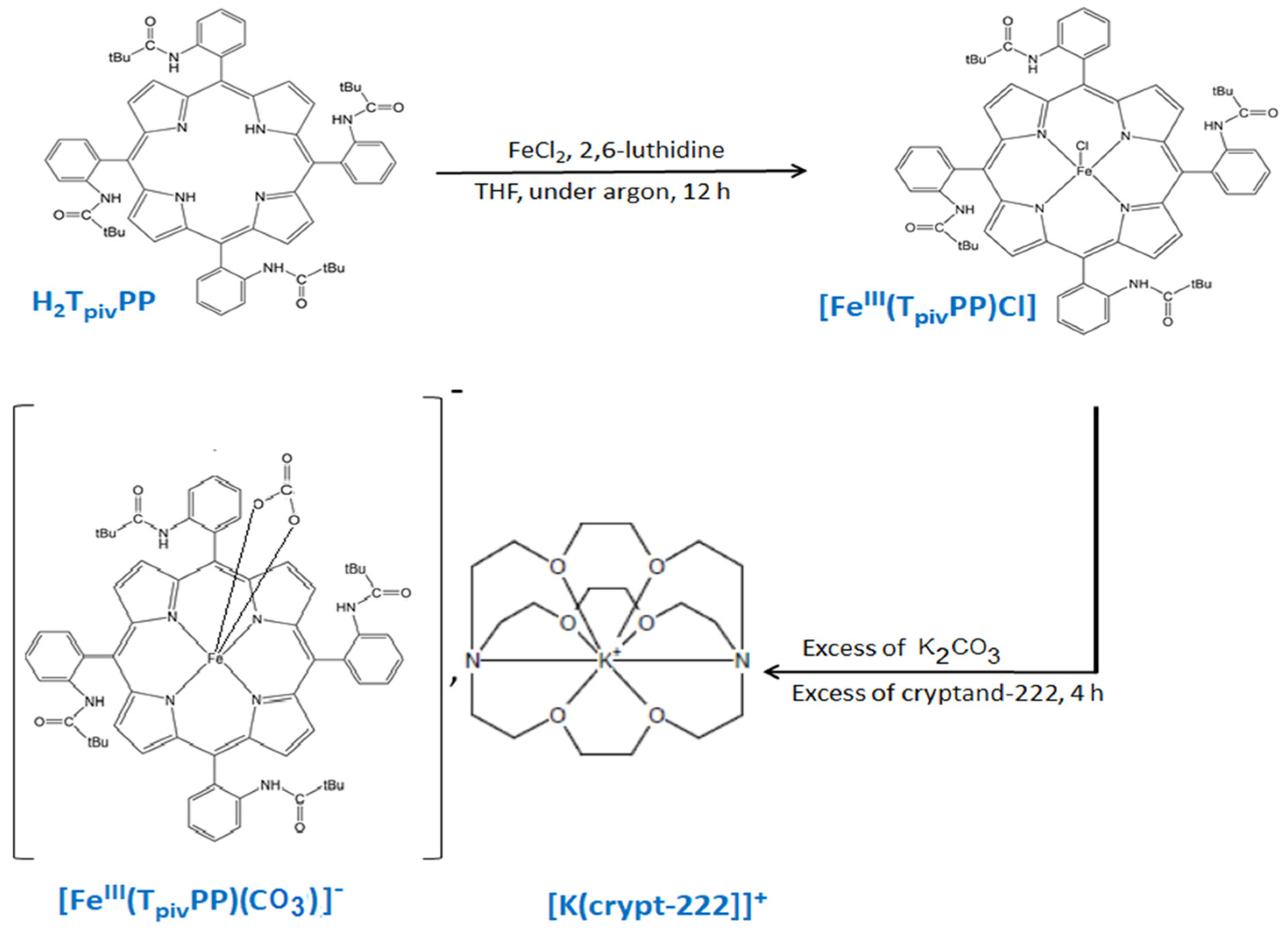
2.1.2. Preparation of [K(2,2,2-crypt)][FeIII(TpivPP)(CO3)]·C6H5Cl·3H2O
2.1.3. X-ray Crystallography
2.2. Synthesis of Complex I
2.3. Spectroscopic Characterizations
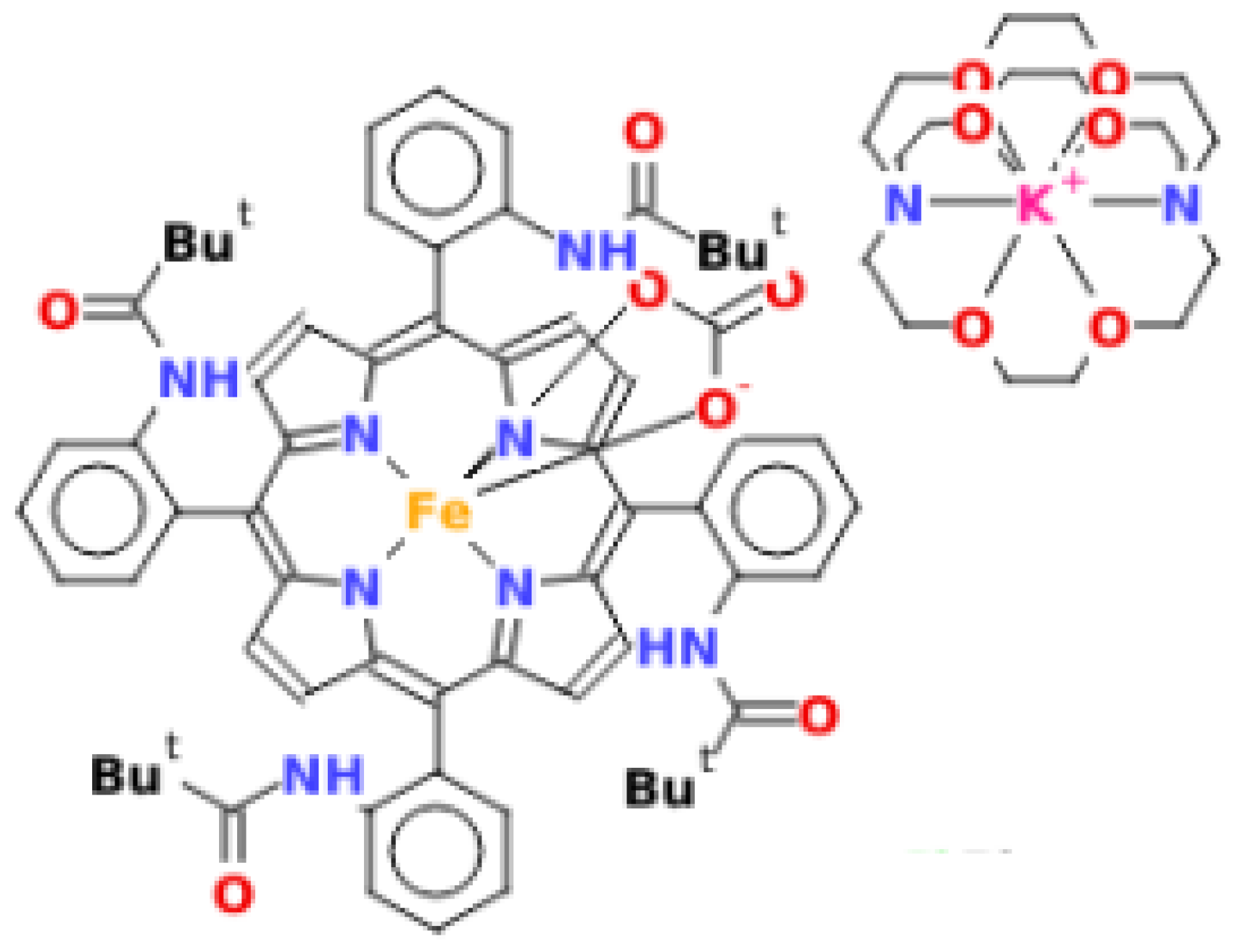
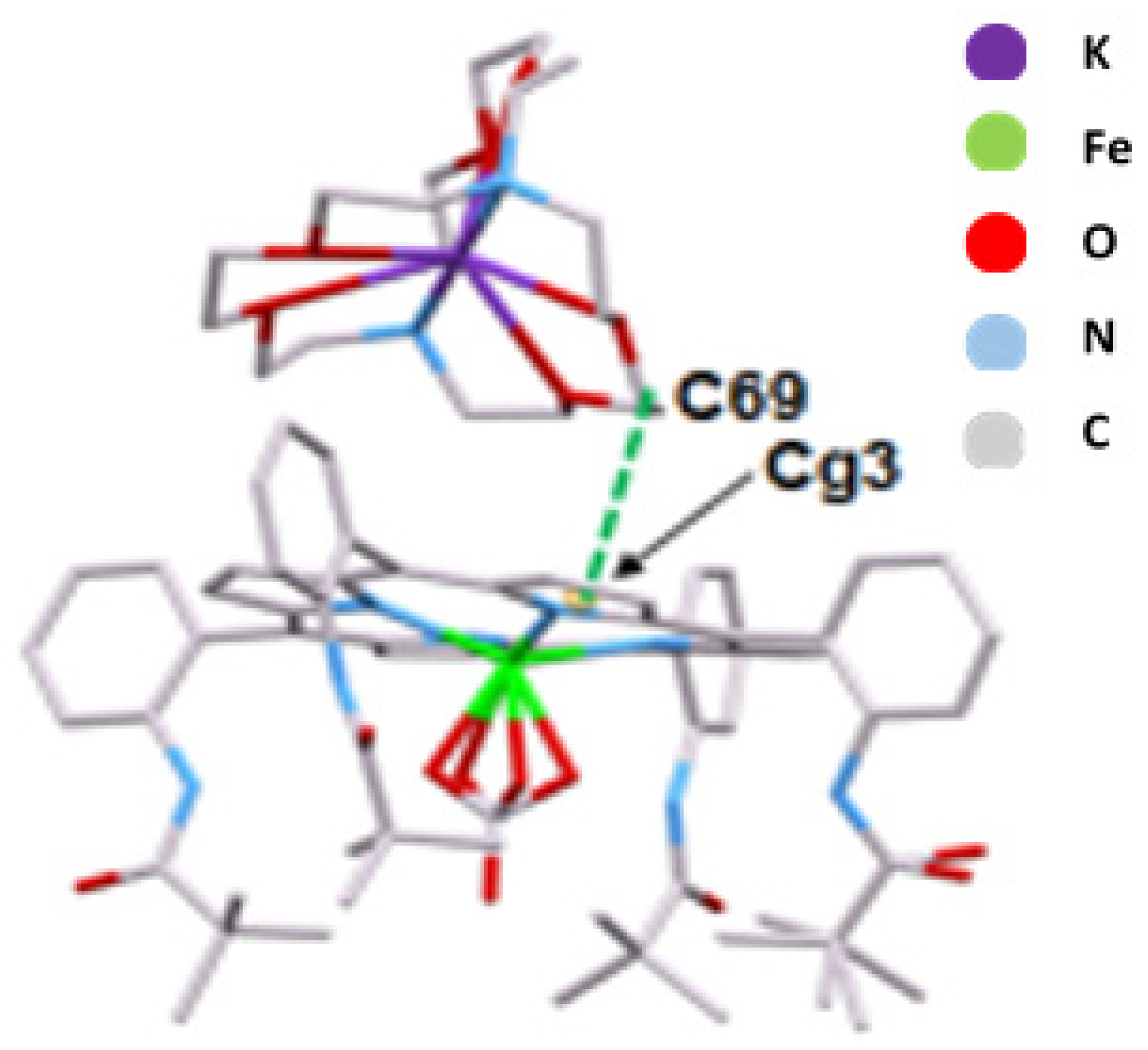
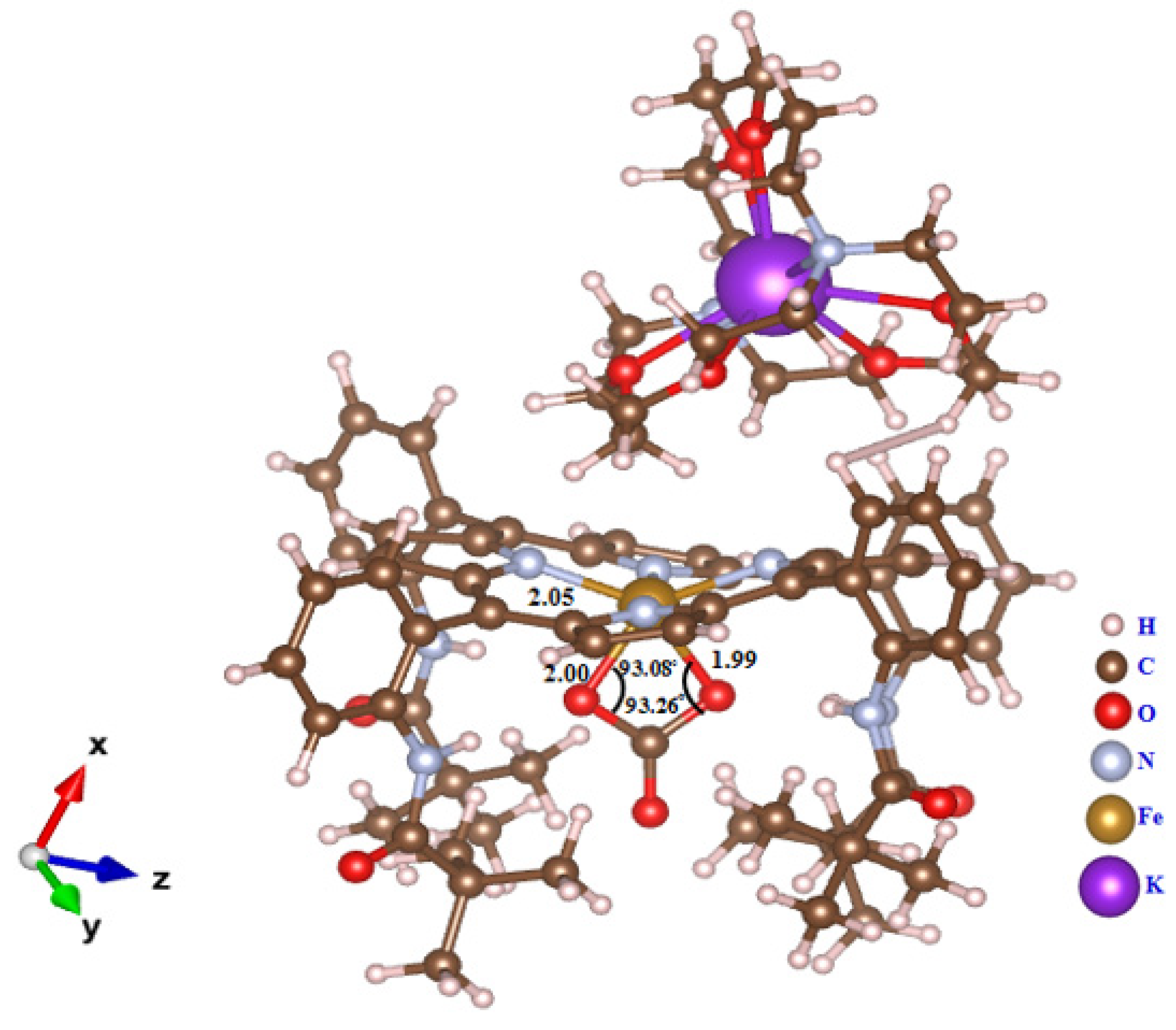
2.4. Structural Properties of [K(2,2,2-crypt)][FeIII(TpivPP)(CO3)] (I)
3. DFT-D3 Investigations of Compound I
3.1. Computational Details
3.2. Optimized Structure of [K(crypt-222)][FeIII(TpivPP)(CO3)] (I)
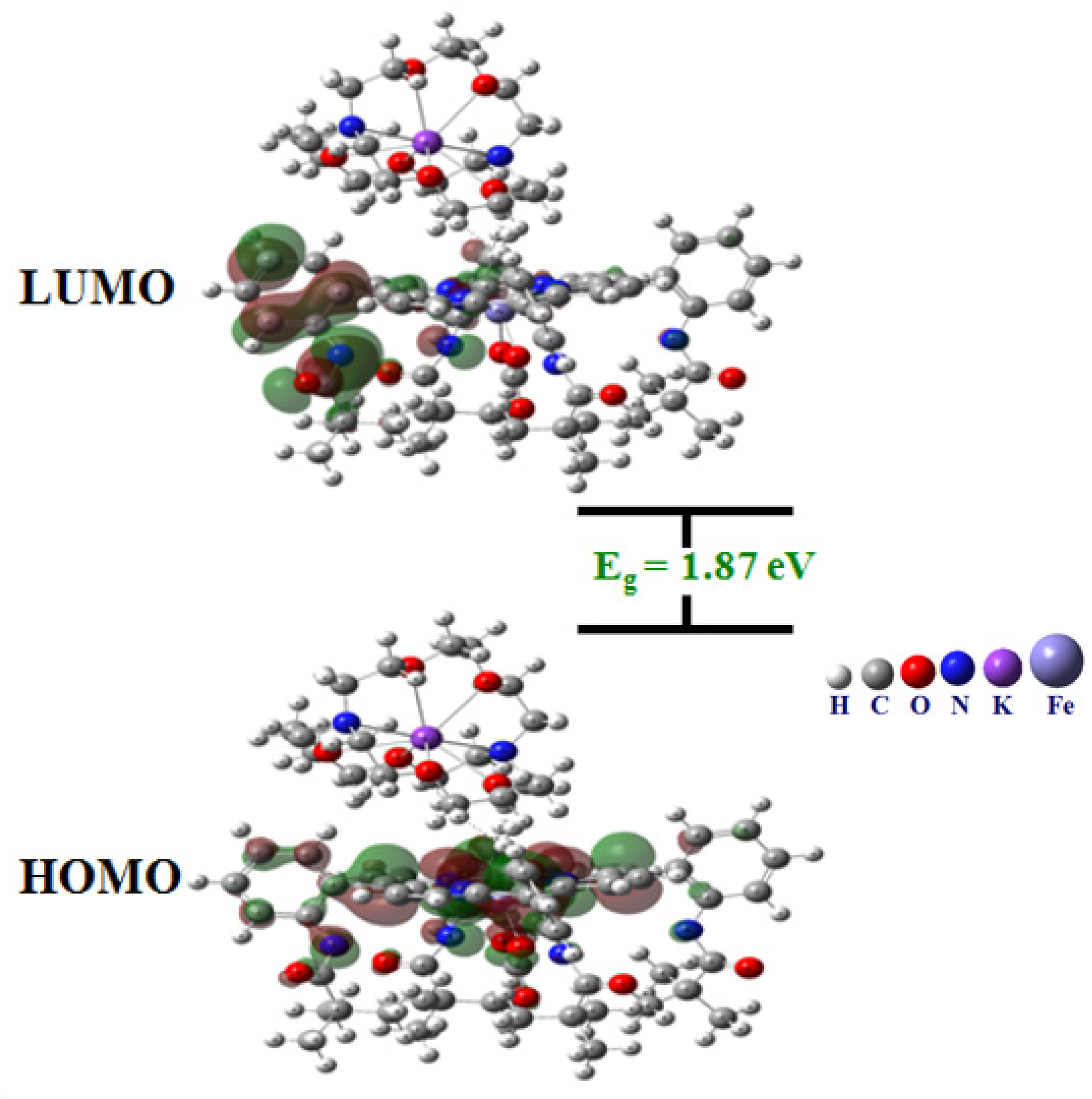
3.3. HOMO/LUMO and Global Reactivity Investigations
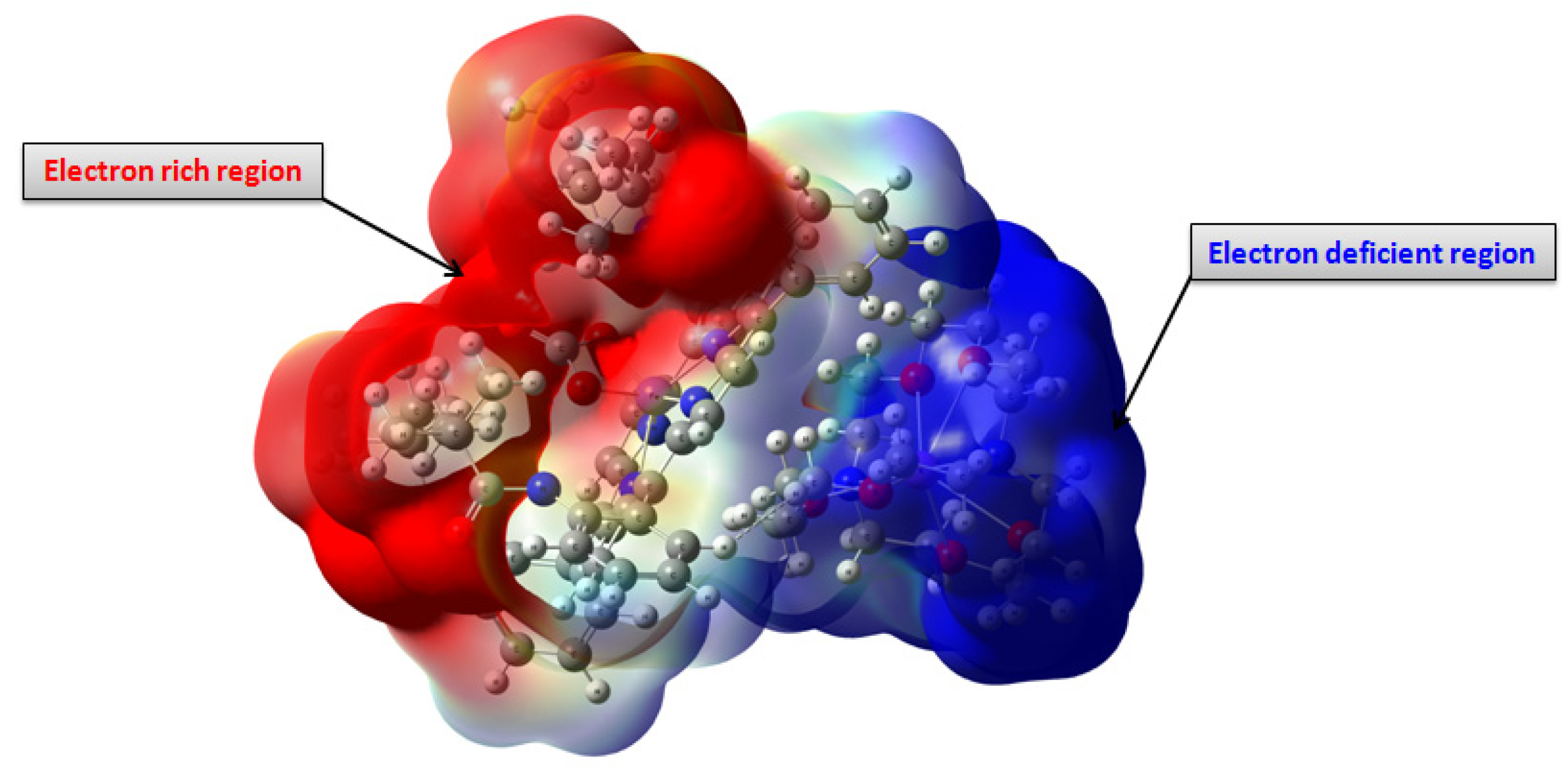
3.4. Molecular Electrostatic Potential (MEP) Analysis
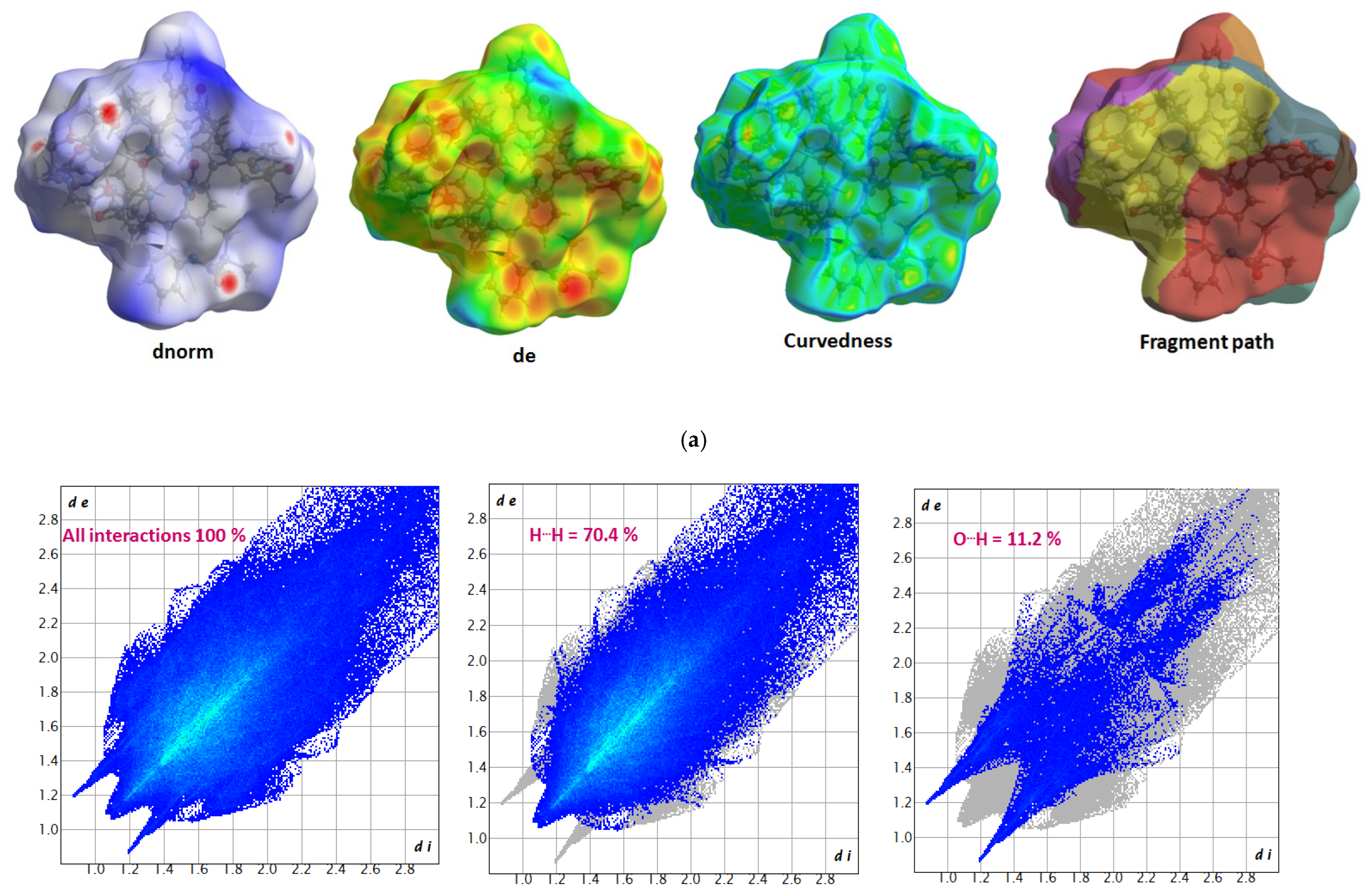
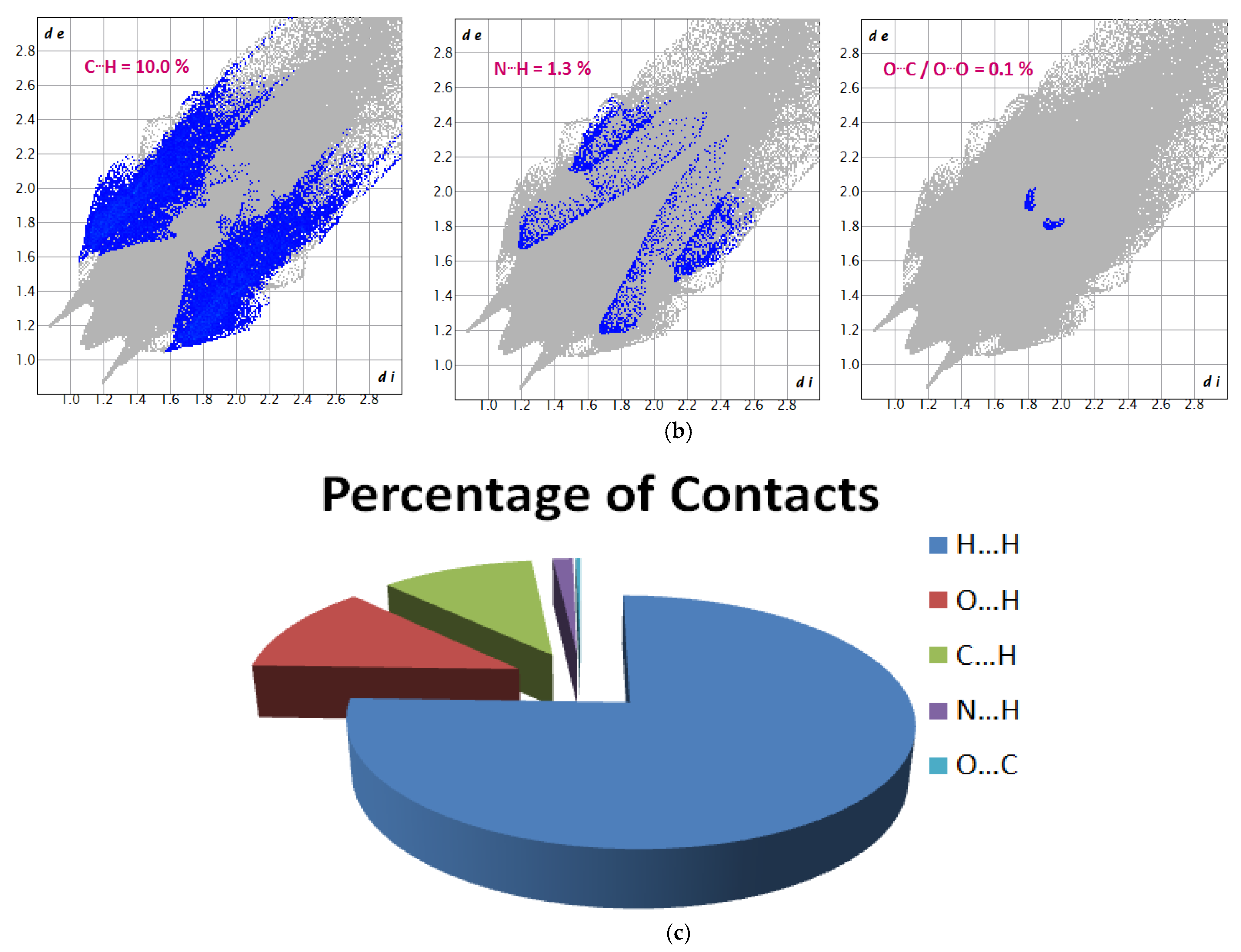
3.5. Hirshfeld Surface (HS) Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Scheidt, W.R.; Hoard, J.L. Hoard, Stereochemistry of low-spin cobalt porphyrins. I. Structure and bonding in a nitrosylcobalt porphyrin and their bearing on one rational model for the oxygenated. J. Am. Chem. Soc. 1973, 95, 8281–8288. [Google Scholar] [CrossRef] [PubMed]
- La Mar, G.N.; Walker, F.A. Dynamics of axial ligation in metalloporphyrins. I. Imidazole exchange in low-spin ferric porphyrins. J. Am. Chem. Soc. 1972, 94, 8607–8608. [Google Scholar] [CrossRef] [PubMed]
- Collman, J.P.; Gagne, R.R.; Halbert, T.R.; Marchon, J.C.; Reed, C.A. Reversible oxygen adduct formation in ferrous complexes derived from a picket fence porphyrin. Model for oxymyoglobin. J. Am. Chem. Soc. 1973, 95, 7868–7870. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, B.M.; Ratner, M.A. Gated electron transfer: When are observed rates controlled by conformational interconversion? J. Am. Chem. Soc. 1980, 109, 6237–6243. [Google Scholar] [CrossRef]
- Patra, R.; Chaudhary, A.; Ghosh, S.K.; Rath, S.P. Modulation of Metal Displacements in a Saddle Distorted Macrocycle: Synthesis, Structure, and Properties of High-Spin Fe(III) Porphyrins and Implications for the Hemoproteins. Inorg. Chem. 2008, 47, 8324–8335. [Google Scholar] [CrossRef] [PubMed]
- Walker, F.A. Models of the bis-histidine-ligated electron-transferring cytochromes. Comparative geometric and electronic structure of low-spin ferro-and ferrihemes. Chem. Rev. 2004, 104, 589–616. [Google Scholar] [CrossRef] [PubMed]
- Montiel-Montoya, R.; Bill, E.; Gonser, U.; Lauer, S.; Trautwein, A.X.; Schappacher, M.; Ricard, L.; Weiss, R. Mössbauer Studies of Synthetic Analogues for the Active Site in Cytochromes P450. In The Coordination Chemistry of Metalloenzymes: The Role of Metals in Reactions Involving Water, Dioxygen and Related Species. Proceedings of the NATO Advanced Study Institute held at San Miniato, Pisa, Italy, 28 May–8 June 1982; Springer: Dordrecht, The Netherlands, 1983. [Google Scholar]
- Hirshfeld, F.L. Bonded-atom fragments for describing molecular charge densities. Theor. Chim. Acta 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Wolff, S.K.; Grimwood, D.J.; McKinnon, J.J.; Turner, M.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A program for Hirshfeld surface analysis, visualization and qu antitative analysis of molecular crystals. J. Appl. Cryst. 2013, 54, 1006–1011. [Google Scholar] [CrossRef]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. Cryst. Eng. Commun. 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Spackman, M.A.; McKinnon, J.J. Fingerprinting intermolecular interactions in molecular crystals. Cryst. Eng. Commun. 2002, 4, 378–392. [Google Scholar] [CrossRef]
- Collman, J.P.; Gagne, R.R.; Reed, C.; Halbert, T.R.; Lang, G.; Robinson, W.T. Robinsonlc, Picket fence porphyrins. Synthetic models for oxygen binding hemoproteins. J. Am. Chem. Soc. 1975, 97, 1427–1439. [Google Scholar] [CrossRef] [PubMed]
- Bruker, A. Instrument Service v4. 2.7, APEX2, SADABS, SAINT-Plus & XPREP; Bruker AXS Inc.: Madison, WI, USA, 2014. [Google Scholar]
- Burla, M.C.; Caliandro, R.; Carrozzini, B.; Cascarano, G.L.; Cuocci, C.; Giacovazzo, C.; Mallamo, M.; Mazzone, A.; Polidori, G. Crystal structure determination and refinement via SIR2014. J. Appl. Cryst. 2005, 48, 306–309. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT–Integrated space-group and crystal-structure determination. Acta Cryst. 2014, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Mcardle, P. SORTX—A program for on-screen stick-model editing and autosorting of SHELX files for use on a PC. J. Appl. Cryst. 1995, 28, 65. [Google Scholar] [CrossRef]
- Spek, A.L. Structure validation in chemical crystallography. Acta. Cryst. 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. ORTEP-3 for Windows-a version of ORTEP-III with a Graphical User Interface (GUI). J. Appl. Cryst. 1997, 30, 565. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0–new features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Spek, A.L. PLATON SQUEEZE: A tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Crystallogr. 2015, 71, 9–18. [Google Scholar] [CrossRef]
- Allen, F.H.; Kennard, O.; Watson, D.G.; Brammer, L.; Orpen, A.G.; Taylor, R. Tables of bond lengths determined by X-ray and neutron diffraction. Part 1. Bond lengths in organic compounds. J. Chem. Soc. Perkin Trans. 1987, 2, S1–S19. [Google Scholar] [CrossRef]
- Dhifet, M.; Belkhiria, M.S.; Giorgi, M.; Nasri, H. Preparation, UV–Vis, IR and 1H NMR spectra, and molecular structure of the complex ion (η2-carbonato)(α,α,α,α-tetrakis (o-pivalamidophenyl) porphinato) ferrate (III). Inorg.Chim. Acta 2009, 362, 2776–2781. [Google Scholar] [CrossRef]
- Phillippi, M.A.; Baenziger, N.; Goff, H.M. Preparation, solution properties, and structure of iron (III) porphyrin oxyanion complexes. Crystal and molecular stereochemistry of a novel bidentate nitrato complex. Inorg. Chem. 1981, 23, 3904–3911. [Google Scholar] [CrossRef]
- Kellett, P.J.; Pawlik, M.J.; Taylor, L.F.; Thompson, R.G.; Levstik, M.A.; Anderson, O.P.; Strauss, S.H. Five-and six-coordinate high-spin iron (III) porphyrin complexes with teflate (OTeF5−) ligands. Inorg. Chem. 1989, 28, 440–447. [Google Scholar] [CrossRef]
- Nasri, H.; Haller, K.J.; Wang, Y.; Hanh, H.B.; Scheidt, W.R. Reactions of bis (nitro)[.alpha.,.alpha.,.alpha.,.alpha.-meso-tetrakis(o-pivalamidophenyl)porphinato]iron(III) with 2,3,5,6-tetrafluorothiophenol and 2,3,5,6-tetrafluorothiophenolate. EPR and Moessbauer spectra and molecular structures. Inorg. Chem. 1992, 31, 3459–3467. [Google Scholar] [CrossRef]
- Doppelt, P.; Fischer, J.; Weiss, R. Synthesis and Structure of a Dimercapto—Iron(III) Porphyrin Derivative: [Fe(SC6HF4)2TPP][Na c 18C6], C6H6. Croat. Chem. Acta 1984, 57, 507–518. [Google Scholar]
- Scheidt, W.R.; Lee, Y. Recent advances in the stereochemistry of metallotetrapyrroles. Struct. Bond. 1987, 64, 1–70. [Google Scholar]
- Grimme, S. Semi empirical hybrid density functional with perturbative second order correlation. J. Chem. Phys. 2006, 124, 034108. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- G09|Gaussian.com. Available online: https://gaussian.com/glossary/g09/ (accessed on 10 June 2024).
- Roy, D.D.; Todd, A.K.; John, M.M. Gauss View, 5.0. 8; Gaussian Inc.: Wallingford, UK; Wallingford, CT, USA, 2009. [Google Scholar]
- Choudhary, V.; Bhatt, A.; Dash, D.; Sharma, N. DFT calculations on molecular structures, HOMO–LUMO study, reactivity descriptors and spectral analyses of newly synthesized diorganotin(IV) 2-chloridophenylacetohydroxamate complexes. J. Comput. Chem. 2019, 40, 2354–2363. [Google Scholar] [CrossRef] [PubMed]
- Fasiuddin, G.; Basha, A.A.; Kubaib, A.; Azam, M.; Muzammil, P.; Bouzid, G.; Ayachi, S.; Khan, F.L.A.; Imran, P.M.; Al-Resayes, S.I. Solvation model, Vibrational analysis, Electronic level, Non-Covalent interactions and Molecular docking investigations of 6-Chloro-2-(4-Aminophenyl)-1H-Benzimidazole. J. Mol. Liq. 2024, 398, 124315. [Google Scholar] [CrossRef]
- Guillén-López, A.; Cabrera, O.G.R.; de la Cruz Arreola, S.; Celaya, C.A.; Sevilla-Camacho, P.Y.; Muñiz, J. Exploring the potential of porphyrin-based materials for organic solar cells supported on carbon: A quantum chemistry approach. J. Photochem. Photobiol. A Chem. 2024, 449, 115401. [Google Scholar] [CrossRef]
- Jumabaev, A.; Holikulov, U.; Hushvaktov, H.; Issaoui, N.; Absanov, A. Intermolecular interactions in ethanol solution of OABA: Raman, FTIR, DFT, M062X, MEP, NBO, FMO, AIM, NCI, RDG analysis. J. Mol. Liq. 2023, 377, 121552. [Google Scholar] [CrossRef]
- Hijazi, S.; Ahmed, N.; Louis, H.; Anna, I.; Shah, W.A. Biological and computational studies of novel scaffolds of gallic acid: Insight from density functional theory and molecular docking studies. Anal. Chem. Lett. 2023, 13, 487–504. [Google Scholar] [CrossRef]
- Owen, A.E.; Anyambula, I.A.; Benson, C.U.; Ojumola, F.O.; Alawa, J.A.; Benjamin, I.; Iyam, S.O.; Ogar, C.U.; Ojong, M.A.; Ojong, R.; et al. Exploration of semi-carbazone derivatives as promising agents against cholera: Insights from spectroscopic analysis, reactivity studies (ELF, HOMO-LUMO, NBO), solvation effects, and molecular docking investigations. Chem. Phys. Impact 2024, 8, 100438. [Google Scholar] [CrossRef]
- Riaz, M.; Ali, A.; Ashfaq, M.; Ibrahim, M.; Akram, N.; Tahir, M.N.; Kuznetsov, A.; Rodríguez, L.; Sameeh, M.Y.; Assiri, M.A.; et al. Polymorphs of Substituted p-Toluenesulfonanilide: Synthesis, Single-Crystal Analysis, Hirshfeld Surface Exploration, and Theoretical Investigation. ACS Omega 2023, 8, 35307–35320. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, S.W.A.; Ashraf, A.; Ashfaq, M.; Tahir, M.N.; Niaz, S. Sulfonamide derived Schiff base Mn (II), Co (II), and Ni (II) complexes: Crystal structures, density functional theory and Hirshfeld surface analysis. Appl. Organomet. Chem. 2023, 37, 7077. [Google Scholar] [CrossRef]
- Fatima, A.; Kumar, A.; Saral, A.K.A.; Muthu, S.; Afzal, M.; Haq, N.; Nazar, I.; Siddiqui, N.; Javed, S. Study of L-tryptophan (a neurotransmitter precursor): Spectral, Hirshfeld surface, molecular docking and dynamics simulations. Z. Phys. Chem. 2023. [Google Scholar] [CrossRef]
- Mackenzie, C.F.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer model energies and energy frameworks: Extension to metal coordination compounds, organic salts, solvates and open-shell systems. IUCrJ 2017, 4, 575–587. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, J.J.; Jayatilaka, D.; Spackman, M.A. Towards quantitative analysis of intermolecular interactions with Hirshfeld surfaces. Chem. Commun. 2007, 3814–3816. [Google Scholar] [CrossRef]
- Ezzayani, K.; Khelifa, A.B.; Saint-Aman, E.; Loiseau, F.; Nasri, H. Synthesis, spectroscopic characterizations, cyclic voltammetry investigation and molecular structure of the di-μ-cyanato-N-bis(μ-1,4,7,10,13,16-hexaoxacyclooctadecane)bis(5,10,15,20-tetraphenylporphyrinato) dimagnesiumdipotassium complex. Polyhedron 2016, 117, 817. [Google Scholar] [CrossRef]
- Amiri, N.; Taheur, F.B.; Chevreux, S.; Wenger, E.; Lemercier, G.; Nasri, H. Synthesis, crystal structure and spectroscopic characterizations of porphyrin-based Mg (II) complexes–Potential application as antibacterial agent. Tetrahedron 2017, 73, 7011. [Google Scholar] [CrossRef]
- Amiri, N.; Hajji, M.; Taheur, F.B.; Chevreux, S.; Roisnel, T.; Lemercier, G.; Nasri, H. Two novel magnesium (II) meso-tetraphenylporphyrin-based coordination complexes: Syntheses, combined experimental and theoretical structures elucidation, spectroscopy, photophysical properties and antibacterial activity. J. Solid State Chem. 2018, 258, 477. [Google Scholar] [CrossRef]
- Fradi, T.; Noureddine, O.; Taheur, F.B.; Guergueb, M.; Nasri, S.; Amiri, N.; Almahri, A.; Roisnel, T.; Guerineau, V.; Issoui, N.; et al. New DMAP meso-arylporphyrin Magnesium(II) complex. Spectroscopic, Cyclic voltammetry and X-ray molecular structure characterization. DFT, DOS and MEP calculations and Antioxidant and Antifungal activities. J. Mol. Struct. 2021, 1236, 130299. [Google Scholar] [CrossRef]
- Bominaar, E.L.; Ding, X.Q.; Gismelseed, A.; Bill, E.; Winkler, H.; Trautwein, A.X.; Nasri, H.; Fischer, J.; Weiss, R. Weiss, Structural, Moessbauer, and EPR investigations on two oxidation states of a five-coordinate, high-spin synthetic heme. Quantitative interpretation of zero-field parameters and large quadrupole splitting. Inorg. Chem. 1992, 31, 1845. [Google Scholar] [CrossRef]
- Nasri, H.; Debbabi, M. Synthesis, spectroscopic and structural characterization of the pentacoordinate high-spin Fe (III) isothiocyanate “picket fence” porphyrin complex. Polyhedron 1998, 17, 3607. [Google Scholar] [CrossRef]
- Belkhiria, M.S.; Dhifet, M.; Nasri, H. Preparation and spectroscopic properties of the (cyanato-N) and (oxalato) iron(III) “picket fence” porphyrins: Structure of the (cyanato-N)(α,α,α,α-tetrakis(o-pivalamidophenyl)porphinato)iron(III) complex. J. Porphyr. Phthalocyanines 2005, 9, 575. [Google Scholar] [CrossRef]
- Nasri, H.; Goodwin, J.A.; Scheidt, W.R. Use of protected binding sites for nitrite binding in iron(III) porphyrinates. Crystal structure of the bis(nitro)(.alpha.,.alpha.,.alpha.,.alpha.-tetrakis(o-pivalamidophenyl)porphinato)iron(III) anion. Inorg. Chem. 1990, 28, 185. [Google Scholar] [CrossRef]
- Nasri, H.; Wang, Y.; Huynh, B.H.; Walker, F.A.; Scheidt, W.R. Reactions of bis(nitro)(.alpha.,.alpha.,.alpha.,.alpha.-tetrakis(o-pivalamidophenyl)porphinato)ferrate(III) with pyridine and imidazole. EPR and Moessbauer spectra and molecular structures of the mixed-ligand species. Inorg. Chem. 1991, 30, 1483. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Empirical Formula | C83H100N10O13KFe |
|---|---|
| Formula weight, M | 1649.2 |
| T (K) | 293 (2) |
| Wavelength(Å) | 0.71073 |
| Crystal system | monoclinic |
| Space group | |
| P21/n | |
| Lattice constants | |
| a (Å); b (Å); c (Å) | 17.932 (2); 21.484 (2); 22.924 (2) |
| β (°) | 100.813 (3) |
| Volume V (Å3) | 8674.8 (14) |
| Z | 4 |
| Dcal, g/cm3 | 1.263 |
| Absorption coefficient, μ (mm−1) | 0.319 |
| F(000) | 3484 |
| Crystal size (mm3) | 0.30 × 0.15 × 0.10 |
| θ-range for data collection | 1.6–19.8 |
| Limiting indices | −17 ≤ h ≤ 17, −20 ≤ k ≤ 20, −21 ≤ l ≤ 21 |
| Reflexions collected/unique | 82,787/7805 |
| R(int)/R(σ) | 0.1122/0.0478 |
| Data/Restraints/Parameters | 7805/17/1028 |
| X (Flack parameter) | 0.18(2) |
| Final R indices [I > 2σ(I)] | a R1= 0.083, wR2= 0.200 |
| R indices (all data) | R1 = 0.103, wR2 = 0.213 |
| Goodness-of-fit on Fo2 | 1.047 |
| Highest peak and deepest hole (eÅ−3) | 1.905 and −0.787 |
| CCDC | 2,367,771 |
| Iron Coordination Polyhedron | |||
| Fe-N1 | 2.115 (6) | N1-Fe-N2 | 85.9 (2) |
| Fe-N2 | 2.083 (5) | N1-Fe-N3 | 146.7 (2) |
| Fe-N3 | 2.120 (6) | N1-Fe-N4 | 85.0 (2) |
| Fe-N4 | 2.101 (6) | N2-Fe-N3 | 85.4 (2) |
| Fe-Np | 2.105 (6) | N2-Fe-N4 | 146.9 (2) |
| Fe-O5A | 2.00 (2) | N1-Fe-O5A | 110.0 (6) |
| Fe-O6A | 2.047 (2) | N2-Fe-O5A | 136.4 (6) |
| N3-Fe-O5A | 98.3 (6) | N3-Fe-O6A | 133.8 (6) |
| N4-Fe-O5A | 76.4 (5) | N4-Fe-O6A | 118.4 (7) |
| N1-Fe-O6A | 78.3 (6) | N2-Fe-O6A | 90.6 (6) |
| Carbonato ligand | |||
| C65-O5A | 1.250 (2) | Fe-O6A-C65 | 91.9 (2) |
| C65-O6A | 1.257 (2) | O5A-C65-O6A | 99.1 (2) |
| C65-O7 | 1.292 (1) | O5A-C65-O7 | 132.8 (2) |
| Fe-O5A-C65 | 95.5 (1) | O6A-C65-O7 | 118.6 (2) |
| Potassium-cryptand-222 | |||
| K-N9 | 3.023 (7) | N9-K-N10 | 179.48 (2) |
| K-N10 | 3.055 (6) | N9-K-O8 | 59.94 (2) |
| K-O8 | 2.807 (6) | N9-K-O9 | 119.91 (2) |
| K-O9 | 2.839 (5) | O8-K-O9 | 60.15 (2) |
| K-O10 | 2.842 (2) | O8-K-O10 | 128.13 (2) |
| K-O11 | 2.823 (5) | O8-K-O11 | 100.93 (2) |
| K-O12 | 2.834 (6) | O9-K-O12 | 121.18 (2) |
| K-O13 | 2.767 (6) | O11-K-O12 | 96.55 (2) |
| Complex | Fe–Np a | Fe–XL b | Fe–PC c | Ref. |
|---|---|---|---|---|
| High-spin (S = 5/2) iron(III) hexacoordinated metalloporphyrins | ||||
| [FeIII(TpivPP)(CO3)]− | 2.105 | 2.047 | 0.654(2) | T.w. |
| 2.000 | ||||
| [FeIII(TpivPP)(η2-O2CO)]− | 2.125 | 2.005 | 0.749(7) | [22] |
| 2.031 | ||||
| [FeIII(TPP)(η2-O2NO)] d | 2.073 | 2.323 | 0.600(1) | [23] |
| 2.019 | ||||
| [FeIII(TPP)(γ2-O2NO)] | 2.085 | 2.124 | 0.620(1) | [23] |
| 2.268 | ||||
| [FeIII(TTP)(OTeF5)(THF)] e | 2.053 | 2.334 | 0.200 | [24] |
| 1.967 | ||||
| Low-spin (S = 1/2) iron(III) hexacoordinated metalloporphyrins | ||||
| [FeIII(TpivPP)(NO2)2]− | 1.992 | 1.985 | 0.020 | [25] |
| 1.985 | ||||
| [FeIII(TpivPP)(SC6HF4)(NO2)]− | 1.980 | 2.277 | 0.050(1) | [25] |
| 1.990 | ||||
| [FeIII(TPP)(SC6HF4)2]− | 1.998 | 2.312 | 0 f | [26] |
| 2.312 | ||||
| Iron Coordination Polyhedron | ||
|---|---|---|
| Experimental Values | Theoretic Values | |
| Fe-N1 | 2.115(6) | 2.263 |
| Fe-N2 | 2.083(5) | 2.021 |
| Fe-N3 | 2.120(6) | 2.061 |
| Fe-N4 | 2.101(6) | 2.052 |
| Fe-O5A | 2.00(2) | 1.992 |
| Fe-O6A | 2.047(2) | 2.001 |
| C65-O5A | 1.250(2) | 1.367 |
| C65-O6A | 1.257(2) | 1.365 |
| C65-O7 | 1.292(1) | 1.244 |
| N1-Fe-O5A | 110.0(6) | 143.44 |
| N1-Fe-O6A | 78.3(6) | 79.30 |
| N2-Fe-O5A | 136.4(6) | 143.43 |
| N2-Fe-O6A | 90.6(6) | 89.90 |
| N3-Fe-O5A | 98.3(6) | 104.44 |
| N3-Fe-O6A | 133.8(6) | 142.59 |
| N4-Fe-O5A | 76.4(5) | 87.67 |
| N4-Fe-O6A | 118.4(7) | 101.87 |
| N1-Fe-N2 | 85.9(2) | 87.16 |
| N1-Fe-N3 | 146.7(2) | 137.55 |
| N1-Fe-N4 | 85.0(2) | 88.04 |
| N2-Fe-N3 | 85.4(2) | 86.88 |
| N2-Fe-N4 | 146.9(2) | 165.89 |
| Fe-O5A-C65 | 95.5(1) | 93.26 |
| Fe-O6A-C65 | 91.9(2) | 93.08 |
| O5A-C65-O6A | 99.1(2) | 107.38 |
| O5A-C65-O7 | 132.8(2) | 126.28 |
| O6A-C65-O7 | 118.6(2) | 126.32 |
| Quantum Chemical Parameters (eV) | |
|---|---|
| εHOMO | −4.84 |
| εLUMO | −2.97 |
| 1.87 | |
| 4.84 | |
| 2.97 | |
| Chemical potential, | −3.90 |
| Mulliken electronegativity, χ = −μ | 3.90 |
| Global hardness, | 0.93 |
| Electrophilicity index, ω = | 8.17 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dhifet, M.; Gassoumi, B.; Lutoshkin, M.A.; Kazachenko, A.S.; Kazachenko, A.S.; Al-Dossary, O.; Issaoui, N.; Nasri, H. Synthesis, X-ray Crystallography, Spectroscopic Characterizations, Density Functional Theory, and Hirshfeld Surface Analyses of a Novel (Carbonato) Picket Fence Iron(III) Complex. Molecules 2024, 29, 3722. https://doi.org/10.3390/molecules29163722
Dhifet M, Gassoumi B, Lutoshkin MA, Kazachenko AS, Kazachenko AS, Al-Dossary O, Issaoui N, Nasri H. Synthesis, X-ray Crystallography, Spectroscopic Characterizations, Density Functional Theory, and Hirshfeld Surface Analyses of a Novel (Carbonato) Picket Fence Iron(III) Complex. Molecules. 2024; 29(16):3722. https://doi.org/10.3390/molecules29163722
Chicago/Turabian StyleDhifet, Mondher, Bouzid Gassoumi, Maxim A. Lutoshkin, Anna S. Kazachenko, Aleksandr S. Kazachenko, Omar Al-Dossary, Noureddine Issaoui, and Habib Nasri. 2024. "Synthesis, X-ray Crystallography, Spectroscopic Characterizations, Density Functional Theory, and Hirshfeld Surface Analyses of a Novel (Carbonato) Picket Fence Iron(III) Complex" Molecules 29, no. 16: 3722. https://doi.org/10.3390/molecules29163722