Sandwich d/f Heterometallic Complexes [(Ln(hfac)3)2M(acac)3] (Ln = La, Pr, Sm, Dy and M = Co; Ln = La and M = Ru)

 , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Results and Discussion
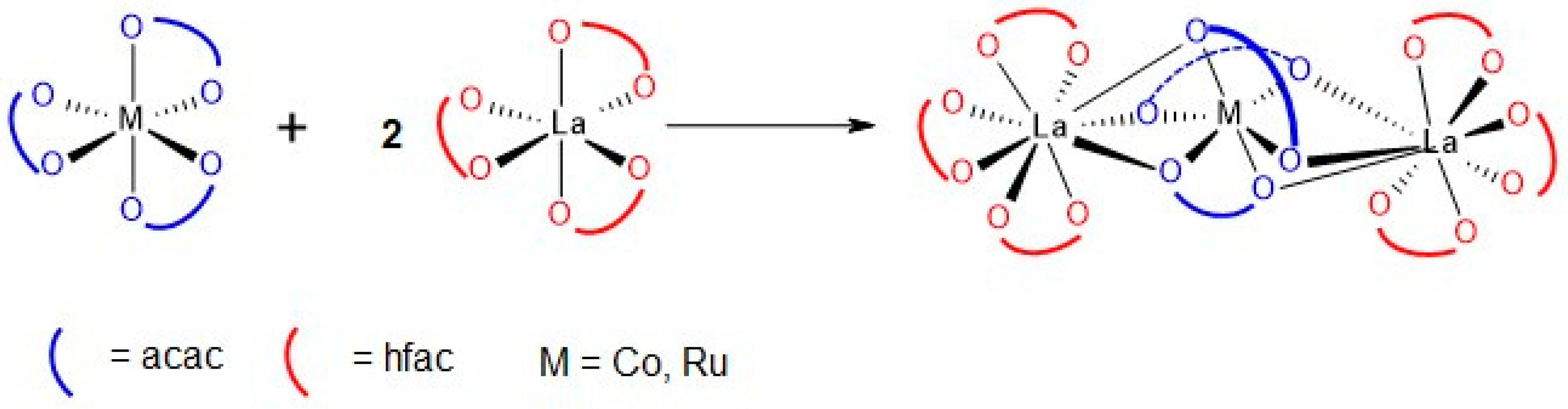
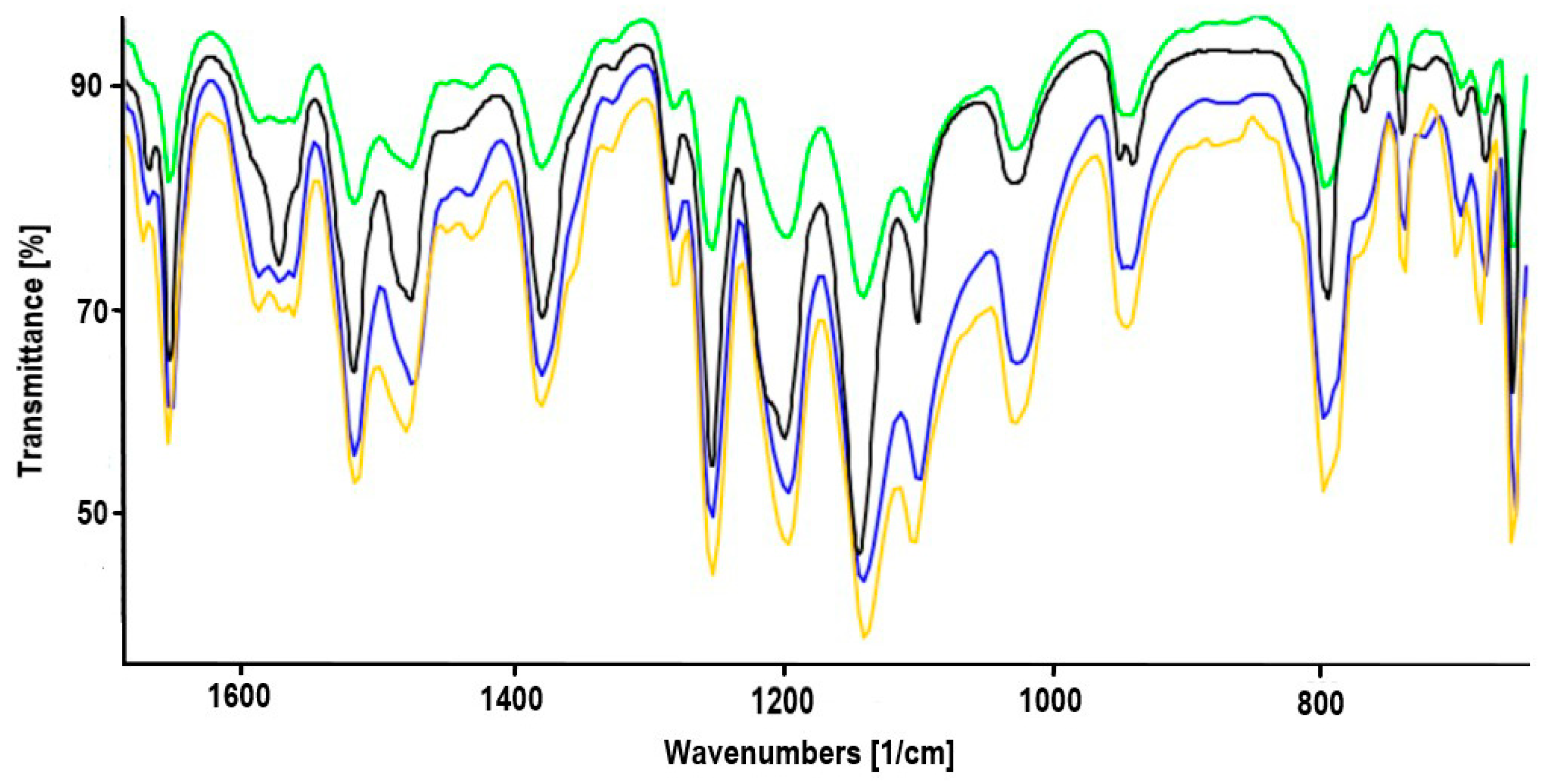
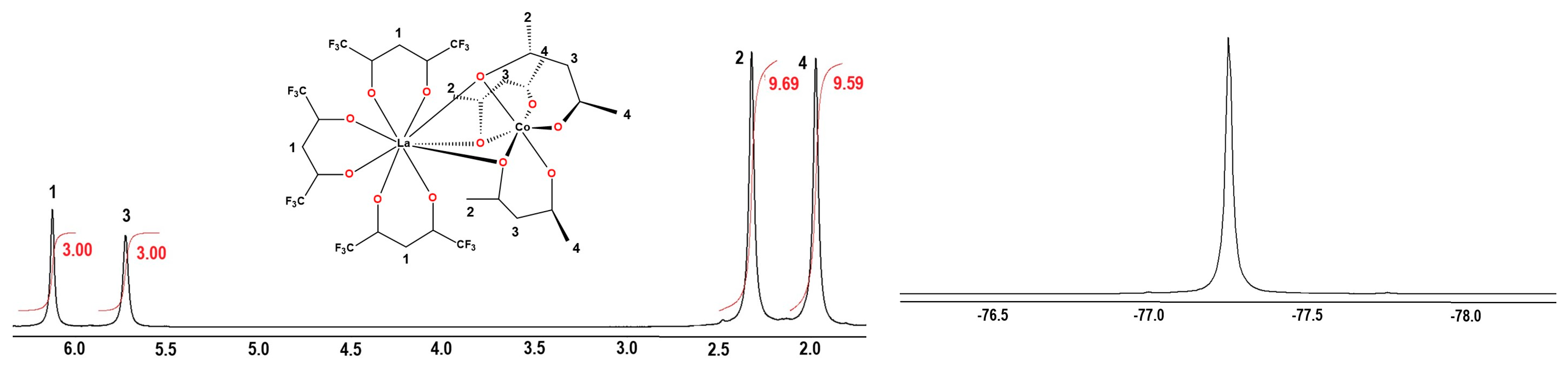
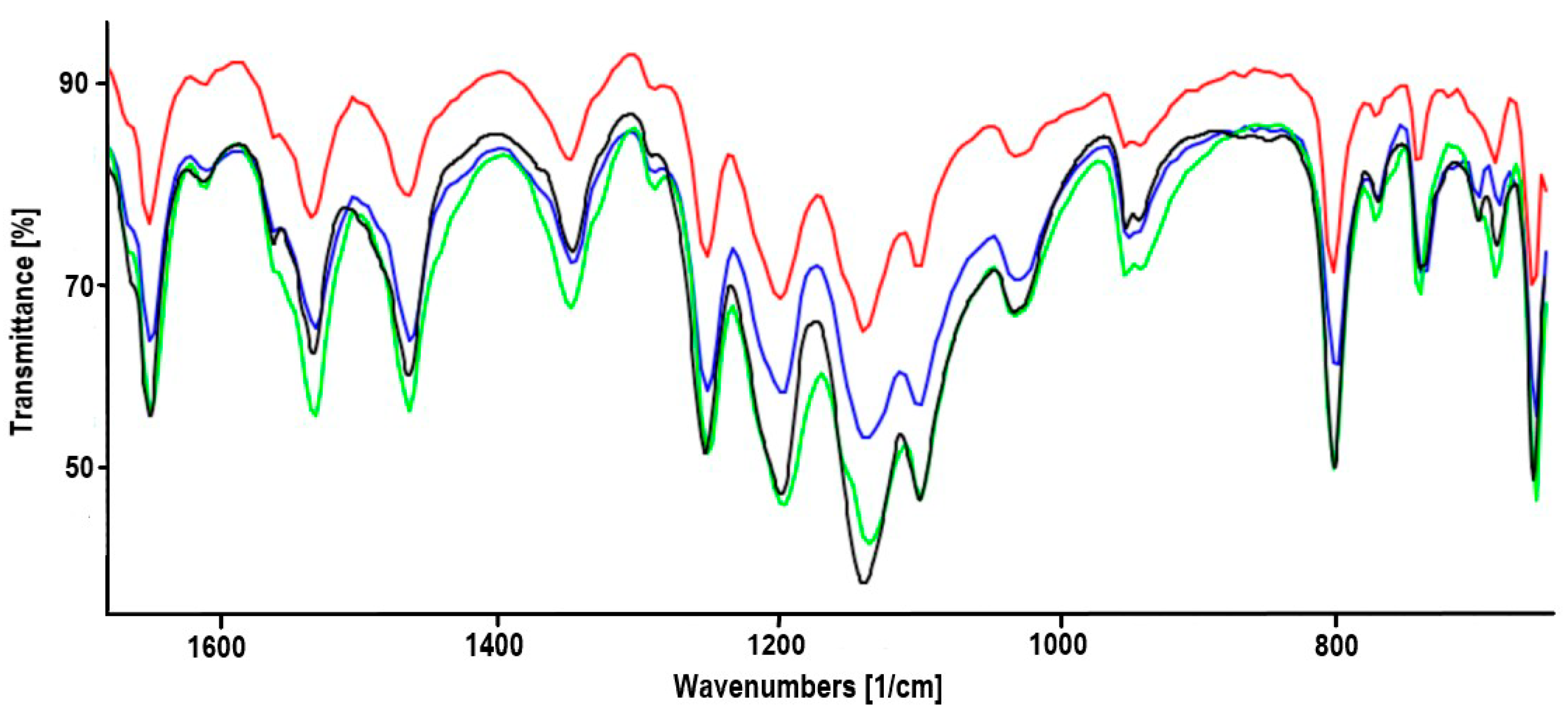
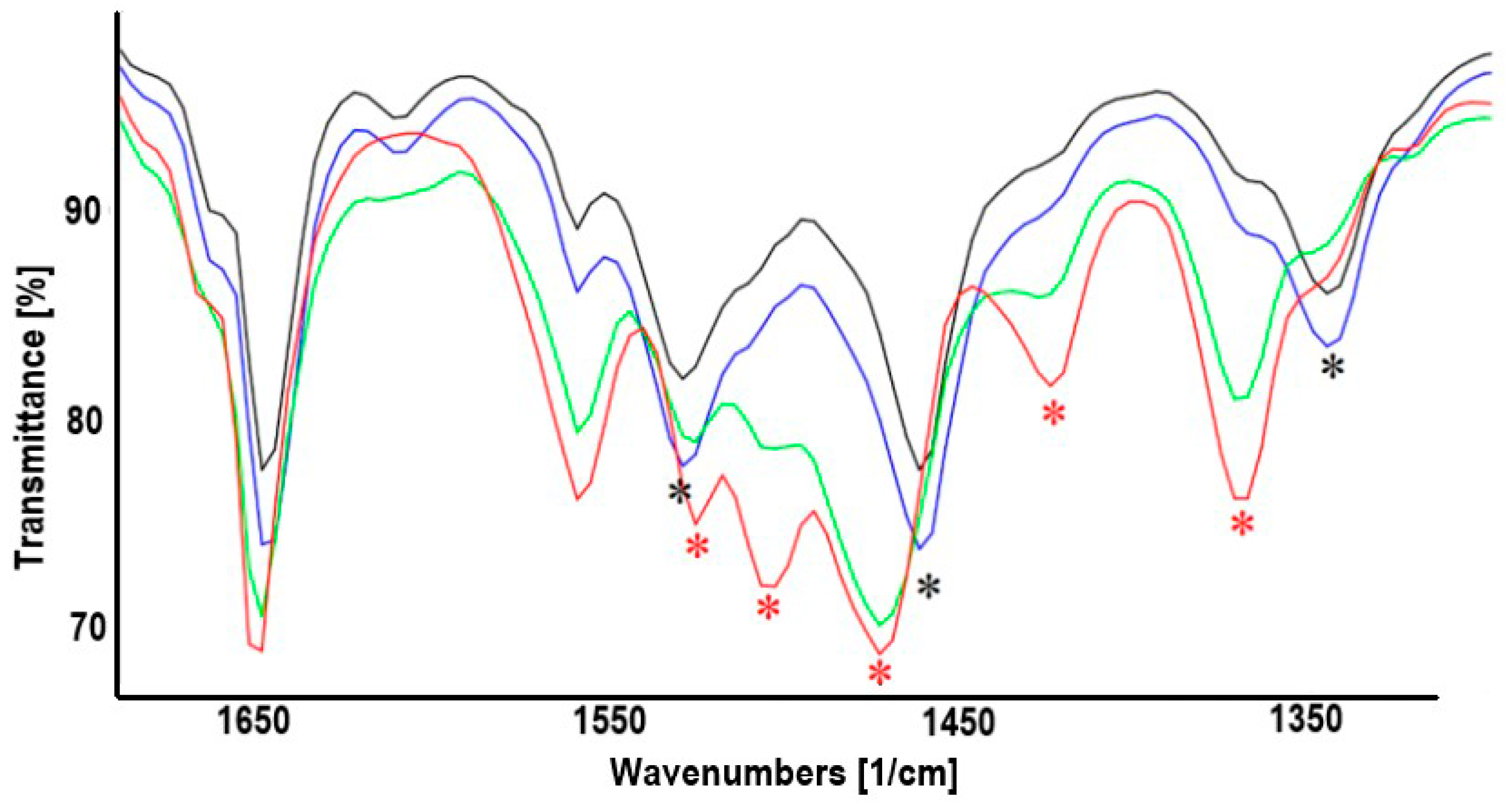
2.1. Synthesis of Heterodinuclear Complexes
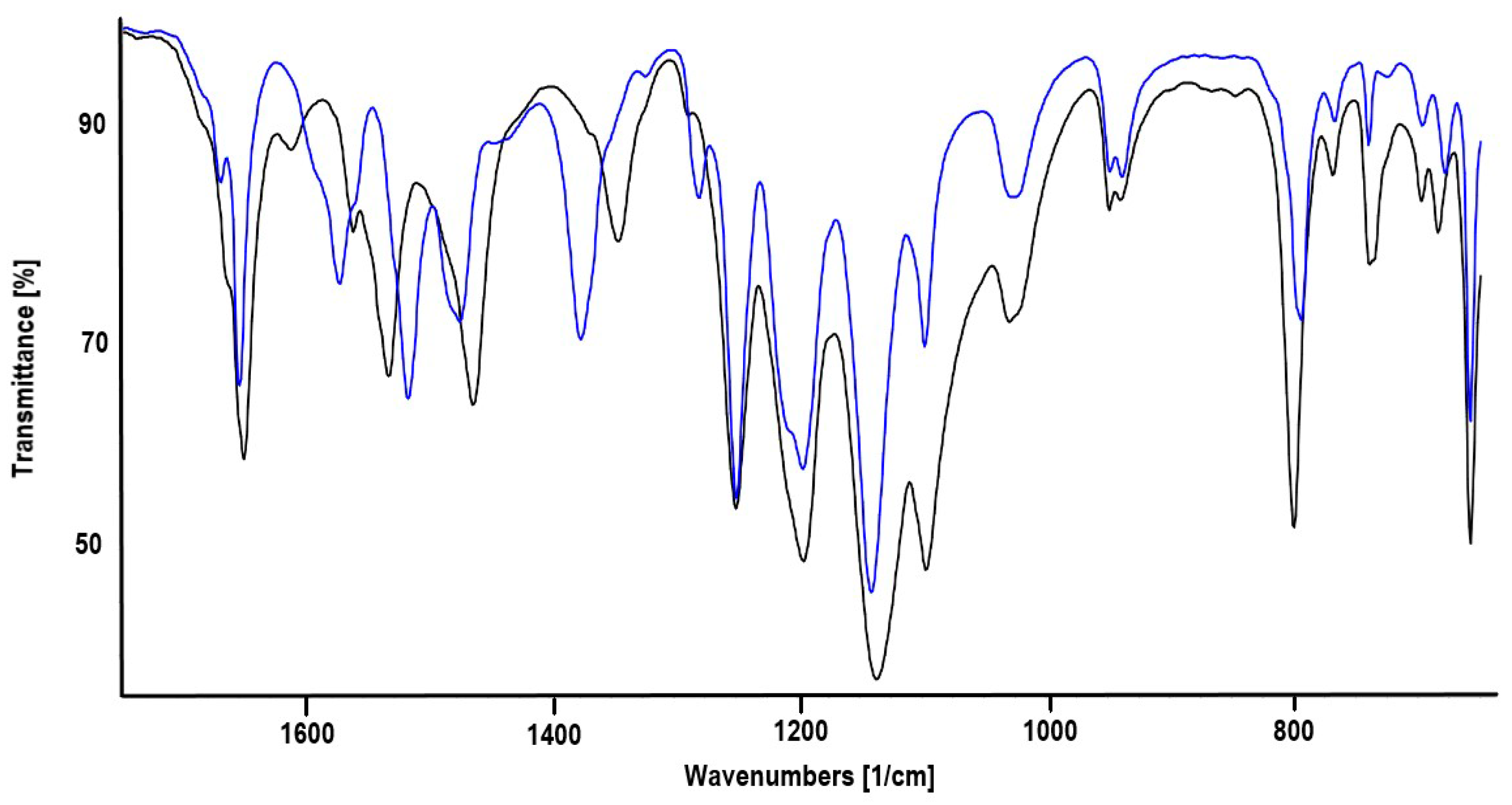
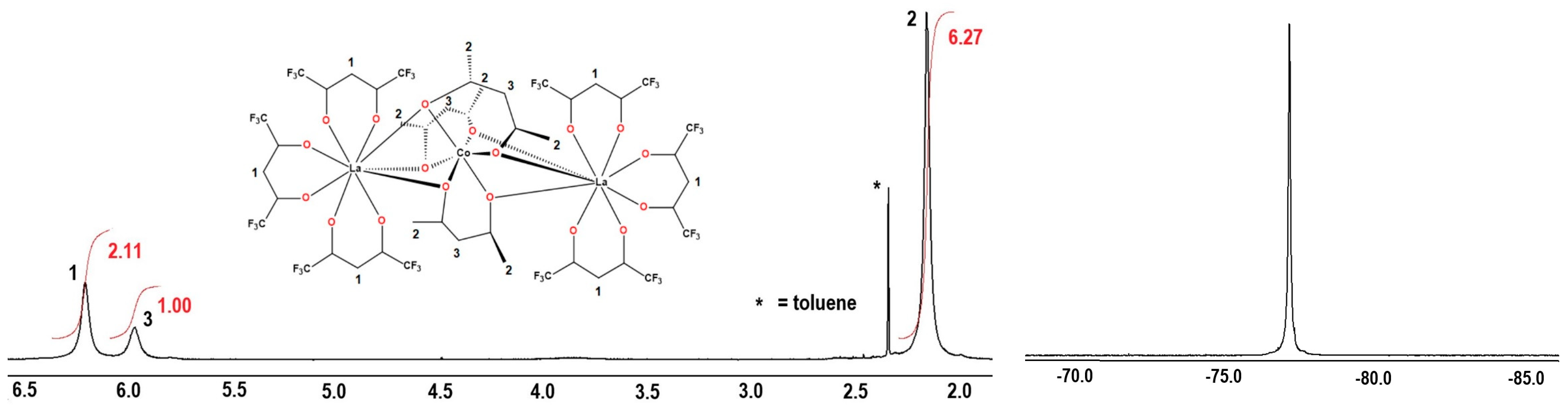
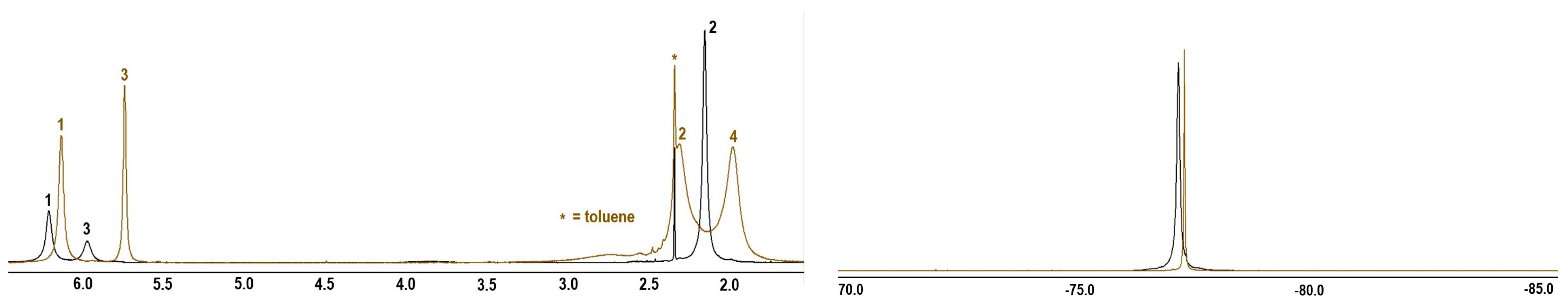
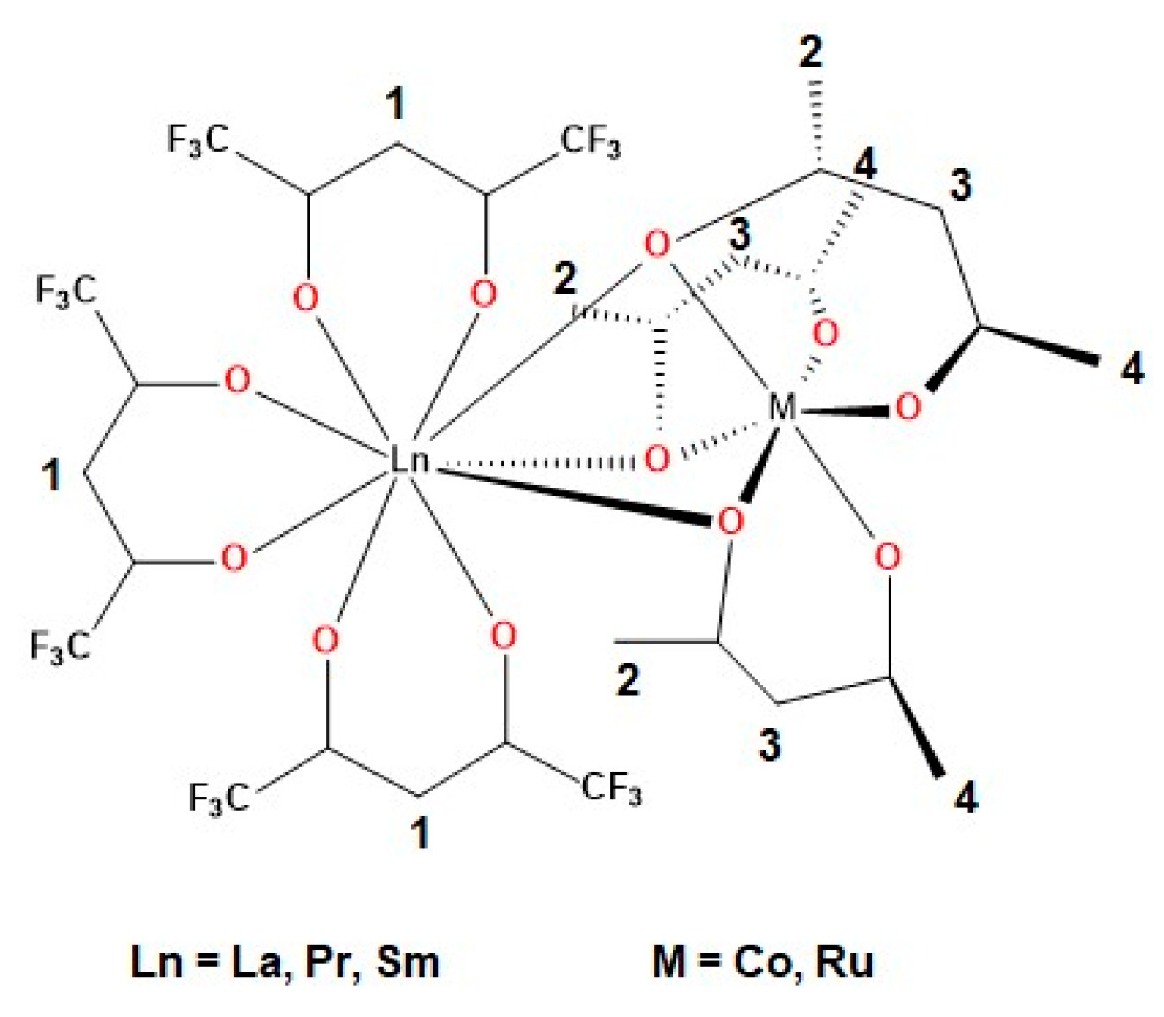
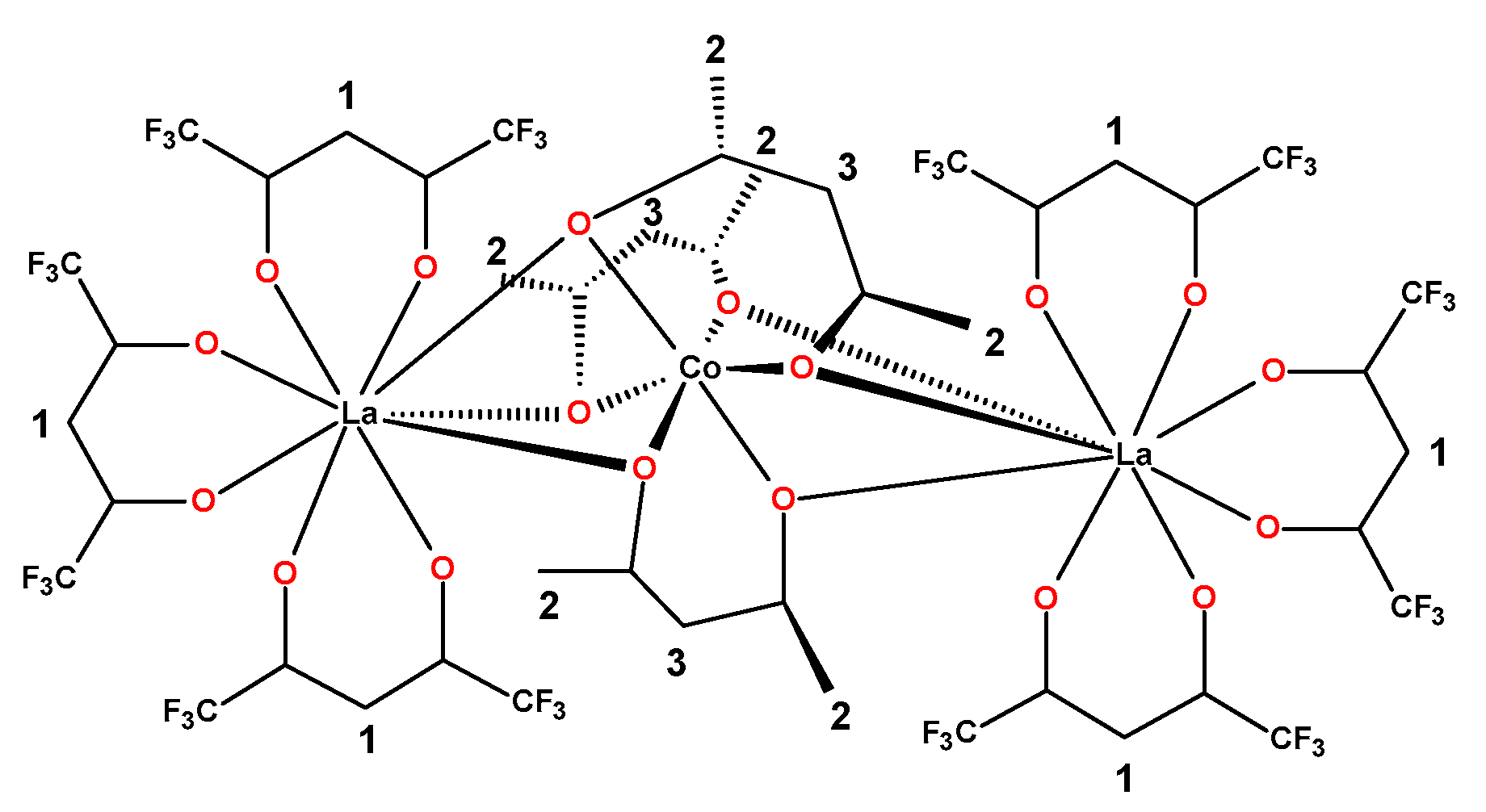
2.2. Synthesis of Heterotrinuclear Sandwich Complexes
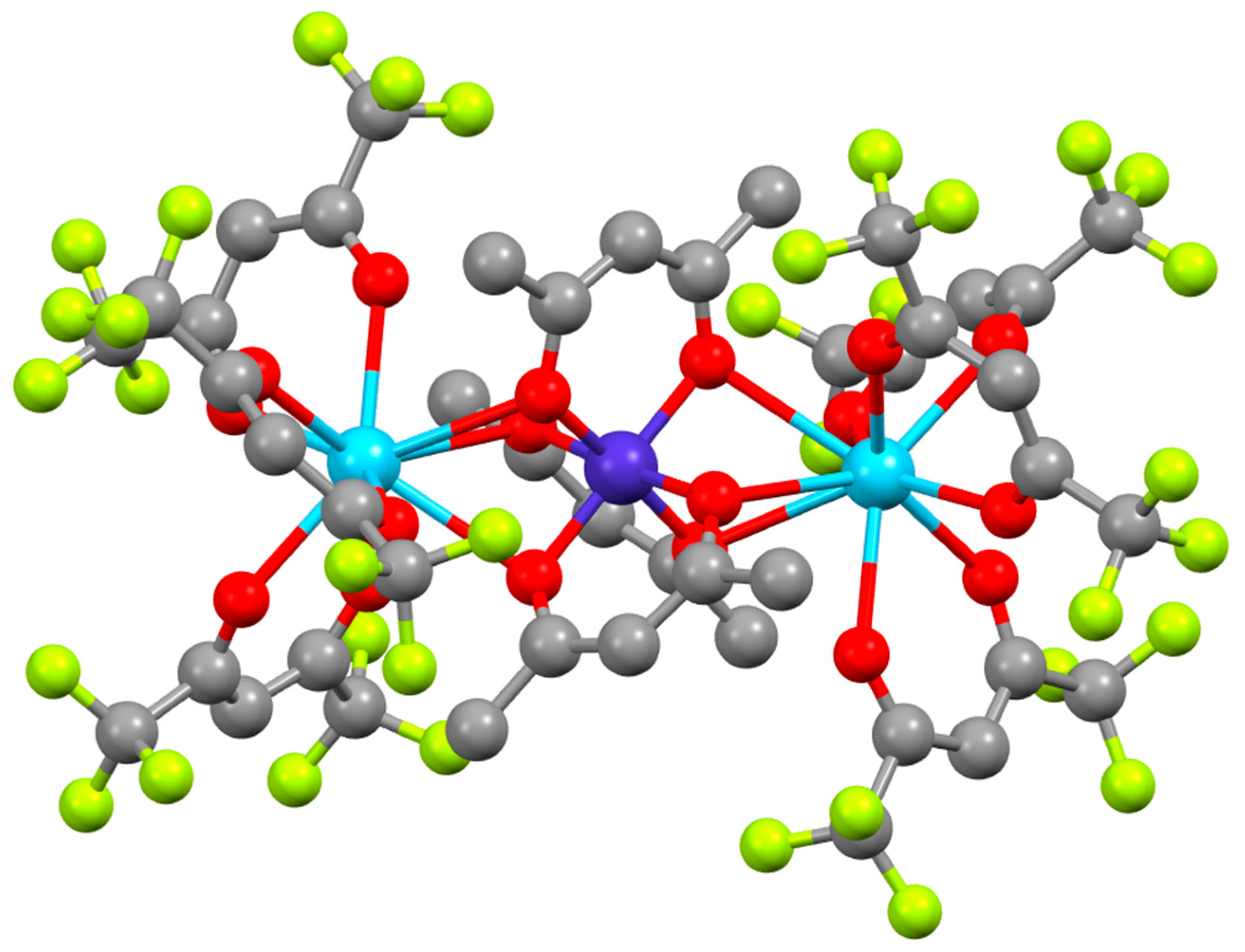
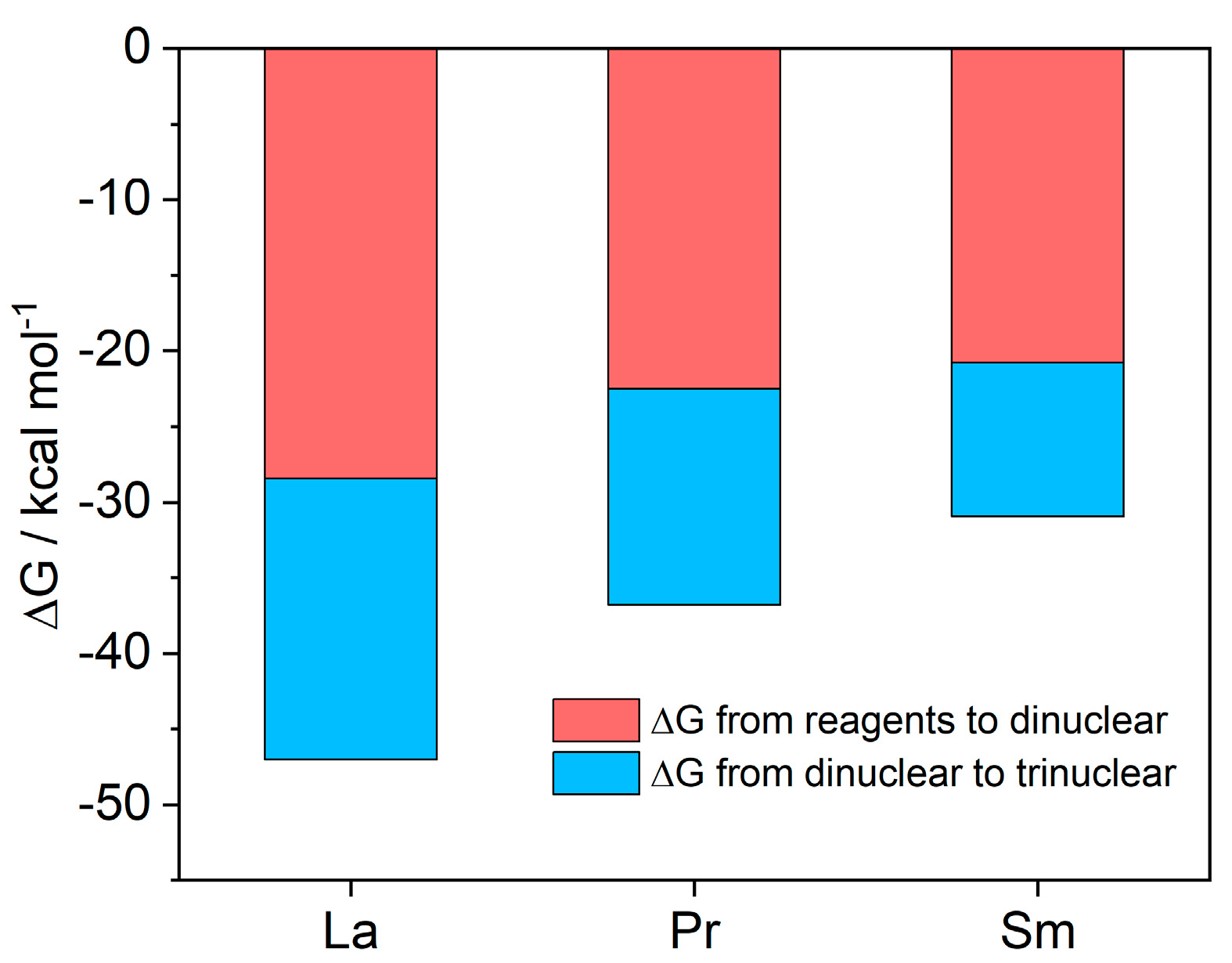
2.3. Quantum Mechanical Calculations
2.4. Studies on the Lanthanum/Ruthenium Analogs
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Griffiths, K.; Kostakis, G.E. Transformative 3d–4f coordination cluster carriers. Dalton Trans. 2018, 47, 12011–12034. [Google Scholar] [CrossRef]
- Petrus, R.; Kowaliński, A.; Utko, J.; Matuszak, K.; Lis, T.; Sobota, P. Heterometallic 3d–4f Alkoxide Precursors for the Synthesis of Binary Oxide Nanomaterials. Inorg. Chem. 2023, 62, 2197–2212. [Google Scholar] [CrossRef] [PubMed]
- López-Hernández, J.E.; Contel, M. Promising heterometallic compounds as anticancer agents: Recent studies in vivo. Curr. Opin. Chem. Biol. 2023, 72, 102250. [Google Scholar] [CrossRef]
- Crowston, B.J.; Shipp, J.D.; Chekulaev, D.; McKenzie, L.K.; Jones, C.; Weinstein, J.A.; Meijer, A.J.H.; Bryant, H.E.; Natrajan, L.; Woodward, A.; et al. Heteronuclear d-d and d-f Ru(II)/M complexes [M = Gd(III), Yb(III), Nd(III), Zn(II) or Mn(II)] of ligands combining phenanthroline and aminocarboxylate binding sites: Combined relaxivity, cell imaging and photophysical studies. Dalton Trans. 2019, 48, 6132–6152. [Google Scholar] [CrossRef]
- Ma, L.; Li, L.; Zhu, G. Platinum-containing heterometallic complexes in cancer therapy: Advances and perspectives. Inorg. Chem. Front. 2022, 9, 2424–2453. [Google Scholar] [CrossRef]
- Pan, M.; Liao, W.-M.; Yin, S.-Y.; Sun, S.-S.; Su, C.-Y. Single-Phase White-Light-Emitting and Photoluminescent Color-Tuning Coordination Assemblies. Chem. Rev. 2018, 118, 8889–8935. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Luo, J.; Gao, L.; Song, B.; Tang, J. Inorganic Lanthanide Compounds with f−d Transition: From Materials to Electroluminescence Devices. J. Phys. Chem. Lett. 2022, 13, 4365–4373. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Bag, P.; Kalita, P.; Chandrasekhar, V. Heterometallic CuII–LnIII complexes: Single molecule magnets and magnetic refrigerants. Coord. Chem. Rev. 2021, 432, 213707. [Google Scholar] [CrossRef]
- Vaz, M.G.F.; Andruh, M. Molecule-based magnetic materials constructed from paramagnetic organic ligands and two different metal ions. Coord. Chem. Rev. 2021, 427, 213611. [Google Scholar] [CrossRef]
- Wang, H.L.; Zhu, Z.H.; Peng, J.M.; Zou, H.H. Heterometallic 3d/4f-Metal Complexes: Structure and Magnetism. J. Clust. Sci. 2022, 33, 1299–1325. [Google Scholar] [CrossRef]
- Chakraborty, A.; Acharya, J.; Chandrasekhar, V. Ferrocene-Supported Compartmental Ligands for the Assembly of 3d/4f Complexes. ACS Omega 2020, 5, 9046–9054. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Shi, W.; Cheng, P. Toward heterometallic single-molecule magnets: Synthetic strategy, structures and properties of 3d–4f discrete complexes. Coord. Chem. Rev. 2015, 289–290, 74–122. [Google Scholar] [CrossRef]
- Jana, A.; Majumder, S.; Carrella, L.; Nayak, M.; Weyhermueller, T.; Dutta, S.; Schollmeyer, D.; Rentschler, E.; Koner, R.; Mohanta, S. Syntheses, Structures, and Magnetic Properties of Diphenoxo-Bridged CuIILnIII and NiII(Low-Spin)LnIII Compounds Derived from a Compartmental Ligand (Ln = Ce-Yb). Inorg. Chem. 2010, 49, 9012–9025. [Google Scholar] [CrossRef]
- Andruh, M. Compartmental Schiff-base ligands—A rich library of tectons in designing magnetic and luminescent materials. Chem. Commun. 2011, 47, 3025–3042. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Gupta, R. Metalloligands to material: Design strategies and network topologies. CrystEngComm 2016, 18, 9185–9208. [Google Scholar] [CrossRef]
- Kumar, G.; Gupta, R. Molecularly designed architectures—The metalloligand way. Chem. Soc. Rev. 2013, 42, 9403–9453. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, M.A.; Weihe, H.; Vinum, M.G.; Mortensen, J.S.; Doerrer, L.H.; Bendix, J. Imposing high-symmetry and tuneable geometry on lanthanide centres with chelating Pt and Pd metalloligands. Chem. Sci. 2017, 8, 3566–3575. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.F.; Ma, J.X.; Liu, W.S. Lanthanide Metalloligand Strategy toward d–f Heterometallic Metal–Organic Frameworks: Magnetism and Symmetric-Dependent Luminescent Properties. Inorg. Chem. 2014, 53, 5922–5930. [Google Scholar] [CrossRef]
- Ma, J.X.; Guo, J.; Wang, H.; Li, B.; Yang, T.; Chen, B. Microporous Lanthanide Metal–Organic Framework Constructed from Lanthanide Metalloligand for Selective Separation of C2H2/CO2 and C2H2/CH4 at Room Temperature. Inorg. Chem. 2017, 56, 7145–7150. [Google Scholar] [CrossRef]
- Qiu, J.Z.; Wang, L.F.; Chen, Y.C.; Zhang, Z.M.; Li, Q.W.; Tong, M.L. Magnetocaloric Properties of Heterometallic 3d–Gd Complexes Based on the [Gd(oda)3]3− Metalloligand. Chem.—A Eur. J. 2016, 22, 802–808. [Google Scholar] [CrossRef]
- Merkens, C.; Englert, U. Ordered bimetallic coordination networks featuring rare earth and silver cations. Dalton Trans. 2012, 41, 4664–4673. [Google Scholar] [CrossRef] [PubMed]
- Armelao, L.; Belli Dell’Amico, D.; Bellucci, L.; Bottaro, G.; Ciattini, S.; Labella, L.; Manfroni, G.; Marchetti, F.; Mattei, C.A.; Samaritani, S. Homodinuclear Lanthanide Complexes with the Divergent Heterotopic 4,4′-Bipyridine N-Oxide (bipyMO) Ligand. Eur. J. Inorg. Chem. 2018, 2018, 4421–4428. [Google Scholar] [CrossRef]
- Bellucci, L.; Bottaro, G.; Labella, L.; Causin, V.; Marchetti, F.; Samaritani, S.; Belli Dell’Amico, D.; Armelao, L. Composition−Thermometric Properties Correlations in Homodinuclear Eu3+ Luminescent Complexes. Inorg. Chem. 2020, 59, 18156–18167. [Google Scholar] [CrossRef]
- Fioravanti, L.; Bellucci, L.; Armelao, L.; Bottaro, G.; Marchetti, F.; Pineider, F.; Poneti, G.; Samaritani, S.; Labella, L. Stoichiometry Controlled Assembly of Lanthanide Molecular Complexes of the heteroditopic divergent ligand 4′-(4-pyridil)-2,2′:6′,2″-terpyridine N-oxide in hypodentate or bridging coordination modes. Structural, Magnetic and Photoluminescence studies. Inorg. Chem. 2022, 61, 265–278. [Google Scholar] [CrossRef]
- Soerensen, T.J.; Faulkner, S. Multimetallic Lanthanide Complexes: Using Kinetic Control to Define Complex Multimetallic Arrays. Acc. Chem. Res. 2018, 51, 2493–2501. [Google Scholar] [CrossRef] [PubMed]
- Bazi, M.; Bracciotti, E.; Fioravanti, L.; Marchetti, F.; Rancan, M.; Armelao, L.; Samaritani, S.; Labella, L. Mononuclear Rare-Earth Metalloligands Exploiting a Divergent Ligand. Inorg. Chem. 2024, 63, 7678–7691. [Google Scholar] [CrossRef]
- Cosquer, G.; Pointillart, F.; Le Gal, Y.; Golhen, S.; Cador, O.; Ouahab, L. Ferromagnetic versus Antiferromagnetic Exchange Interactions in Tetrathiafulvalene-Based 3d/4f Heterobimetallic Complexes. Chem.—Eur. J. 2011, 17, 12502–12511. [Google Scholar] [CrossRef] [PubMed]
- Kuz’mina, N.P.; Malkerova, I.P.; Alikhanyan, A.S.; Gleizes, A.N. The use of 3d-metal complexes as ligands to prepare volatile 4f–3d heterobimetallic complexes. J. Alloys Compd. 2004, 374, 315–319. [Google Scholar] [CrossRef]
- Benelli, C.; Caneschi, A.; Gatteschi, D.; Guillou, O.; Pardi, L. Synthesis, Crystal Structure, and Magnetic Properties of Tetranuclear Complexes Containing Exchange-Coupled Ln2Cu2 (Ln = Gd, Dy) Species. Inorg. Chem. 1990, 29, 1750–1755. [Google Scholar] [CrossRef]
- Jia, R.; Li, H.-F.; Chen, P.; Gao, T.; Sun, W.-B.; Li, G.-M.; Yan, P.-F. Synthesis, structure, and tunable white light emission of heteronuclear Zn2Ln2 arrays using a zinc complex as ligand. CrystEngComm 2016, 18, 917–923. [Google Scholar] [CrossRef]
- Chen, L.; Breeze, S.R.; Rousseau, R.J.; Wang, S.; Thompson, L.K. Polynuclear Copper-Lanthanide Complexes with Amino Alcohol Ligands. Syntheses, Structures, and Magnetic and Spectroscopic Studies of CuII(bdmmp)2(H2O), PrIIICuII(bdmmp)(bdmmpH)(µ-OH)(hfacac)3, [LaIIICuII(bdmmp)(bdmmpH)(µ-OH)(O2CCF3)3]2, and CuII4(bdmmp)2(µ4-O)(O2CCF3)4, (bdmmpH= 2,6-Bis[(dimethylamino)methyl]-4-methylphenol; hfacac = Hexafluoroacetylacetonato). Inorg. Chem. 1995, 34, 454–465. [Google Scholar] [CrossRef]
- Armelao, L.; Belli Dell’Amico, D.; Bottaro, G.; Bellucci, L.; Labella, L.; Marchetti, F.; Mattei, C.A.; Mian, F.; Pineider, F.; Poneti, G.; et al. 1D hetero-bimetallic regularly alternated 4f–3d coordination polymers based on N-oxide-4,4′-bipyridine (bipyMO) as a linker: Photoluminescence and magnetic properties. Dalton Trans. 2018, 47, 8337–8345. [Google Scholar] [CrossRef] [PubMed]
- Ramade, I.; Kahn, O.; Jeannin, Y.; Robert, F. Design and Magnetic Properties of a Magnetically Isolated GdIIICuII Pair. Crystal Structures of [Gd(hfa)3Cu(salen)], [Y(hfa)3Cu(salen)], [Gd(hfa)3Cu(salen)(Meim)], and [La(hfa)3(H2O)Cu(salen)] [hfa = Hexafluoroacetylacetonato, salen = N,N′-Ethylenebis(salicylideneaminato), Meim = 1-Methylimidazole]. Inorg. Chem. 1997, 36, 930–936. [Google Scholar] [CrossRef]
- Gleizes, A.N.; Julve, M.; Kuz’mina, N.P.; Alikhanyan, A.S.; Lloret, F.; Malkerova, I.P.; Sanz, J.L.; Senocq, F. Heterobimetallic d2f Metal Complexes as Potential Single-Source Precursors for MOCVD: Structure and Thermodynamic Study of the Sublimation of [Ni(salen)Ln(hfa)3], Ln = Y, Gd. Eur. J. Inorg. Chem. 1998, 1998, 1169–1174. [Google Scholar] [CrossRef]
- Pointillart, F.; Bernot, K. Determination of the Nature of Exchange Interactions in the 3d–4f Magnetic Chain {[Cu(salen)Pr(hfac)3]2(L)}n (L = 4,4′-Bipyridine, Pyrazine). Eur. J. Inorg. Chem. 2010, 2010, 952–964. [Google Scholar] [CrossRef]
- Sasaki, M.; Horiuchi, H.; Kumagai, M.; Sakamoto, M.; Sakiyama, H.; Nishida, Y.; Sadaoka, Y.; Ohba, M.; Ōkawa, H. A Novel Discrete d-f Heterobinuclear Complex Designed from Tetrahedrally Distorted [Cu(salabza)](H2salabza: N,N′-Bis(salicylidene)-2-aminobenzylamine) and [Gd(hfac)3]. Chem. Lett. 1998, 27, 911–912. [Google Scholar] [CrossRef]
- Sasaki, M.; Manseki, K.; Horiuchi, H.; Kumagai, M.; Sakamoto, M.; Sakiyama, H.; Nishida, Y.; Saki, M.; Sadaoka, Y.; Ohba, M.; et al. Synthesis, structure, and magnetic properties of discrete d–f heterodinuclear complexes designed from tetrahedrally distorted [Cu(salabza)] (H2salabza= N,N′-bis(salicylidene)-2-aminobenzylamine) and [Ln(hfac)3] (Hhfac = 1,1,1,5,5,5-hexafluoroacetylacetone, Ln = Gd or Lu). J. Chem. Soc. Dalton Trans. 2000, 259, 263. [Google Scholar] [CrossRef]
- Ryazanov, M.; Nikiforov, V.; Lloret, F.; Julve, M.; Kuz’mina, N.P.; Gleizes, A. Magnetically Isolated CuIIGdIII Pairs in the Series [Cu(acacen)Gd(pta)3], [Cu(acacen)Gd(hfa)3], [Cu(salen)Gd(pta)3], and [Cu(salen)Gd(hfa)3], [acacen = N,N′-Ethylenebis(acetylacetoniminate(−)), salen = N,N′-Ethylenebis(salicylideniminate(−)), hfa = 1,1,1,5,5,5-Hexafluoropentane-2,4-dionate(−), pta = 1,1,1-Trifluoro-5,5-dimethylhexane-2,4-dionate(−)]. Inorg. Chem. 2002, 41, 1816–1823. [Google Scholar] [CrossRef]
- Cosquer, G.; Pointillart, F.; Le Guennic, B.; Le Gal, Y.; Golhen, S.; Cador, O.; Ouahab, L. 3d4f Heterobimetallic Dinuclear and Tetranuclear Complexes Involving Tetrathiafulvalene as Ligands: X-ray Structures and Magnetic and Photophysical Investigations. Inorg. Chem. 2012, 51, 8488–8501. [Google Scholar] [CrossRef]
- Brewer, C.; Brewer, G.; Scheidt, W.R.; Shang, M.; Carpenter, E.E. Synthesis and structural and magnetic characterization of discrete phenolato and imidazolate bridged Gd(III)–M(II) [M=Cu, Ni] dinuclear complexes. Inorg. Chim. Acta 2001, 313, 65–70. [Google Scholar] [CrossRef]
- Ohba, M.; Ohtsubo, N.; Shiga, T.; Sakamoto, M.; Ōkawa, H. Synthesis, structure and magnetic properties of a linear Cu(II)Cu(II)Gd(III) complex. Polyhedron 2003, 22, 1905–1910. [Google Scholar] [CrossRef]
- Rogachev, A.Y.; Mironov, A.V.; Nemukhin, A.V. Experimental and theoretical studies of the products of reaction between Ln(hfa)3 and Cu(acac)2 (Ln = La, Y; acac = acetylacetonate, hfa = hexafluoroacetylacetonate). J. Mol. Struct. 2007, 831, 46–54. [Google Scholar] [CrossRef]
- Bellucci, L.; Giordano, L.; Carlotto, S.; Poneti, G.; Rancan, M.; Samaritani, S.; Armelao, L.; Labella, L. Heterometallic [Ln(hfac)3Cu(acac)2] complexes with late 4f ions. Inorg. Chim. Acta 2023, 556, 121665. [Google Scholar] [CrossRef]
- Øwre, A.; Vinum, M.; Kern, M.; van Slageren, J.; Bendix, J.; Perfetti, M. Chiral, Heterometallic Lanthanide–Transition Metal Complexes by Design. Inorganics 2018, 6, 72. [Google Scholar] [CrossRef]
- Bellucci, L.; Babetto, L.; Bottaro, G.; Carlotto, S.; Labella, L.; Gallo, E.; Marchetti, F.; Samaritani, S.; Armelao, L. Competing excitation paths in Luminescent Heterobimetallic Ln-Al complexes: Unravelling interactions via experimental and theoretical investigations. iScience 2023, 26, 106614. [Google Scholar] [CrossRef] [PubMed]
- Gallo, E.; Bellucci, L.; Carlotto, S.; Bottaro, G.; Babetto, L.; Giordano, L.; Marchetti, F.; Samaritani, S.; Armelao, L.; Labella, L. Aluminium 8-hydroxyquinolinate N-oxide as a precursor to heterometallic aluminium-lanthanide complexes. Molecules 2024, 29, 451. [Google Scholar] [CrossRef]
- Dalal, A.; Nehra, K.; Hooda, A.; Singh, D.; Kumar, P.; Kumar, S.; Malik, R.S.; Rathi, B. Luminous lanthanide diketonates: Review on synthesis and optoelectronic characterizations. Inorg. Chim. Acta 2023, 550, 121406. [Google Scholar] [CrossRef]
- Eliseeva, S.V.; Pleshkov, D.N.; Lyssenko, K.A.; Lepney, L.S.; Bunzli, J.G.; Kuzmina, N.P.J. Highly luminescent and triboluminescent coordination polymers assembled from lanthanide β-diketonates and aromatic bidentate O-donor ligands. Inorg. Chem. 2010, 49, 9300–9311. [Google Scholar] [CrossRef] [PubMed]
- Quadrelli, E.A. Lanthanide contraction over the 4f series follows a quadratic decay. Inorg. Chem. 2002, 41, 167–169. [Google Scholar] [CrossRef]
- Baisch, U.; Belli Dell’Amico, D.; Calderazzo, F.; Labella, L.; Marchetti, F.; Merigo, A. N,N-Dialkylcarbamato Lanthanide Complexes, a Series of Isotypical Coordination Compounds. Eur. J. Inorg. Chem. 2004, 2004, 1219–1224. [Google Scholar] [CrossRef]
- Baisch, U.; Belli Dell’Amico, D.; Calderazzo, F.; Conti, R.; Labella, L.; Marchetti, F.; Quadrelli, E.A. The mononuclear and dinuclear dimethoxyethane adducts of lanthanide trichlorides [LnCl3(DME)2]n, n = 1 or 2, fundamental starting materials in lanthanide chemistry: Preparation and structures. Inorg. Chim. Acta 2004, 357, 1538–1548. [Google Scholar] [CrossRef]
- Bridgeman, A.J.; Cavigliasso, G.; Irelanda, L.R.; Rothery, J. The Mayer bond order as a tool in inorganic chemistry. J. Chem. Soc. Dalton Trans. 2001, 2095–2108. [Google Scholar] [CrossRef]
- Armelao, L.; Belli Dell’Amico, D.; Bellucci, L.; Bottaro, G.; Labella, L.; Marchetti, F.; Samaritani, S. A convenient synthesis of highly luminescent lanthanide 1D-zigzag coordination chains based only on 4,4′-bipyridine as connector. Polyhedron 2016, 119, 371–376. [Google Scholar] [CrossRef]
- Bellucci, L.; Fioravanti, L.; Armelao, L.; Bottaro, G.; Marchetti, F.; Pineider, F.; Poneti, G.; Samaritani, S.; Labella, L. Size Selectivity in Heterolanthanide Molecular Complexes with a Ditopic Ligand. Chem.—Eur. J. 2023, 29, e202202823. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system, version 4.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 1992, 45, 13244–13249. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Hartree–Fock Exchange Fitting Basis Sets for H to Rn. J. Comput Chem. 2008, 29, 167–175. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Bruker. APEX4 V2021.10-0; Bruker AXS Inc.: Madison, WI, USA, 2021. [Google Scholar]
- Bruker. SAINT v8.30A; Bruker AXS Inc.: Madison, WI, USA, 2012. [Google Scholar]
- Bruker. XPREP V2014/2; Bruker AXS Inc.: Madison, WI, USA, 2014. [Google Scholar]
- Bruker. SADABS V2016/2; Bruker AXS Inc.: Madison, WI, USA, 2016. [Google Scholar]
- Sheldrick, G.M. SHELXL-2019/1; Bruker AXS Inc.: Madison, WI, USA, 2019. [Google Scholar]
- Lindoy, L.F.; Lip, H.C.; Louie, H.W.; Drew, M.G.B.; Hudson, M.J. Interaction of lanthanide shift reagents with co-ordination complexes; direct observation of nuclear magnetic resonance signals for free and complexed tris(pentane-2,4-dionato)cobalt(III) at ambient temperature, and X-ray crystal and molecular structure. J. Chem. Soc. Chem. Commun. 1977, 778–780. [Google Scholar] [CrossRef]
- Chaabene, M.; Agren, S.; Allouche, A.R.; Lahcinie, M.; Chaabane, R.B.; Baouab, M.H.V. Theoretical and experimental investigations of complexation with BF3.Et2O effects on electronic structures, energies and photophysical properties of Anil and tetraphenyl (hydroxyl) imidazole. Appl. Organometal. Chem. 2019, 33, e5218. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Distance | Bond Lengths | Mayer Bond Order (MBO) | |||
|---|---|---|---|---|---|
| Ln⋯Co | Ln-Obridge | Co-Obridge | Ln-Obridge | Co-Obridge | |
| [Co(acac)3] | // | // | 1.901 | // | 0.665 |
| [La(hfac)3Co(acac)3] | 3.419 | 2.701 | 1.914 | 0.160 | 0.530 |
| [Pr(hfac)3Co(acac)3] | 3.383 | 2.670 | 1.911 | 0.190 | 0.551 |
| [Sm(hfac)3Co(acac)3] | 3.367 | 2.654 | 1.908 | 0.188 | 0.557 |
| [(La(hfac)3)2Co(acac)3] | 3.471 | 2.759 | 1.904 | 0.126 | 0.587 |
| [(Pr(hfac)3)2Co(acac)3] | 3.437 | 2.732 | 1.901 | 0.170 | 0.597 |
| [(Sm(hfac)3)2Co(acac)3] | 3.427 | 2.720 | 1.900 | 0.173 | 0.604 |
| Product | Reagents | ∆E | ∆G |
|---|---|---|---|
| [La(hfac)3Co(acac)3] | [Co(acac)3] and [La(hfac)3] | −45.2 | −28.4 |
| [Pr(hfac)3Co(acac)3] | [Co(acac)3] and [Pr(hfac)3] | −39.2 | −22.5 |
| [Sm(hfac)3Co(acac)3] | [Co(acac)3] and [Sm(hfac)3] | −37.5 | −20.8 |
| [(La(hfac)3)2Co(acac)3] | [Co(acac)3] and 2[La(hfac)3] | −81.7 | −47.0 |
| [(Pr(hfac)3)2Co(acac)3] | [Co(acac)3] and 2[Pr(hfac)3] | −71.5 | −36.8 |
| [(Sm(hfac)3)2Co(acac)3] | [Co(acac)3] and 2[Sm(hfac)3] | −65.6 | −30.9 |
| [(La(hfac)3)2Co(acac)3] | [La(hfac)3Co(acac)3] and [La(hfac)3] | −36.5 | −18.6 |
| [(Pr(hfac)3)2Co(acac)3] | [Pr(hfac)3Co(acac)3] and [Pr(hfac)3] | −32.3 | −14.3 |
| [(Sm(hfac)3)2Co(acac)3] | [Sm(hfac)3Co(acac)3] and [Sm(hfac)3] | −28.1 | −10.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grechi, C.; Carlotto, S.; Guelfi, M.; Samaritani, S.; Armelao, L.; Labella, L. Sandwich d/f Heterometallic Complexes [(Ln(hfac)3)2M(acac)3] (Ln = La, Pr, Sm, Dy and M = Co; Ln = La and M = Ru). Molecules 2024, 29, 3927. https://doi.org/10.3390/molecules29163927
Grechi C, Carlotto S, Guelfi M, Samaritani S, Armelao L, Labella L. Sandwich d/f Heterometallic Complexes [(Ln(hfac)3)2M(acac)3] (Ln = La, Pr, Sm, Dy and M = Co; Ln = La and M = Ru). Molecules. 2024; 29(16):3927. https://doi.org/10.3390/molecules29163927
Chicago/Turabian StyleGrechi, Cristian, Silvia Carlotto, Massimo Guelfi, Simona Samaritani, Lidia Armelao, and Luca Labella. 2024. "Sandwich d/f Heterometallic Complexes [(Ln(hfac)3)2M(acac)3] (Ln = La, Pr, Sm, Dy and M = Co; Ln = La and M = Ru)" Molecules 29, no. 16: 3927. https://doi.org/10.3390/molecules29163927