
Nicotinamide Riboside: What It Takes to Incorporate It into RNA

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Stability of Nicotinamide Riboside
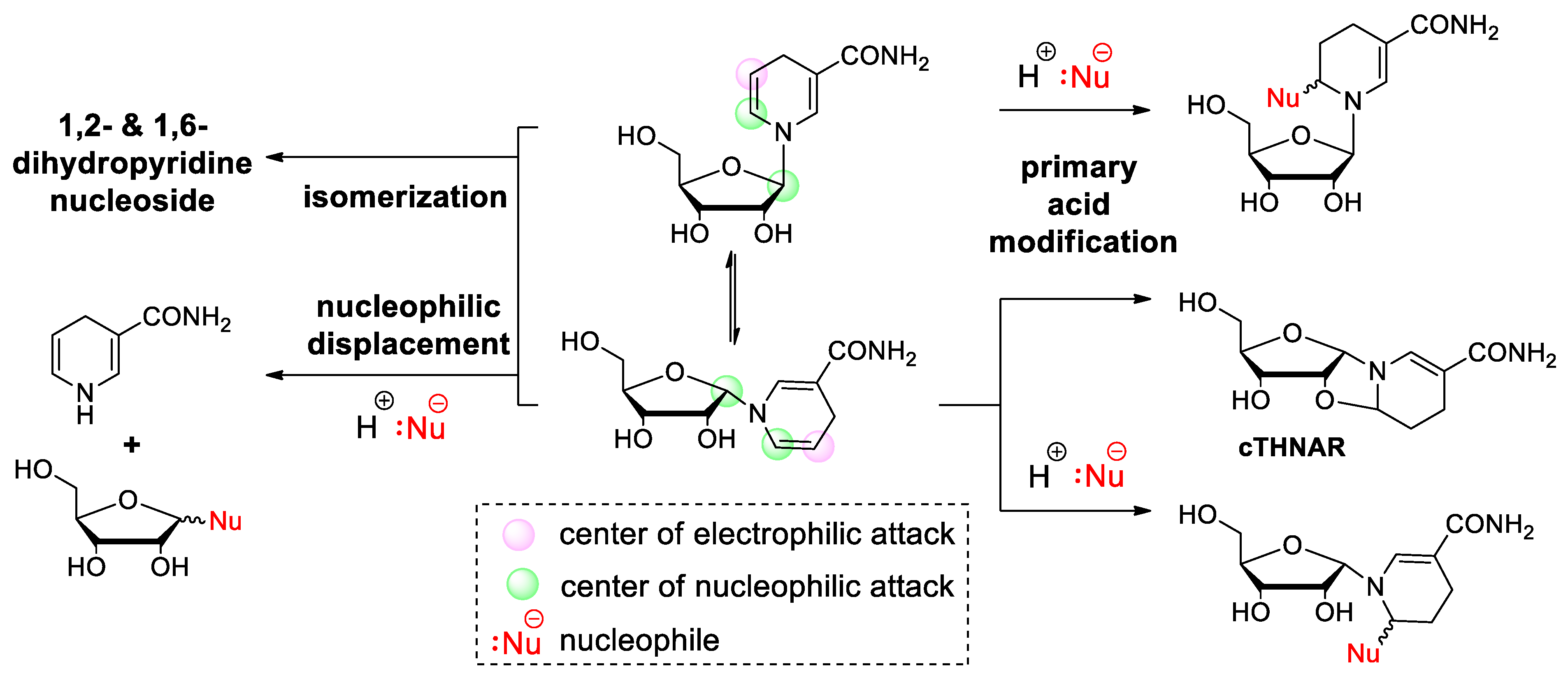
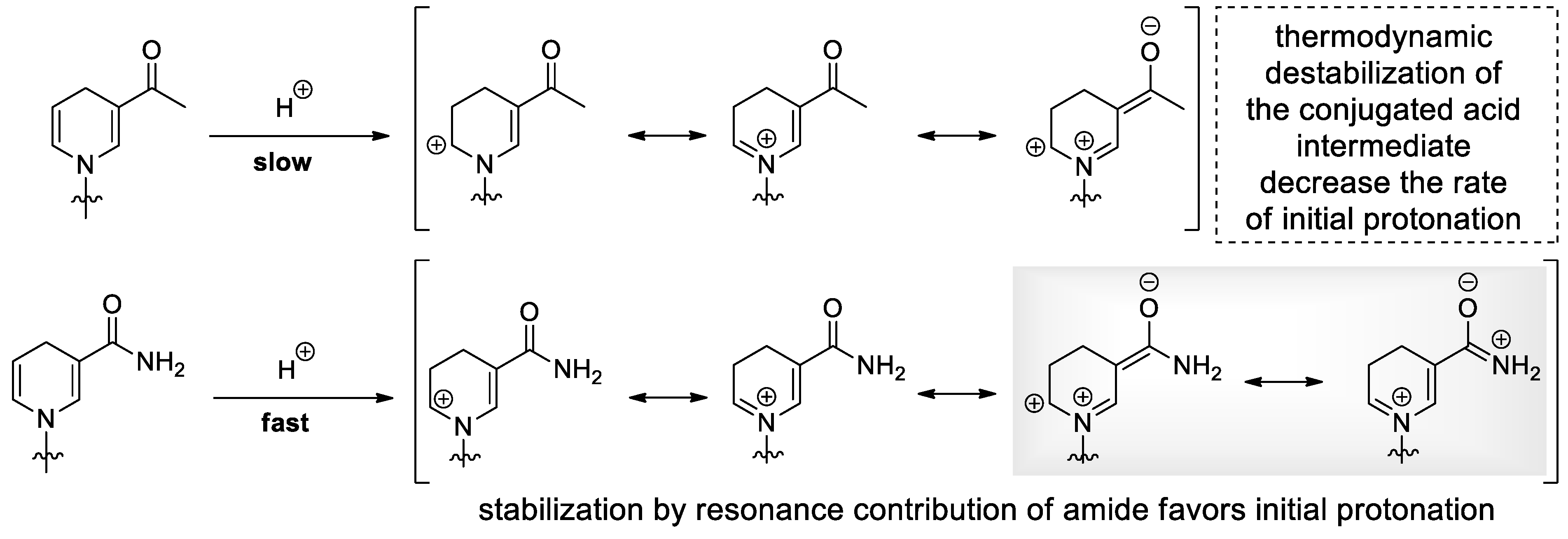
2.1. Nucleophilic Attack on the Anomeric Carbon and the Pyridinium Moiety Leads to Decomposition of Nicotinamide Riboside under Alkaline Conditions
2.2. Dihydronicotinamide Riboside (NARH) Is Prone to Degradation by Acid-Catalyzed Addition and Nucleophilic Displacement of the Dihydropyridine Moiety in Alkaline Media
3. Synthesis of Nicotinamide Riboside
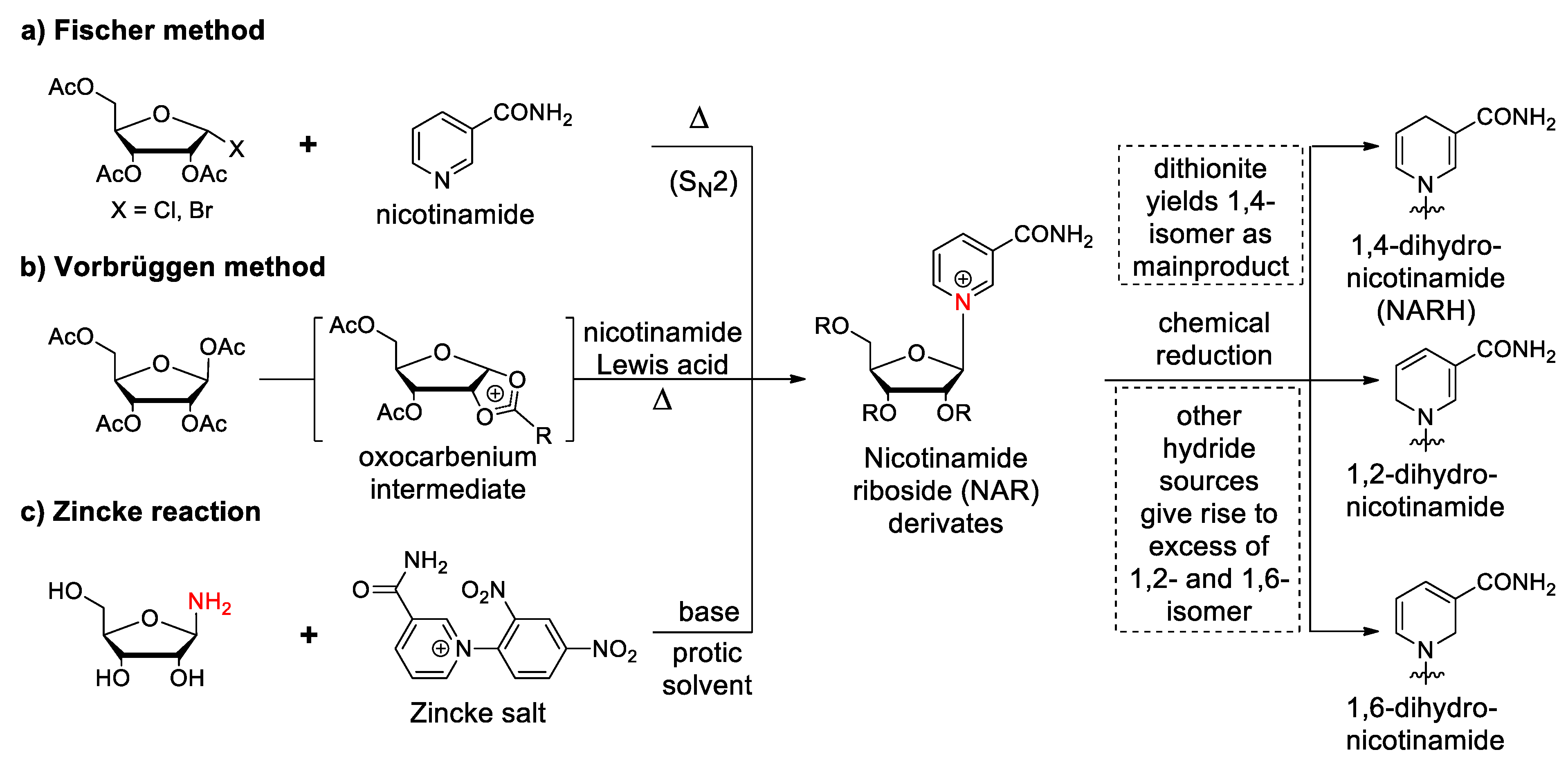
3.1. Synthetic Paths to Nicotinamide Riboside
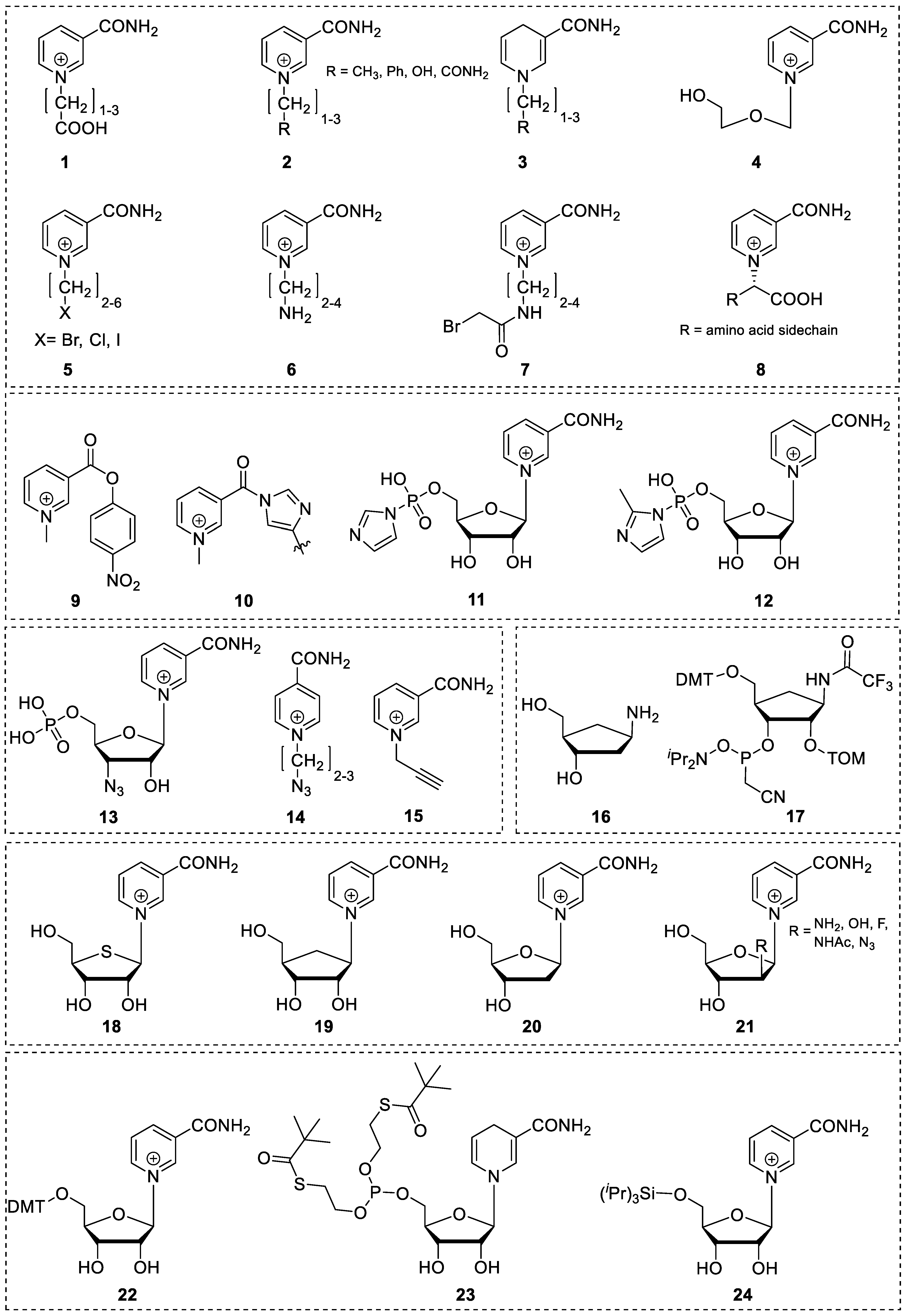
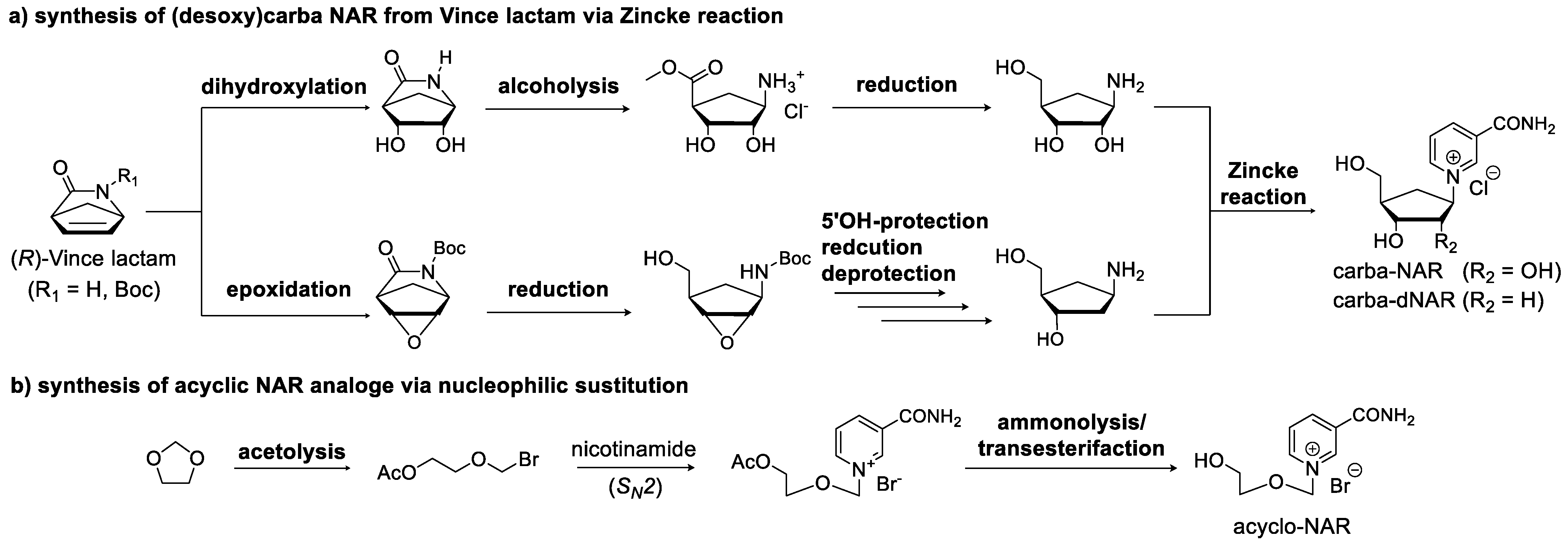
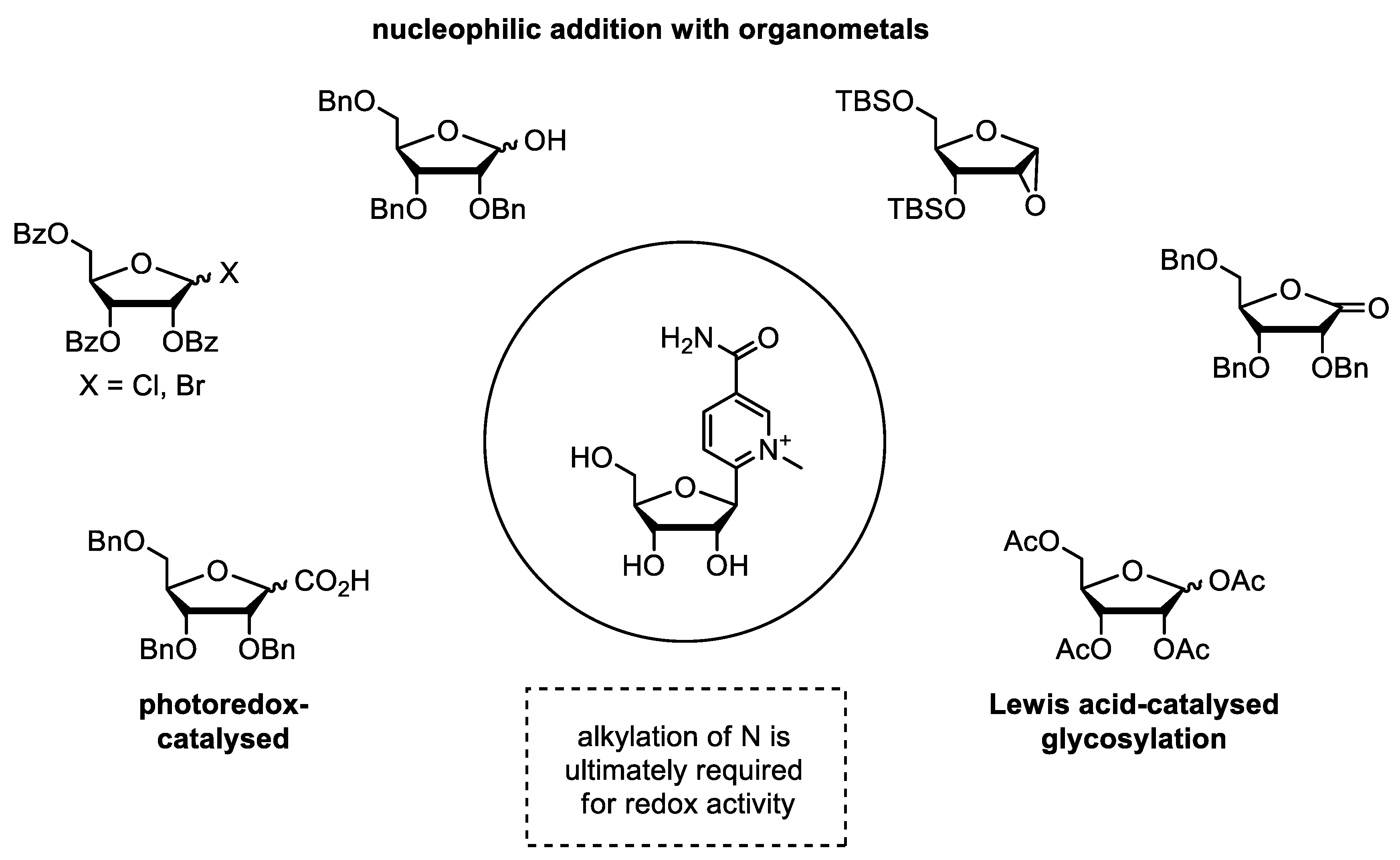
3.2. Synthetic Modification of the Structure of the Ribose Moiety
3.3. Synthetic Modifications of the Nicotinamide Base
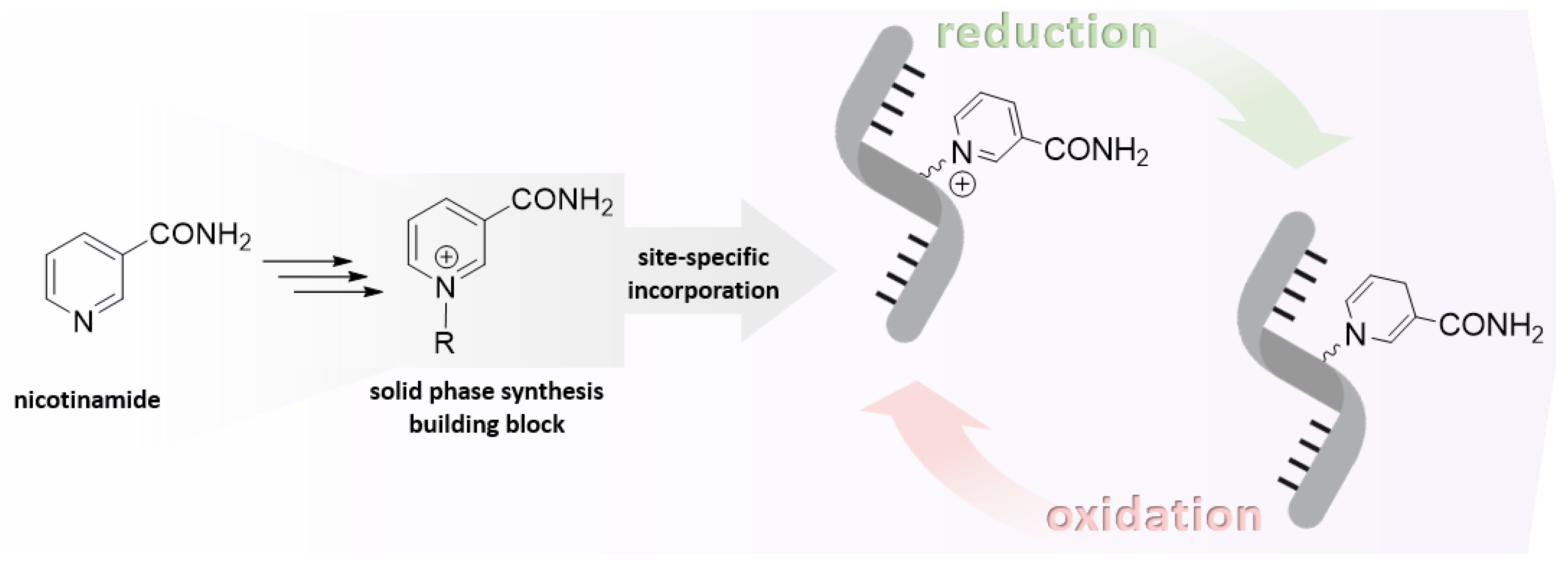
4. Attempts to Incorporate Nicotinamide Riboside in RNA
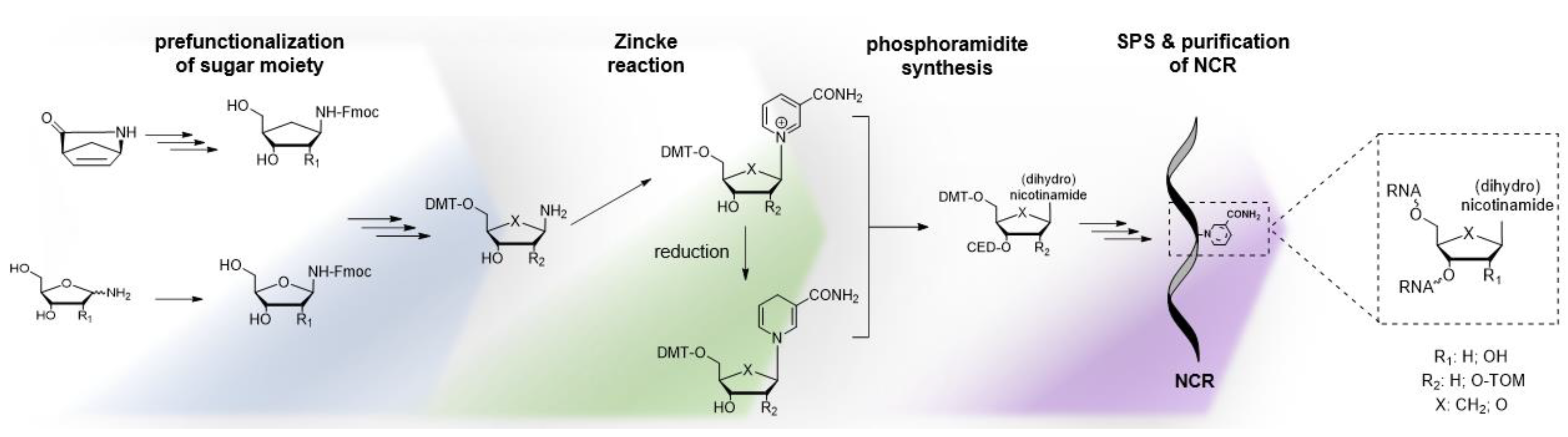
4.1. Synthesis of NAR/NARH Building Blocks
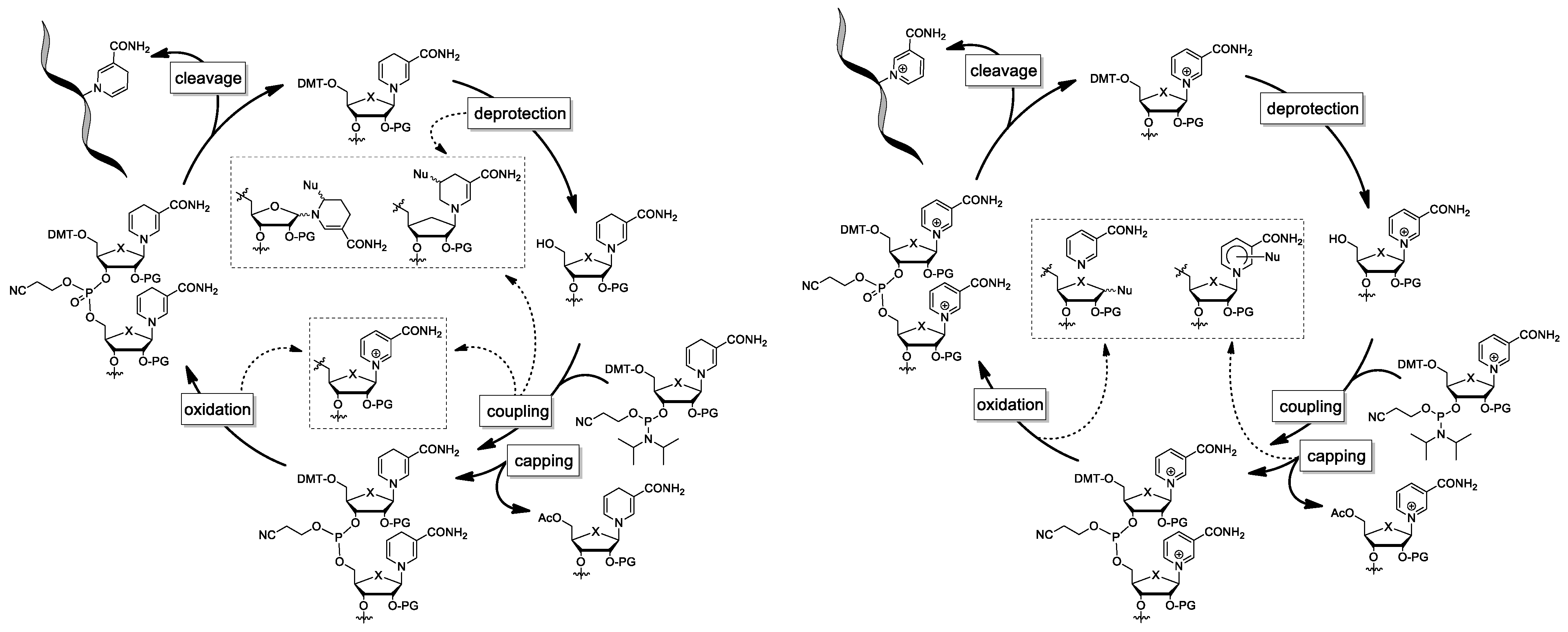
4.2. Application of a NAR/NARH Phosphoramidite
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Panchapakesan, S.S.S.; Breaker, R.R. The Case of the Missing Allosteric Ribozymes. Nat. Chem. Biol. 2021, 17, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Robertson, M.P.; Joyce, G.F. The Origins of the RNA World. Cold Spring Harb. Perspect. Biol. 2012, 4, a003608. [Google Scholar] [CrossRef] [PubMed]
- Vitreschak, A. Riboswitches: The Oldest Mechanism for the Regulation of Gene Expression? Trends Genet. 2004, 20, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Breaker, R.R. Riboswitches and the RNA World. Cold Spring Harb. Perspect. Biol. 2012, 4, a003566. [Google Scholar] [CrossRef] [PubMed]
- Breaker, R.R. Riboswitches: From Ancient Gene-Control Systems to Modern Drug Targets. Future Microbiol. 2009, 4, 771–773. [Google Scholar] [CrossRef] [PubMed]
- Goldman, A.D.; Kacar, B. Cofactors Are Remnants of Life’s Origin and Early Evolution. J. Mol. Evol. 2021, 89, 127–133. [Google Scholar] [CrossRef]
- Huang, F.; Bugg, C.W.; Yarus, M. RNA-Catalyzed Coa, NAD, and FAD Synthesis from Phosphopantetheine, NMN, and FMN. Biochemistry 2000, 39, 15548–15555. [Google Scholar] [CrossRef] [PubMed]
- Kirschning, A. Coenzymes and Their Role in the Evolution of Life. Angew. Chem. Int. Ed. 2021, 60, 6242–6269. [Google Scholar] [CrossRef] [PubMed]
- Breaker, R.R.; Joyce, G.F. Self-Incorporation of Coenzymes by Ribozymes. J. Mol. Evol. 1995, 40, 551–558. [Google Scholar] [CrossRef]
- Fujita, Y.; Furuta, H.; Ikawa, Y. Construction of an Artificial Ribozyme Which Ligates an RNA Fragment Activated by Nicotinamide Mononucleotide. Nucleic Acids Symp. Ser. 2006, 50, 231–232. [Google Scholar] [CrossRef]
- Fujita, Y.; Furuta, H.; Ikawa, Y. Tailoring RNA Modular Units on a Common Scaffold: A Modular Ribozyme with a Catalytic Unit for β-Nicotinamide Mononucleotide-Activated RNA Ligation. RNA 2009, 15, 877–888. [Google Scholar] [CrossRef] [PubMed]
- Cleaves, H.J.; Miller, S.L. The Nicotinamide Biosynthetic Pathway Is a By-Product of the RNA World. J. Mol. Evol. 2001, 52, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Benner, S.A. A Direct Prebiotic Synthesis of Nicotinamide Nucleotide. Chem. Eur. J. 2018, 24, 581–584. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, H.B.; Williams, C.; King, S.J.; Allison, S.J. Nicotinamide Adenine Dinucleotide (NAD+): Essential Redox Metabolite, Co-Substrate and an Anti-Cancer and Anti-Ageing Therapeutic Target. Biochem. Soc. Trans. 2020, 48, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Emahi, I.; Gruenke, P.R.; Baum, D.A. Effect of Aptamer Binding on the Electron-Transfer Properties of Redox Cofactors. J. Mol. Evol. 2015, 81, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Tsukiji, S.; Ramaswamy, K.; Suga, H. Ribozymes That Use Redox Cofactors. Pure Appl. Chem. 2004, 76, 1525–1536. [Google Scholar] [CrossRef]
- Tsukiji, S.; Pattnaik, S.B.; Suga, H. An Alcohol Dehydrogenase Ribozyme. Nat. Struct. Mol. Biol. 2003, 10, 713–717. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.T.; Jones, D.S.; Andrews, G.P.; Li, S. Understanding the Physicochemical Properties and Degradation Kinetics of Nicotinamide Riboside, a Promising Vitamin B(3)Nutritional Supplement. Food Nutr. Res. 2019, 63. [Google Scholar] [CrossRef] [PubMed]
- Oppenheimer, N.J. NAD Hydrolysis: Chemical and Enzymatic Mechanisms. Mol. Cell Biochem. 1994, 138, 245–251. [Google Scholar] [CrossRef]
- Handlon, A.L.; Oppenheimer, N.J. Substituent Effects on the pH-Independent Hydrolysis of 2’-Substituted Nicotinamide Arabinosides. J. Org. Chem. 1991, 56, 5009–5010. [Google Scholar] [CrossRef]
- Minato, H.; Yamazaki, E.; Kobayashi, M. Reactions of Several Nucleophiles with Quaternary Salts of Nicotinamide. Chem. Lett. 1976, 5, 525–530. [Google Scholar] [CrossRef]
- Engbersen, J.F.J.; Koudijs, A.; Sleiderink, H.M.; Franssen, M.C.R. Addition of Cyanide Ion to Nicotinamide Cations in Acetonitrile. Formation of Non-Productive Charge-Transfer Complexes. J. Chem. Soc. Perkin Trans. 1990, 2, 79–83. [Google Scholar] [CrossRef]
- Makarov, M.V.; Hayat, F.; Graves, B.; Sonavane, M.; Salter, E.A.; Wierzbicki, A.; Gassman, N.R.; Migaud, M.E. Chemical and Biochemical Reactivity of the Reduced Forms of Nicotinamide Riboside. ACS Chem. Biol. 2021, 16, 604–614. [Google Scholar] [CrossRef] [PubMed]
- You, W.; Hugar, K.M.; Selhorst, R.C.; Treichel, M.; Peltier, C.R.; Noonan, K.J.T.; Coates, G.W. Degradation of Organic Cations under Alkaline Conditions. J. Org. Chem. 2021, 86, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.; Morrison, D.L. The Alkaline Reaction of Nicotinamide Adenine Dinucleotide, a New Transient Intermediate. J. Biol. Chem. 1970, 245, 4519–4524. [Google Scholar] [CrossRef]
- Johnson, S.L.; Guilbert, C.C. Isolation and Characterization of the Fluorescent Alkali Product from Diphosphopyridine Nucleotide. Biochemistry 1971, 10, 2313–2316. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Orgel, L.E. Enzymatic Synthesis of Polymers Containing Nicotinamide Mononucleotide. Nucleic Acids Res. 1995, 23, 3742–3749. [Google Scholar] [CrossRef] [PubMed]
- Zeynizadeh, B.; Dilmaghani, K.A.; Roozijoy, A. Aromatization of Hantzsch Ester 1,4-Dihydropyridines with Iodine under Normal Conditions and Ultrasound Irradiation. J. Chin. Chem. Soc. 2005, 52, 1001–1004. [Google Scholar] [CrossRef]
- Cai, X.-H.; Yang, H.-J.; Zhang, G.-L. Aromatization of 1,4-Dihydropyridines with Selenium Dioxide. Can. J. Chem. 2005, 83, 273–275. [Google Scholar] [CrossRef]
- Hayashi, M.; Nakamichi, N.; Kawashita, Y. Activated Carbon-Promoted Oxidative Aromatization of Hantzsch 1,4-Dihydropyridines and 1,3,5-Trisubstituted Pyrazolines Using Molecular Oxygen. Synthesis 2004, 7, 1015–1020. [Google Scholar] [CrossRef]
- Abdoli-Senejani, M.; Karami, K. Ultrasound-Assisted Heterogeneous Oxidation of 1,4-Dihydropyridines. Org. Prep. Proced. Int. 2020, 52, 274–281. [Google Scholar] [CrossRef]
- Boecker, R.H.; Guengerich, F.P. Oxidation of 4-Aryl- and 4-Alkyl-Substituted 2,6-Dimethyl-3,5-Bis(Alkoxycarbonyl)-1,4-Dihydropyridines by Human Liver Microsomes and Immunochemical Evidence for the Involvement of a Form of Cytochrome P-450. J. Med. Chem. 1986, 29, 1596–1603. [Google Scholar] [CrossRef]
- Zarei, A.; Khazdooz, L.; Enayati, M.; Madarshahian, S.; Wooster, T.J.; Ufheil, G.; Abbaspourrad, A. Dihydronicotinamide Riboside: Synthesis from Nicotinamide Riboside Chloride, Purification and Stability Studies. RSC Adv. 2021, 11, 21036–21047. [Google Scholar] [CrossRef] [PubMed]
- Sollenberger, P.Y.; Martin, R.B. Mechanism of Enamine Hydrolysis. J. Am. Chem. Soc. 1970, 92, 4261–4270. [Google Scholar] [CrossRef]
- Maas, W.; Janssen, M.J.; Stamhuis, E.J.; Wynberg, H. Mechanism of Enamine Reactions. IV. The Hydrolysis of Tertiary Enamines in Acidic Medium. J. Org. Chem. 1967, 32, 1111–1115. [Google Scholar] [CrossRef]
- Johnston, C.C.; Gardner, J.L.; Suelter, C.H.; Metzler, D.E. Acid-Catalyzed Addition of Water to 1 4-Dihydronicotinamide Derivatives. Biochemistry 1963, 2, 689–696. [Google Scholar] [CrossRef] [PubMed]
- Oppenheimer, N.J.; Kaplan, N.O. The Alpha Beta Epimerization of Reduced Nicotinamide Adenine Dinucleotide. Arch. Biochem. Biophys. 1975, 166, 526–535. [Google Scholar] [CrossRef]
- Johnson, S.L.; Tuazon, P.T. Acid-Catalyzed Hydration of Reduced Nicotinamide Adenine Dinucleotide and Its Analogues. Biochemistry 1977, 16, 1175–1183. [Google Scholar] [CrossRef]
- Branlant, G.; Eiler, B.; Biellmann, J. A Word of Caution: 1,4,5,6-Tetrahydronicotinamide Adenine Dinucleotide (Phosphate) Should Be Used with Care in Acidic and Neutral Media. Anal. Biochem. 1982, 125, 264–268. [Google Scholar] [CrossRef]
- Ammon, H.L.; Jensen, L.H. The “Dimeric Acid Product” of 1-Methyl-1,4-Dihydronicotinamide. J. Am. Chem. Soc. 1966, 88, 613–614. [Google Scholar] [CrossRef]
- Ilic, S.; Kadel, U.P.; Basdogan, Y.; Keith, J.A.; Glusac, K.D. Thermodynamic Hydricities of Biomimetic Organic Hydride Donors. J. Am. Chem. Soc. 2018, 140, 4569–4579. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.-Q.; Tan, Y.; Cao, C.-T. Thermodynamic Diagnosis of the Properties and Mechanism of Dihydropyridine-Type Compounds as Hydride Source in Acetonitrile with “Molecule ID Card”. J. Phys. Chem. B 2010, 114, 2058–2075. [Google Scholar] [CrossRef]
- Anderson, A.G.; Berkelhammer, G. A Study of the Primary Acid Reaction on Model Compounds of Reduced Diphosphopyridine Nucleotide. J. Am. Chem. Soc. 1958, 80, 992–999. [Google Scholar] [CrossRef]
- Nowak, C.; Pick, A.; Csepei, L.; Sieber, V. Characterization of Biomimetic Cofactors According to Stability, Redox Potentials, and Enzymatic Conversion by Nadh Oxidase from Lactobacillus Pentosus. ChemBioChem 2017, 18, 1944–1949. [Google Scholar] [CrossRef]
- Zachos, I.; Döring, M.; Tafertshofer, G.; Simon, R.C.; Sieber, V. Carba Nicotinamide Adenine Dinucleotide Phosphate: Robust Cofactor for Redox Biocatalysis. Angew. Chem. Int. Ed. 2021, 60, 14701–14706. [Google Scholar] [CrossRef] [PubMed]
- Haynes, L.J.; Todd, A.R. 66. Codehydrogenases. Part I. The Synthesis of Dihydronicotinamide-D-Ribofuranoside [N-D-Ribofuranosidyl-1:2(or 6)-Dihydronicotinamide]. J. Chem. Soc. 1950, 0, 303–308. [Google Scholar] [CrossRef]
- Haykes, L.J.; Hughes, N.A.; Kenner, G.W.; Todd, A. 734. Codehydrogenases. Part II. A Synthesis of Nicotinamide Nucleotide. J. Chem. Soc. 1957, 0, 3727–3732. [Google Scholar] [CrossRef]
- Mikhailopulo, I.A.; Miroshnikov, A.I. New Trends in Nucleoside Biotechnology. Acta Nat. 2010, 2, 36–58. [Google Scholar] [CrossRef]
- Lee, J.; Churchil, H.; Choi, W.-B.; Lynch, J.E.; Roberts, F.E.; Volante, R.P.; Reider, P.J. A Chemical Synthesis of Nicotinamide Adenine Dinucleotide (NAD+). Chem. Commun. 1999, 8, 729–730. [Google Scholar] [CrossRef]
- Jarman, M.; Ross, W.C.J. 4-Substituted Nicotinic Acids and Nicotinamides. Part II. The Preparation of 4-Methylnicotinamide Riboside. J. Chem. Soc. C Org. 1969, 2, 199–203. [Google Scholar] [CrossRef]
- Tanimori, S.; Ohta, T.; Kirihata, M. An Efficient Chemical Synthesis of Nicotinamide Riboside (NAR) and Analogues. Bioorg. Med. Chem. Lett. 2002, 12, 1135–1137. [Google Scholar] [CrossRef] [PubMed]
- Franchetti, P.; Pasqualini, M.; Petrelli, R.; Ricciutelli, M.; Vita, P.; Cappellacci, L. Stereoselective Synthesis of Nicotinamide β-Riboside and Nucleoside Analogs. Bioorg. Med. Chem. Lett. 2004, 14, 4655–4658. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Chan, N.Y.-K.; Sauve, A.A. Syntheses of Nicotinamide Riboside and Derivatives: Effective Agents for Increasing Nicotinamide Adenine Dinucleotide Concentrations in Mammalian Cells. J. Med. Chem. 2007, 50, 6458–6461. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Sauve, A.A. Synthesis of β-Nicotinamide Riboside Using an Efficient Two-Step Methodology. Curr. Prot. Nucleic Acid. Chem. 2017, 71, 14.14.1–14.14.9. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, M.R.; Morton, R.K.; Naylor, R. 98. Synthesis of Glycosylpyridinium Compounds from Glycosylamines and from Glycosyl Halides. J. Chem. Soc. 1965, 0, 610–615. [Google Scholar] [CrossRef]
- Makarov, M.V.; Harris, N.W.; Rodrigues, M.; Migaud, M.E. Scalable Syntheses of Traceable Ribosylated NAD+ Precursors. Org. Biomol. Chem. 2019, 17, 8716–8720. [Google Scholar] [CrossRef] [PubMed]
- Carelli, V.; Liberatore, F.; Scipione, L.; Di Rienzo, B.; Tortorella, S. Dithionite Adducts of Pyridinium Salts: Regioselectivity of Formation and Mechanisms of Decomposition. Tetrahedron 2005, 61, 10331–10337. [Google Scholar] [CrossRef]
- Paul, C.E.; Arends, I.W.C.E.; Hollmann, F. Is Simpler Better? Synthetic Nicotinamide Cofactor Analogues for Redox Chemistry. ACS Catal. 2014, 4, 788–797. [Google Scholar] [CrossRef]
- Szczepankiewicz, B.G.; Dai, H.; Koppetsch, K.J.; Qian, D.; Jiang, F.; Mao, C.; Perni, R.B. Synthesis of Carba-NAD and the Structures of Its Ternary Complexes with Sirt3 and Sirt5. J. Org. Chem. 2012, 77, 7319–7329. [Google Scholar] [CrossRef]
- Akabane-Nakata, M.; Chickering, T.; Harp, J.M.; Schlegel, M.K.; Matsuda, S.; Egli, M.; Manoharan, M. RNAs Containing Carbocyclic Ribonucleotides. Org. Lett. 2022, 24, 525–530. [Google Scholar] [CrossRef]
- Largy, E.; Liu, W.; Hasan, A.; Perrin, D.M. Base-Pairing Behavior of a Carbocyclic Janus-AT Nucleoside Analogue Capable of Recognizing A and T within a DNA Duplex. ChemBioChem 2013, 14, 2199–2208. [Google Scholar] [CrossRef] [PubMed]
- Rydzik, A.M.; Balk, R.; Koegler, M.; Steinle, T.; Riether, D.; Gottschling, D. Access to 1′-Amino Carbocyclic Phosphoramidite to Enable Postsynthetic Functionalization of Oligonucleotides. Org. Lett. 2021, 23, 6735–6739. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Zhang, X.-N.; Nasertorabi, F.; Cheng, Q.; Pei, H.; Louie, S.G.; Stevens, R.C.; Zhang, Y. Facile Chemoenzymatic Synthesis of a Novel Stable Mimic of NAD+. Chem. Sci. 2018, 9, 8337–8342. [Google Scholar] [CrossRef] [PubMed]
- Zang, H.; Lou, J.; Jiao, S.; Li, H.; Du, Y.; Wang, J. Valorization of Chitin Derived N-Acetyl-D-Glucosamine into High Valuable N-Containing 3-Acetamido-5-Acetylfuran Using Pyridinium-Based Ionic Liquids. J. Mol. Liq. 2021, 330, 115667. [Google Scholar] [CrossRef]
- Craig, J.H.; Huang, P.C.; Scott, T.G.; Leonard, N.J. Synthetic Spectroscopic Models Related to Coenzymes and Base Pairs. X. Quaternized Bisnicotinamides. J. Am. Chem. Soc. 1972, 94, 5872–5879. [Google Scholar] [CrossRef] [PubMed]
- Knox, R.J.; Jenkins, T.C.; Hobbs, S.M.; Chen, S.; Melton, R.G.; Burke, P.J. Bioactivation of 5-(Aziridin-1-Yl)-2,4-Dinitrobenzamide (CB 1954) by Human NAD(P)H Quinone Oxidoreductase 2: A Novel Co-Substrate-Mediated Antitumor Prodrug Therapy1. Cancer Res 2000, 60, 4179–4186. [Google Scholar] [PubMed]
- Malver, O.; Sebastian, M.J.; Oppenheimer, N.J. Alteration in Substrate Specificity of Horse Liver Alcohol Dehydrogenase by an Acyclic Nicotinamide Analog of NAD+. DNA Repair. 2014, 23, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Bušić, V.; Vrandečić, K.; Siber, T.; Roca, S.; Tomičić, V.; Sokač, D.G. Novel Synthetic Routes to Quaternary Pyridinium Salts and Their Antifungal Activity. Croat. Chem. Acta 2022, 95, 31–38. [Google Scholar] [CrossRef]
- Chennamaneni, S.R.; Vobalaboina, V.; Garlapati, A. Quaternary Salts of 4,3′ and 4,4′ Bis-Pyridinium Monooximes: Synthesis and Biological Activity. Bioorg. Med. Chem. Lett. 2005, 15, 3076–3080. [Google Scholar] [CrossRef]
- Friedman, O.M.; Pollak, K.; Khedouri, E. 1-(β-Chloroethyl)-3-Carbamylpyridinium Chloride. Prototype of a New Class of Latently Cytotoxic Potential Antitumor Agents. J. Med. Chem. 1963, 6, 462–463. [Google Scholar] [CrossRef]
- Plapp, B.V.; Woenckhaus, C.; Pfleiderer, G. Evaluation of N1-(Ω-Bromoacetamidoalkyl)Nicotinamides as Inhibitors of Dehydrogenases. Arch. Biochem. Biophys. 1968, 128, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Robins, M.J.; Hatfield, P.W. Nucleic Acid Related Compounds. 37. Convenient and High-Yield Syntheses of N-[(2-Hydroxyethoxy)Methyl] Heterocycles as “Acyclic Nucleoside” Analogues. Can. J. Chem. 1982, 60, 547–553. [Google Scholar] [CrossRef]
- Štefko, M.; Slavětínská, L.; Klepetářová, B.; Hocek, M. General and Modular Synthesis of Isomeric 5-Substituted Pyridin-2-yl and 6-Substituted Pyridin-3-yl C-Ribonucleosides Bearing Diverse Alkyl, Aryl, Hetaryl, Amino, Carbamoyl, and Hydroxy Groups. J. Org. Chem. 2011, 76, 6619–6635. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Liu, S.; Xi, Y.; Li, H.; Yang, K.; Cheng, Z.; Wang, W.; Zhang, Y. Highly Stereoselective Synthesis of Aryl/Heteroaryl-C-Nucleosides Via the Merger of Photoredox and Nickel Catalysis. Chem. Commun. 2019, 55, 14657–14660. [Google Scholar] [CrossRef]
- Maeba, I.; Iwata, K.; Usami, F.; Furukawa, H. C-Nucleosides. 1. Synthesis of 3-(β-D-Ribofuranosyl)Pyridazines. J. Org. Chem. 1983, 48, 2998–3002. [Google Scholar] [CrossRef]
- Singh, I.; Seitz, O. Diastereoselective Synthesis of β-Aryl-C-Nucleosides from 1,2-Anhydrosugars. Org. Lett. 2006, 8, 4319–4322. [Google Scholar] [CrossRef] [PubMed]
- Brotschi, C.; Häberli, A.; Leumann, C.J. A Stable DNA Duplex Containing a Non-Hydrogen-Bonding and Non-Shape-Complementary Base Couple: Interstrand Stacking as the Stability Determining Factor. Angew. Chem. Int. Ed. 2001, 40, 3012–3014. [Google Scholar] [CrossRef]
- Franchetti, P.; Cappellacci, L.; Grifantini, M.; Barzi, A.; Nocentini, G.; Yang, H.; O’Connor, A.; Jayaram, H.N.; Carrell, C.; Goldstein, B.M. Furanfurin and Thiophenfurin: Two Novel Tiazofurin Analogs. Synthesis, Structure, Antitumor Activity, and Interactions with Inosine Monophosphate Dehydrogenase. J. Med. Chem. 1995, 38, 3829–3837. [Google Scholar] [CrossRef]
- Kabat, M.M.; Pankiewicz, K.W.; Watanabe, K.A. Nucleosides. 141. Synthesis of 5-β-D-Ribofuranosylnicotinamide and Its N-Methyl Derivative. The Isosteric and Isoelectronic Analogs of Nicotinamide Nucleoside. J. Med. Chem. 1987, 30, 924–927. [Google Scholar] [CrossRef]
- Pankiewicz, K.W.; Watanabe, K.A.; Lesiak-Watanabe, K.; Goldstein, B.M.; Jayaram, H.N. The Chemistry of Nicotinamide Adenine Dinucleotide (NAD) Analogues Containing C-Nucleosides Related to Nicotinamide Riboside. Curr. Med. Chem. 2002, 9, 733–741. [Google Scholar] [CrossRef]
- Höfer, K.; Abele, F.; Schlotthauer, J.; Jäschke, A. Synthesis of 5′-NAD-Capped RNA. Bioconjugate Chem. 2016, 27, 874–877. [Google Scholar] [CrossRef]
- Göckel, A.; Richert, C. Synthesis of an Oligonucleotide with a Nicotinamide Mononucleotide Residue and Its Molecular Recognition in DNA Helices. Org. Biomol. Chem. 2015, 13, 10303–10309. [Google Scholar] [CrossRef]
- Kjellstrand, M.; Broo, K.; Andersson, L.; Farre, C.; Nilsson, Å.; Baltzer, L. The Site-Selective Incorporation of a NAD+ Cofactor Mimic into a Folded Helix–Loop–Helix Polypeptide Motif. J. Chem. Soc. Perkin Trans. 1997, 2, 2745–2750. [Google Scholar] [CrossRef]
- Zhang, B.; Zhu, X.-Q.; Lu, J.-Y.; He, J.; Wang, P.G.; Cheng, J.-P. Polysiloxane-Supported NAD(P)H Model 1-Benzyl-1,4-Dihydronicotinamide: Synthesis and Application in the Reduction of Activated Olefins. J. Org. Chem. 2003, 68, 3295–3298. [Google Scholar] [CrossRef]
- Chen, Z.; Kwong, A.K.Y.; Yang, Z.; Zhang, L.; Lee, H.C.; Zhang, L. Cheminform Abstract: Studies of the Synthesis of Nicotinamide Nucleoside and Nucleotide Analogues and Their Inhibitions Towards CD38 NADase. ChemInform 2012, 43. [Google Scholar] [CrossRef]
- Kovarik, Z.; Kalisiak, J.; Hrvat, N.M.; Katalinić, M.; Zorbaz, T.; Žunec, S.; Green, C.; Radić, Z.; Fokin, V.V.; Sharpless, K.B.; et al. Reversal of Tabun Toxicity Enabled by a Triazole-Annulated Oxime Library—Reactivators of Acetylcholinesterase. Chem. Eur. J. 2019, 25, 4100–4114. [Google Scholar] [CrossRef] [PubMed]
- Brunner, H.; Ackermann, S.; Lucks, S.; Wu, J.; Adolf, J. Aqueous Composition for Depositing a Cobalt Deposit and Method for Electrolytically Depositing Such a Deposit. WO Patent 2019/013762 A1, January 17, 2019. [Google Scholar]
- Mutschler, H.; Holliger, P. Non-Canonical 3′-5′ Extension of RNA with Prebiotically Plausible Ribonucleoside 2′,3′-Cyclic Phosphates. J. Am. Chem. Soc. 2014, 136, 5193–5196. [Google Scholar] [CrossRef] [PubMed]
- Tereshko, V.; Gryaznov, S.; Egli, M. Consequences of Replacing the DNA 3‘-Oxygen by an Amino Group: High-Resolution Crystal Structure of a Fully Modified N3‘ → P5‘ Phosphoramidate DNA Dodecamer Duplex. J. Am. Chem. Soc. 1998, 120, 269–283. [Google Scholar] [CrossRef]
- Kam, B.L.; Oppenheimer, N.J. Synthesis of a New Class of D-Aldopentofuranosylamines, the 5-O-Trityl-D-Aldopentofuranosylamines. Carbohyd Res. 1979, 77, 275–280. [Google Scholar] [CrossRef]
- Livingston, D.; McKearin, J.M.; Szczepankiewicz, B.; Kremsky, J.N. Nicotinamide Mononucleotide Derivatives and Their Uses for Treating Diseases and Improving Cell and Tissue Survival. WO Patent 2017/024255 A1, 9 February 2017. [Google Scholar]
- Hayat, F.; Migaud, M.E. Nicotinamide Riboside-Amino Acid Conjugates That Are Stable to Purine Nucleoside Phosphorylase. Org. Biomol. Chem. 2020, 18, 2877–2885. [Google Scholar] [CrossRef]
- Xu, H.; Chen, F. Method for Preparing Beta-Nicotinamide Mononucleotide. CN Patent 114685582 A, 7 January 2022. [Google Scholar]
- Matteucci; Caruthers, M. The Use of Zinc Bromide for Removal of Dimethoxytrityl Ethers from Deoxynucleosides. Tetrahedron Lett. 1980, 21, 3243–3246. [Google Scholar] [CrossRef]
- Berridge, M.; Tan, A. Characterization of the Cellular Reduction of 3-(4,5-Dimethylthiazol-2-Yl)-2,5-Diphenyltetrazolium Bromide (MTT): Subcellular Localization, Substrate Dependence, and Involvement of Mitochondrial Electron Transport in MTT Reduction. Arch. Biochem. Biophys. 1993, 303, 474–482. [Google Scholar] [CrossRef]
- Alul, R.H.; Singman, C.N.; Zhang, G.; Letsinger, R.L. Oxalyl-CPG: A Labile Support for Synthesis of Sensitive Oligonucleotide Derivatives. Nucleic Acids Res. 1991, 19, 1527–1532. [Google Scholar] [CrossRef]
- Sambandam, A.; Greenberg, M.M. The Effects of 5R-5,6-Dihydro-5-Hydroxythymidine on Duplex DNA Stability and Structure. Nucleic Acids Res. 1999, 27, 3597–3602. [Google Scholar] [CrossRef]
- Guerlavais-Dagland, T.; Meyer, A.; Imbach, J.; Morvan, F. Fluoride-Labile Protecting Groups for the Synthesis of Base-Sensitive Methyl-SATE Oligonucleotide Prodrugs. Eur. J. Org. Chem. 2003, 2003, 2327–2335. [Google Scholar] [CrossRef]
- Araki, M.; Okuno, Y.; Hara, Y.; Sugiura, Y. Allosteric Regulation of a Ribozyme Activity through Ligand-Induced Conformational Change. Nucleic Acids Res. 1998, 26, 3379–3384. [Google Scholar] [CrossRef]
- Strohbach, D.; Novak, N.; Müller, S. Redox-Active Riboswitching: Allosteric Regulation of Ribozyme Activity by Ligand-Shape Control. Angew. Chem. Int. Ed. 2006, 45, 2127–2129. [Google Scholar] [CrossRef]
- Chen, A.G.; Sudarsan, N.; Breaker, R.R. Mechanism for Gene Control by a Natural Allosteric Group I Ribozyme. RNA 2011, 17, 1967–1972. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wenzek, F.; Biallas, A.; Müller, S. Nicotinamide Riboside: What It Takes to Incorporate It into RNA. Molecules 2024, 29, 3788. https://doi.org/10.3390/molecules29163788
Wenzek F, Biallas A, Müller S. Nicotinamide Riboside: What It Takes to Incorporate It into RNA. Molecules. 2024; 29(16):3788. https://doi.org/10.3390/molecules29163788
Chicago/Turabian StyleWenzek, Felix, Alexander Biallas, and Sabine Müller. 2024. "Nicotinamide Riboside: What It Takes to Incorporate It into RNA" Molecules 29, no. 16: 3788. https://doi.org/10.3390/molecules29163788