
Exploring Aromaticity Effects on Electronic Transport in Cyclo[n]carbon Single-Molecule Junctions

 , , ,
, , ,
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
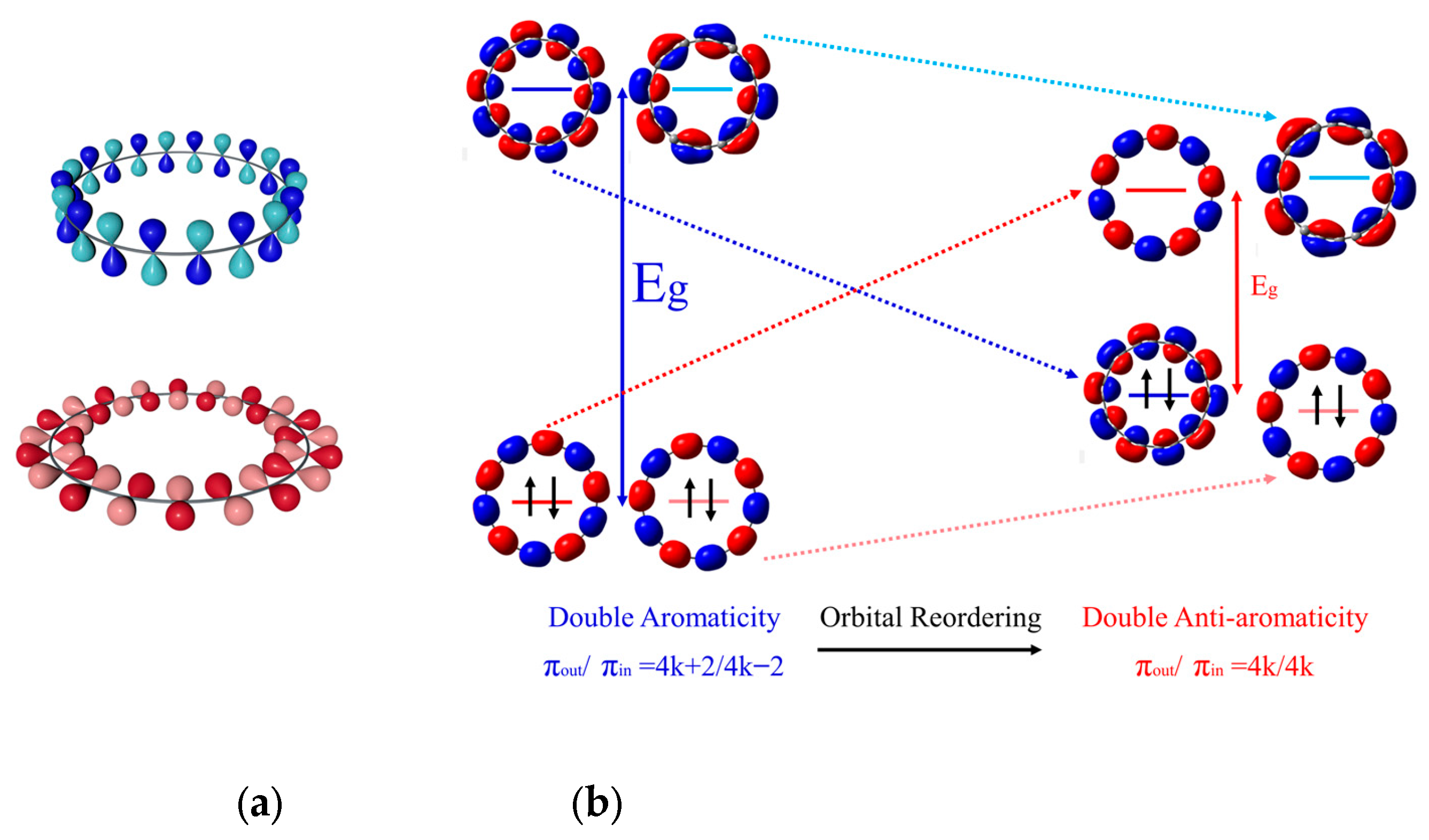
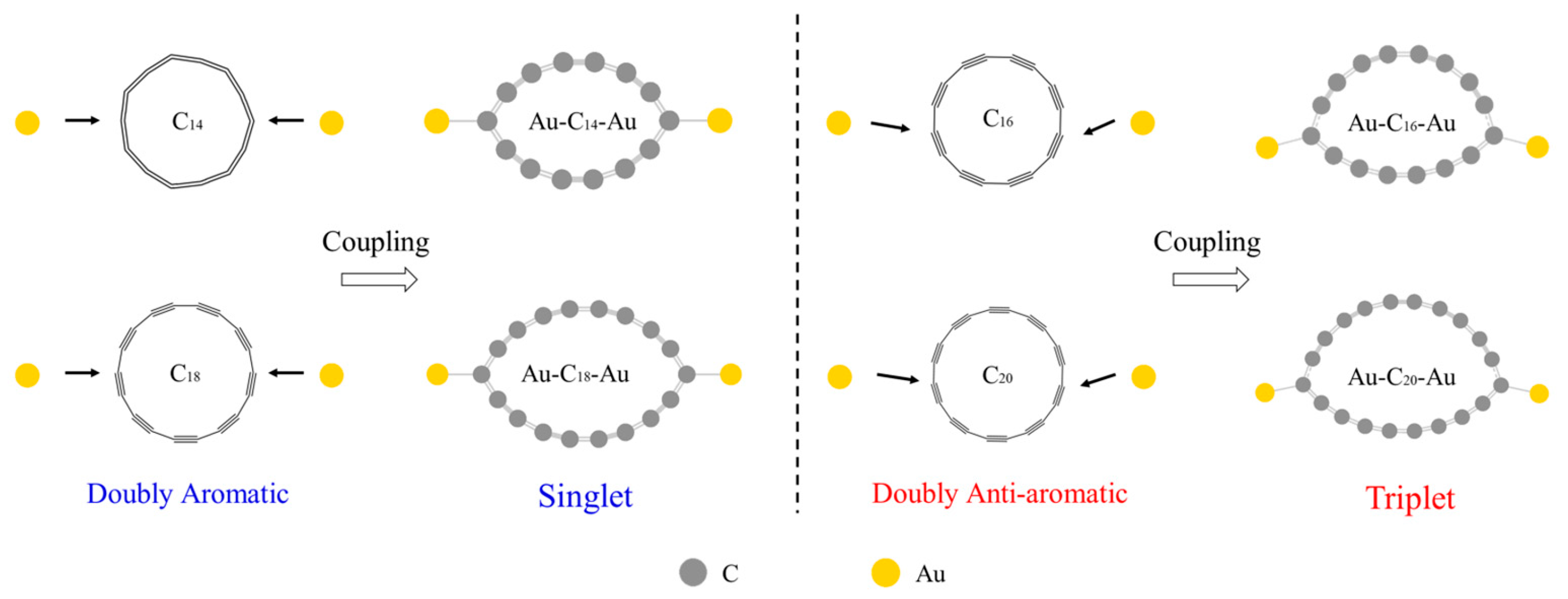
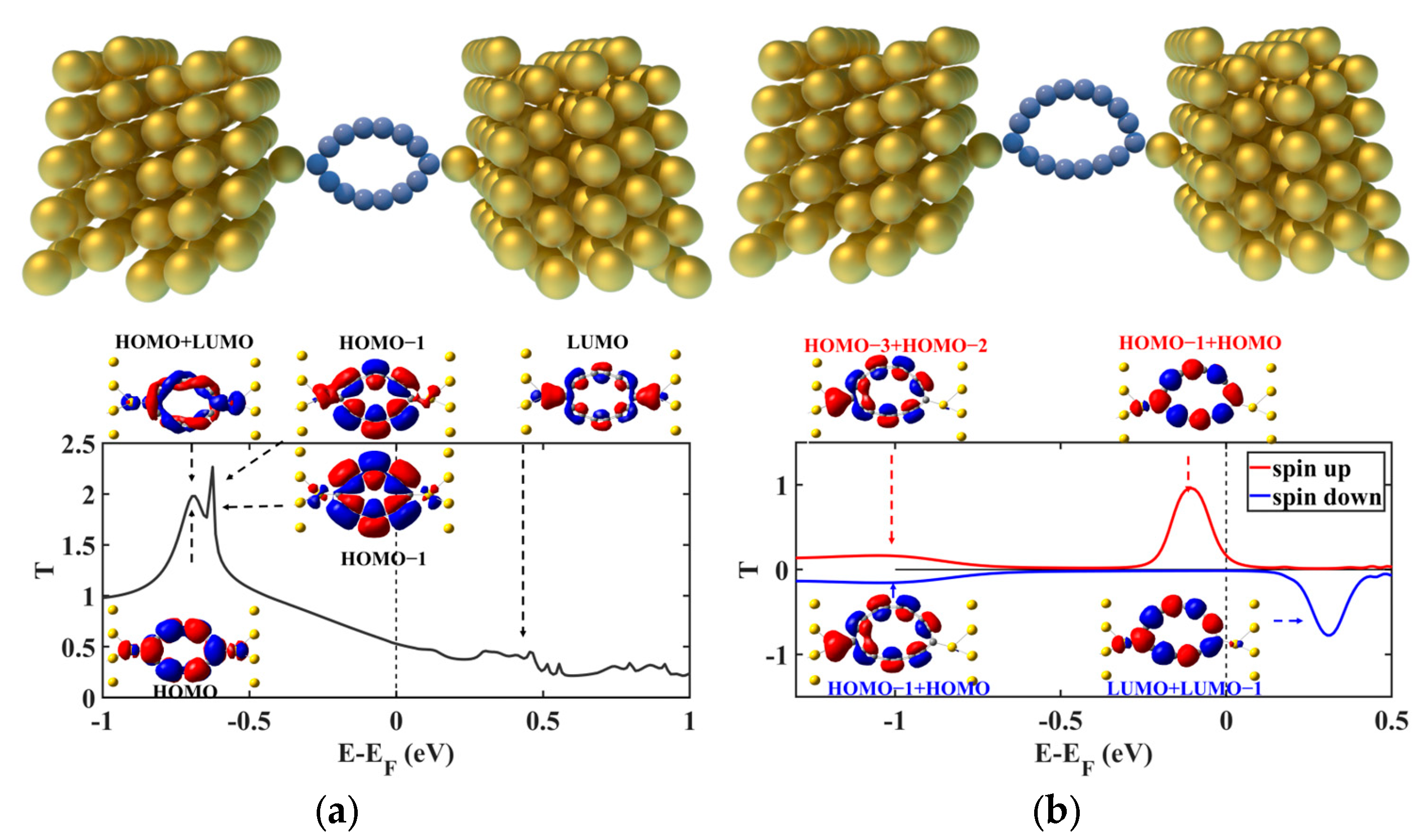
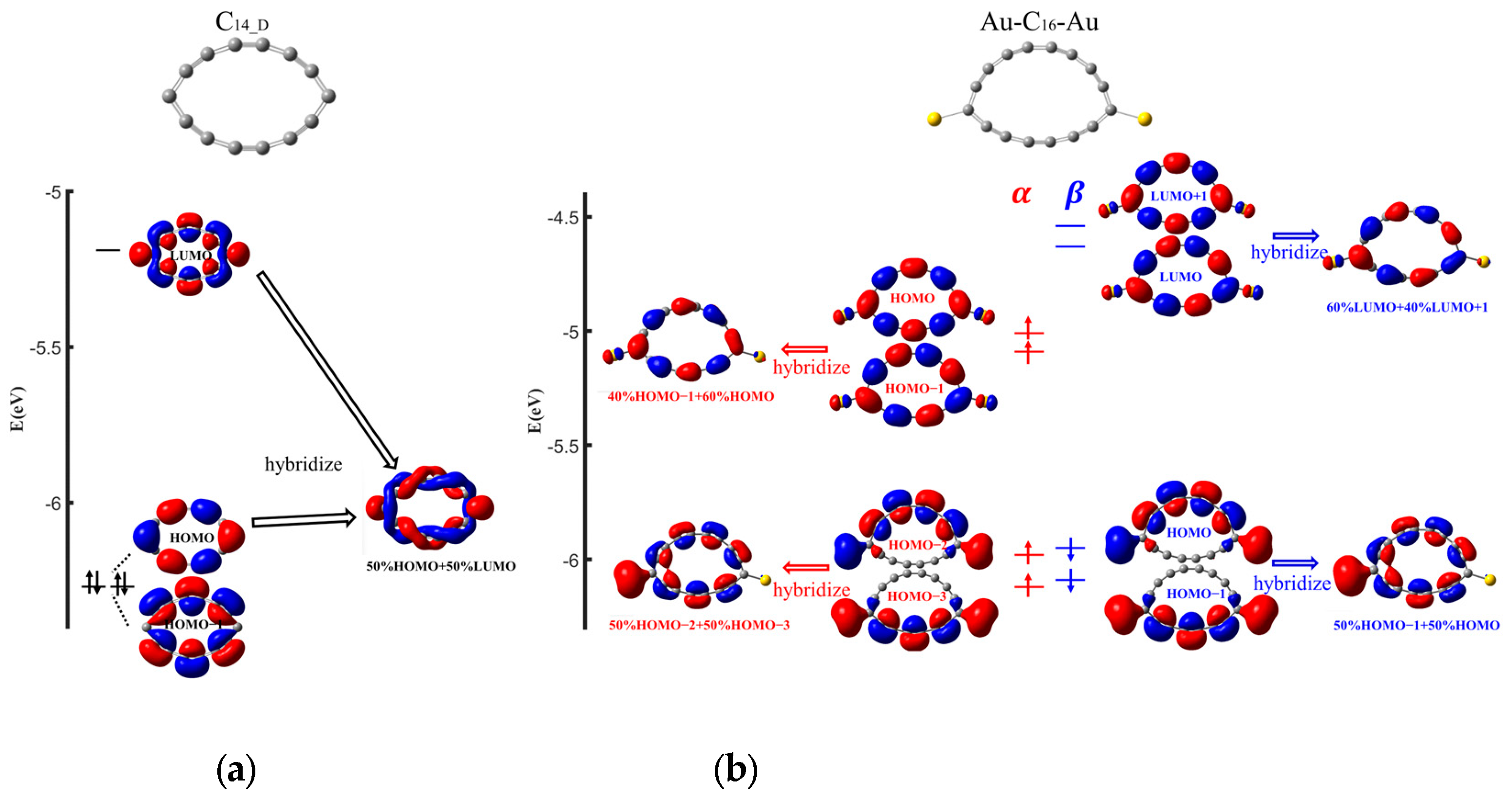
2. Results and Discussion
3. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric field effect in atomically thin carbon films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [PubMed]
- Kroto, H.W.; Heath, J.R.; O’Brien, S.C.; Curl, R.F.; Smalley, R.E. C60: Buckminsterfullerene. Nature 1985, 318, 162–163. [Google Scholar] [CrossRef]
- Iijima, S. Helical microtubules of graphitic carbon. Nature 1991, 354, 56–58. [Google Scholar] [CrossRef]
- Diederich, F.; Rubin, Y.; Knobler, C.B.; Whetten, R.L.; Schriver, K.E.; Houk, K.N.; Li, Y. All-carbon molecules: Evidence for the generation of cyclo[18]carbon from a stable organic precursor. Science 1989, 245, 1088–1090. [Google Scholar] [CrossRef] [PubMed]
- von Helden, G.; Gotts, N.G.; Bowers, M.T. Experimental evidence for the formation of fullerenes by collisional heating of carbon rings in the gas phase. Nature 1993, 363, 60–63. [Google Scholar] [CrossRef]
- McElvany, S.W.; Ross, M.M.; Goroff, N.S.; Diederich, F. Cyclocarbon coalescence: Mechanisms for tailor-made fullerene formation. Science 1993, 259, 1594–1596. [Google Scholar] [CrossRef] [PubMed]
- Diederich, F.; Kivala, M. All-carbon scaffolds by rational design. Adv. Mater. 2010, 22, 803–812. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, K.; Scriven, L.M.; Schulz, F.; Gawel, P.; Gross, L.; Anderson, H.L. An sp-hybridized molecular carbon allotrope, cyclo[18]carbon. Science 2019, 365, 1299–1301. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Albrecht, F.; Rončević, I.; Ettedgui, I.; Kumar, P.; Scriven, L.M.; Christensen, K.E.; Mishra, S.; Righetti, L.; Rossmannek, M.; et al. On-surface synthesis of a doubly anti-aromatic carbon allotrope. Nature 2023, 623, 977–981. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Zheng, W.; Kang, F.; Gao, W.; Xu, W. On-surface synthesis and characterization of anti-aromatic cyclo[12]carbon and cyclo[20]carbon. Res. Sq. 2023. [Google Scholar] [CrossRef]
- Sun, L.; Zheng, W.; Gao, W.; Kang, F.; Zhao, M.; Xu, W. On-surface synthesis of aromatic cyclo[10]carbon and cyclo[14]carbon. Nature 2023, 623, 972–976. [Google Scholar] [CrossRef]
- Brémond, E.; Pérez-Jiménez, A.J.; Adamo, C.; Sancho-García, J.C. Stability of the polyynic form of C18, C22, C26, and C30 nanorings: A challenge tackled by range-separated double-hybrid density functionals. Phys. Chem. Chem. Phys. 2022, 24, 4515–4525. [Google Scholar] [CrossRef]
- Pereira, Z.S.; da Silva, E.Z. Spontaneous symmetry breaking in cyclo[18]carbon. J. Phys. Chem. A 2020, 124, 1152–1157. [Google Scholar] [CrossRef]
- Zhu, B.-C.; Liu, C.-J.; Deng, P.-J.; Zhao, J.; Zhang, J.; Zeng, L.; Liao, Y.-H.; Bao, L.; Bao, J. DFT-based study on the differences between odd and even Cn (n = 6–31) ring clusters. Results Phys. 2023, 52, 106852. [Google Scholar] [CrossRef]
- Zheng, P.; Zhang, L.; Zhang, X.; Ma, Y.; Jiang, Y.; Li, H. Parallel-self-assembling stack, center-capture effect, and reactivity-enhancing effect of n-layer (n = 1, 2, 3) cyclo[18]carbon. ACS Nano 2022, 16, 21345–21355. [Google Scholar] [CrossRef]
- Baryshnikov, G.V.; Valiev, R.R.; Nasibullin, R.T.; Sundholm, D.; Kurten, T.; Ågren, H. Aromaticity of even-number cyclo[n]carbons (n = 6–100). J. Phys. Chem. A 2020, 124, 10849–10855. [Google Scholar] [CrossRef]
- Fowler, P.W.; Mizoguchi, N.; Bean, D.E.; Havenith, R.W.A. Double aromaticity and ring currents in all-carbon rings. Chem.—Eur. J. 2009, 15, 6964–6972. [Google Scholar] [CrossRef]
- Baryshnikov, G.V.; Valiev, R.R.; Valiulina, L.I.; Kurtsevich, A.E.; Kurtén, T.; Sundholm, D.; Pittelkow, M.; Zhang, J.; Ågren, H. Odd-number cyclo[n]carbons sustaining alternating aromaticity. J. Phys. Chem. A 2022, 126, 2445–2452. [Google Scholar] [CrossRef]
- Dai, C.; Chen, D.; Zhu, J. Achieving adaptive aromaticity in cyclo[10]carbon by screening cyclo[n]carbon (n=8−24). Chem.—Asian J. 2020, 15, 2187–2191. [Google Scholar] [CrossRef]
- Baryshnikov, G.V.; Valiev, R.R.; Kuklin, A.V.; Sundholm, D.; Ågren, H. Cyclo[18]carbon: Insight into electronic structure, aromaticity, and surface coupling. J. Phys. Chem. Lett. 2019, 10, 6701–6705. [Google Scholar] [CrossRef]
- Liu, Z.; Lu, T.; Chen, Q. An sp-hybridized all-carboatomic ring, cyclo[18]carbon: Bonding character, electron delocalization, and aromaticity. Carbon 2020, 165, 468–475. [Google Scholar] [CrossRef]
- Lu, T.; Chen, Q. Ultrastrong regulation effect of the electric field on the all-carbon atomic ring cyclo[18]Carbon. ChemPhysChem 2021, 22, 386–395. [Google Scholar] [CrossRef]
- Liu, Z.; Lu, T.; Yuan, A.; Wang, X.; Chen, Q.; Yan, X. Remarkable size effect on photophysical and nonlinear optical properties of all-carboatomic rings, cyclo[18]carbon and its analogues. Chem.—Asian J. 2021, 16, 2267–2271. [Google Scholar] [CrossRef]
- Nandi, A.; Solel, E.; Kozuch, S. Carbon tunneling in the automerization of cyclo[18]carbon. Chem.—Eur. J. 2020, 26, 625–628. [Google Scholar] [CrossRef]
- Iyakutti, K.; Surya, V.J.; Lakshmi, I.; Rajeswarapalanichamy, R.; Kawazoe, Y. 18 and 12—Member carbon rings (cyclo[n]carbons)—A density functional study. Mater. Sci. Eng. B 2021, 263, 114895. [Google Scholar] [CrossRef]
- Hassani, N.; Hassani, M.R.; Neek-Amal, M. Catalytic properties of cyclo-carbon clusters: An investigation on o2 activation and CO oxidation. Surf. Sci. 2022, 720, 122050. [Google Scholar] [CrossRef]
- Tang, C.; Xu, D.; Ouyang, G. Cross-plane transport in cyclo[18]carbon-based molecular devices. Appl. Phys. Lett. 2023, 122, 044101. [Google Scholar] [CrossRef]
- Xiong, S.; Dong, X.; Xie, L.; Guan, Z.; Long, M.; Chen, T. Spin-resolved transport of multifunctional C(18)molecule-based nanodevices: A first-principles study. J. Phys. Condens. Matter 2023, 35, 395302. [Google Scholar] [CrossRef]
- Zhang, L.; Li, H.; Feng, Y.P.; Shen, L. Diverse transport behaviors in cyclo[18]carbon-based molecular devices. J. Phys. Chem. Lett. 2020, 11, 2611–2617. [Google Scholar] [CrossRef]
- Rojas, C.; León, A.; Pacheco, M.; Chico, L.; Orellana, P.A. Transport signatures of few-atom carbon rings. Phys. Chem. Chem. Phys. 2022, 24, 15973–15981. [Google Scholar] [CrossRef]
- Hou, L.; Hu, H.; Yang, G.; Ouyang, G. Giant switching effect and spintronic transport properties in cyclo[18]carbon-based molecular devices. Phys. Status Solidi—Rapid Res. Lett. 2021, 15, 2000582. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Meir, Y.; Wingreen, N.S. Landauer formula for the current through an interacting electron region. Phys. Rev. Lett. 1992, 68, 2512–2515. [Google Scholar] [CrossRef]
- Xue, Y.; Datta, S.; Ratner, M.A. First-principles based matrix Green’s function approach to molecular electronic devices: General formalism. J. Chem. Phys. 2002, 281, 151–170. [Google Scholar] [CrossRef]
- Brandbyge, M.; Mozos, J.-L.; Ordejón, P.; Taylor, J.; Stokbro, K. Density-functional method for nonequilibrium electron transport. Phys. Rev. B 2002, 65, 165401. [Google Scholar] [CrossRef]
- Rocha, A.R.; García-suárez, V.M.; Bailey, S.W.; Lambert, C.J.; Ferrer, J.; Sanvito, S. Towards molecular spintronics. Nat. Mater. 2005, 4, 335–339. [Google Scholar] [CrossRef]
- Zhao, D.; Liu, S.; Rong, C.; Zhong, A.; Liu, S. Toward understanding the isomeric stability of fullerenes with density functional theory and the information-theoretic approach. ACS Omega 2018, 3, 17986–17990. [Google Scholar] [CrossRef]
- Luo, C.; He, X.; Zhong, A.; Liu, S.; Zhao, D. What dictates alkane isomerization? A combined density functional theory and information-theoretic approach study. Theor. Chem. Acc. 2023, 142, 78. [Google Scholar] [CrossRef]
- Plattner, D.A.; Houk, K.N. C18 Is a polyyne. J. Am. Chem. Soc. 1995, 117, 4405–4406. [Google Scholar] [CrossRef]
- Arulmozhiraja, S.; Ohno, T. CCSD calculations on C14, C18, and C22 carbon clusters. J. Chem. Phys. 2008, 128, 114301. [Google Scholar] [CrossRef]
- Gaweł, P.; Foroutan-Nejad, C. Carbon rings push limits of chemical theories. Nature 2023, 623, 922–924. [Google Scholar] [CrossRef]
- Lin, J.; Wang, S.; Zhang, F.; Yang, B.; Du, P.; Chen, C.; Zang, Y.; Zhu, D. Highly efficient charge transport across carbon nanobelts. Sci. Adv. 2022, 8, eade4692. [Google Scholar] [CrossRef]
- Lv, Y.; Lin, J.; Song, K.; Song, X.; Zang, H.; Zang, Y.; Zhu, D. Single cycloparaphenylene molecule devices: Achieving large conductance modulation via tuning radial π-conjugation. Sci. Adv. 2021, 7, eabk3095. [Google Scholar] [CrossRef]
- Xin, N.; Guan, J.; Zhou, C.; Chen, X.; Gu, C.; Li, Y.; Ratner, M.A.; Nitzan, A.; Stoddart, J.F.; Guo, X. Concepts in the design and engineering of single-molecule electronic devices. Nat. Rev. Phys. 2019, 1, 211–230. [Google Scholar] [CrossRef]
- Xu, Y.; Wu, W. High-efficiency switching effect and negative differential conductance in cyclo[18]carbon–graphene nanoribbon junction. J. Appl. Phys. 2020, 128, 194303. [Google Scholar] [CrossRef]
- Paulsson, M.; Brandbyge, M. Transmission eigenchannels from nonequilibrium Green’s functions. Phys. Rev. B 2007, 76, 115117. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Andrae, D.; Häußermann, U.; Dolg, M.; Stoll, H.; Preuß, H. Energy-adjustedab initio pseudopotentials for the second and third row transition elements. Theoret. Chim. Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Binning, R.C., Jr.; Curtiss, L.A. Compact contracted basis sets for third-row atoms: Ga–Kr. J. Comput. Chem. 1990, 11, 1206–1216. [Google Scholar] [CrossRef]
- Fuentealba, P.; Preuss, H.; Stoll, H.; Von Szentpály, L. A proper account of core-polarization with pseudopotentials: Single valence-electron alkali compounds. Chem. Phys. Lett. 1982, 89, 418–422. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Chem. Phys. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- García, A.; Papior, N.; Akhtar, A.; Artacho, E.; Blum, V.; Bosoni, E.; Brandimarte, P.; Brandbyge, M.; Cerdá, J.I.; Corsetti, F.; et al. Siesta: Recent developments and applications. J. Chem. Phys. 2020, 152, 204108–204131. [Google Scholar] [CrossRef] [PubMed]
- Papior, N.; Lorente, N.; Frederiksen, T.; García, A.; Brandbyge, M. Improvements on non-equilibrium and transport Green function techniques: The next-generation transiesta. Comput. Phys. Commun. 2017, 212, 8–24. [Google Scholar] [CrossRef]
- Soler, J.; Artacho, E.; Gale, J.; Garcia, A.; Junquera, J.; Ordejón, P.; Sánchez-Portal, D. The SIESTA method for ab initio order-N materials simulation. J. Phys. Condens. Matter 2002, 14, 2745–2779. [Google Scholar] [CrossRef]
- Troullier, N.; Martins, J.L. Efficient pseudopotentials for plane-wave calculations. Phys. Rev. B 1991, 43, 1993–2006. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Pan, H.; Lin, D.; Li, S.; Wang, Y.; Sanvito, S.; Hou, S. Robust covalent pyrazine anchors forming highly conductive and polarity-tunable molecular junctions with carbon electrodes. Phys. Chem. Chem. Phys. 2022, 24, 21337–21347. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, P.; Pan, H.; Wang, Y.; Li, J.; Dong, Y.; Wang, Y.; Hou, S. Exploring Aromaticity Effects on Electronic Transport in Cyclo[n]carbon Single-Molecule Junctions. Molecules 2024, 29, 3827. https://doi.org/10.3390/molecules29163827
Yang P, Pan H, Wang Y, Li J, Dong Y, Wang Y, Hou S. Exploring Aromaticity Effects on Electronic Transport in Cyclo[n]carbon Single-Molecule Junctions. Molecules. 2024; 29(16):3827. https://doi.org/10.3390/molecules29163827
Chicago/Turabian StyleYang, Peiqi, Haoyang Pan, Yudi Wang, Jie Li, Yangyu Dong, Yongfeng Wang, and Shimin Hou. 2024. "Exploring Aromaticity Effects on Electronic Transport in Cyclo[n]carbon Single-Molecule Junctions" Molecules 29, no. 16: 3827. https://doi.org/10.3390/molecules29163827
APA StyleYang, P., Pan, H., Wang, Y., Li, J., Dong, Y., Wang, Y., & Hou, S. (2024). Exploring Aromaticity Effects on Electronic Transport in Cyclo[n]carbon Single-Molecule Junctions. Molecules, 29(16), 3827. https://doi.org/10.3390/molecules29163827