
Arylboronic Acid Pinacol Esters as Stable Boron Sources for Dihydrodibenzoborepin Derivatives and a Dibenzoborole


Abstract

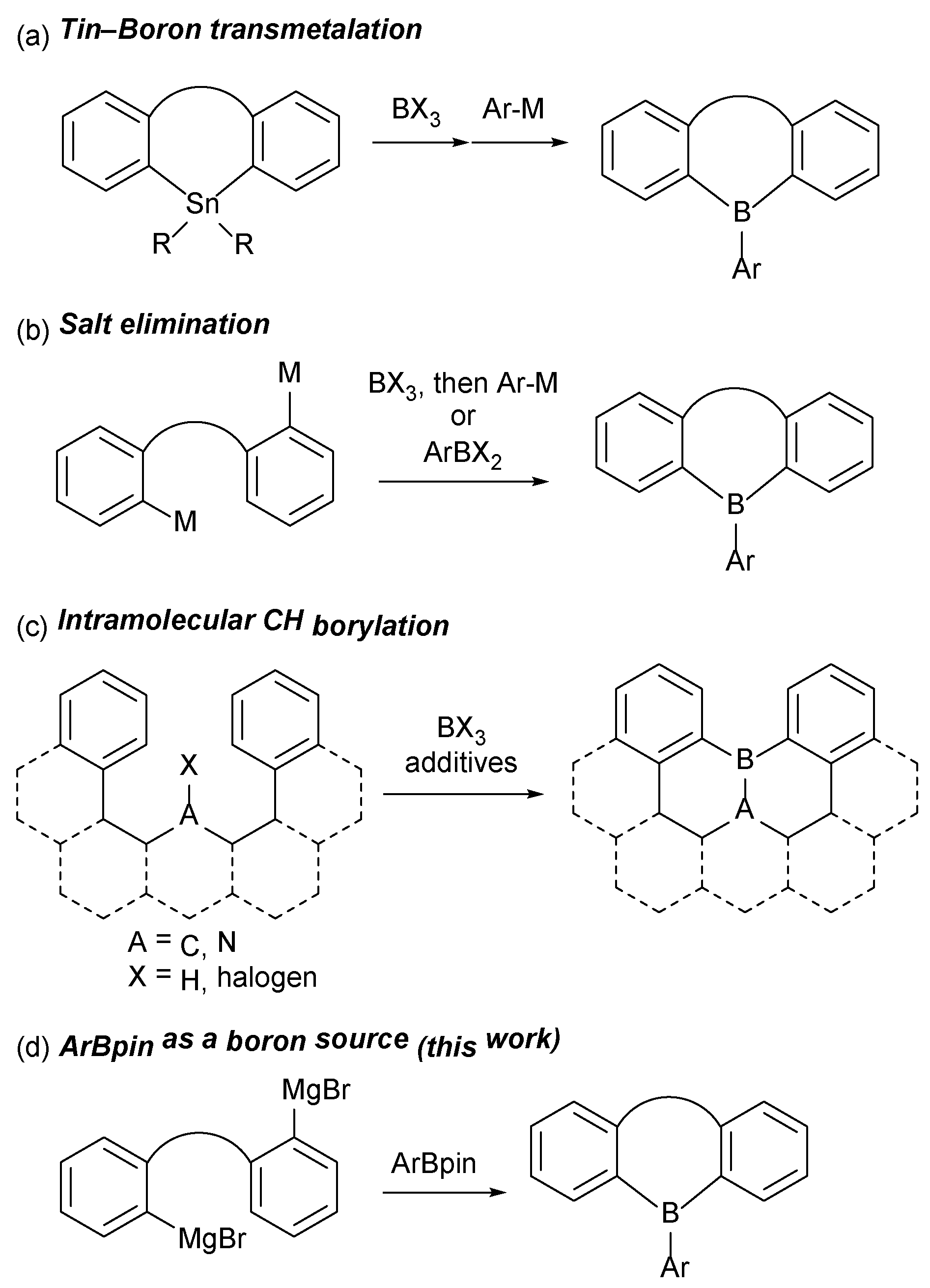
1. Introduction
2. Results and Discussion
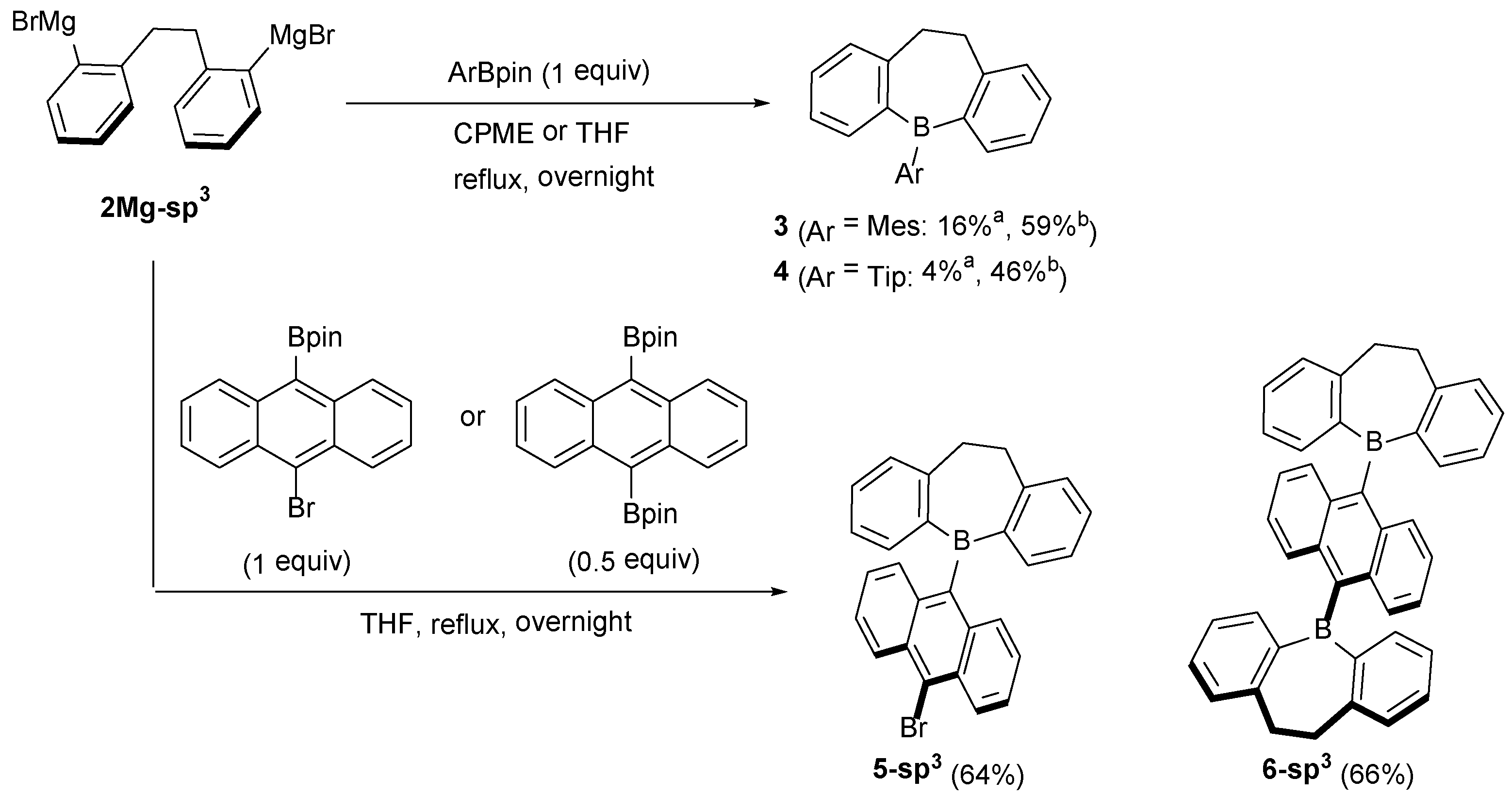
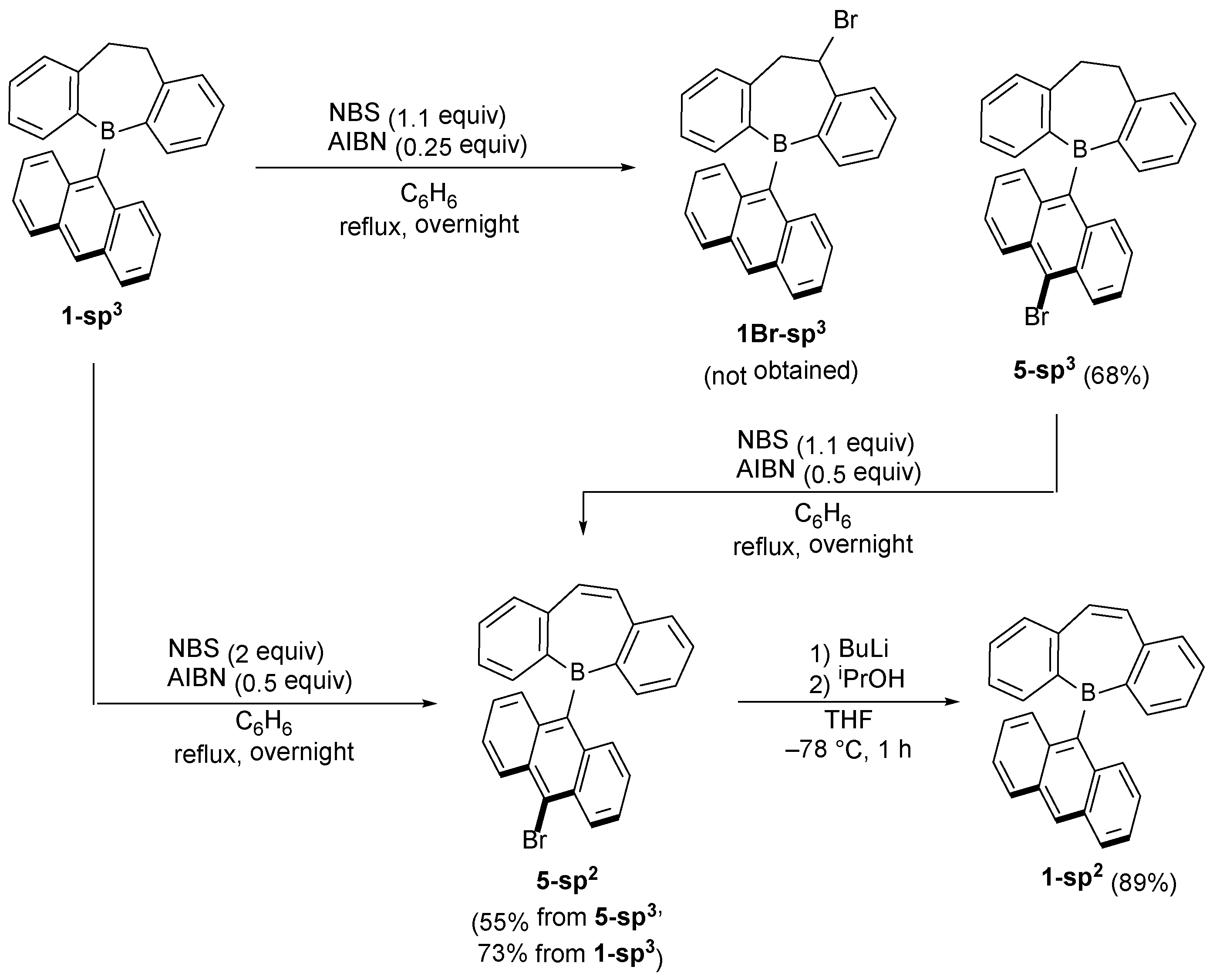
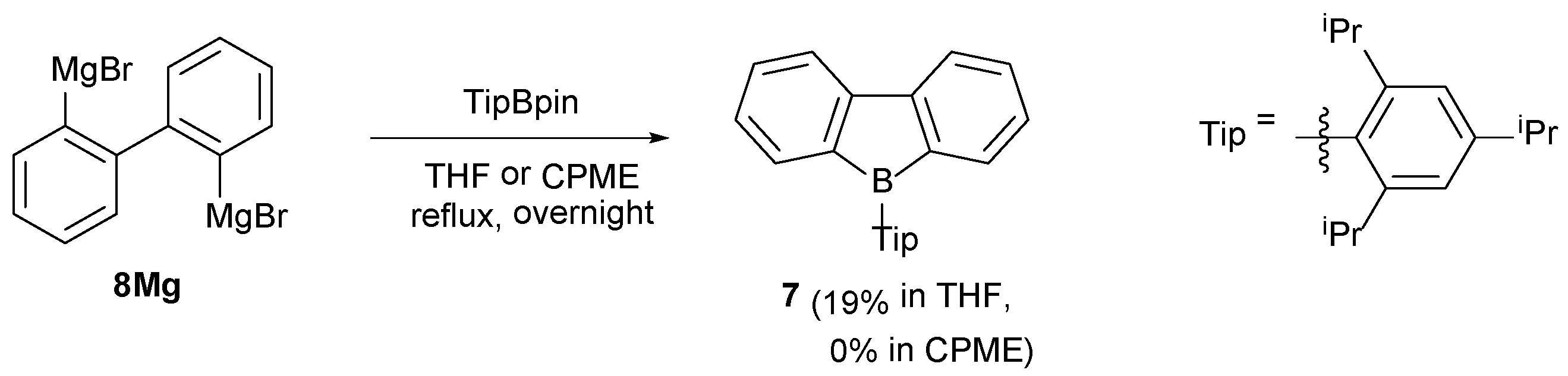
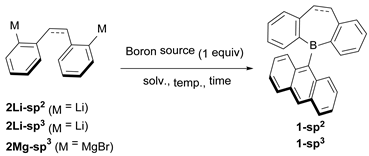
2.1. Synthetic Studies
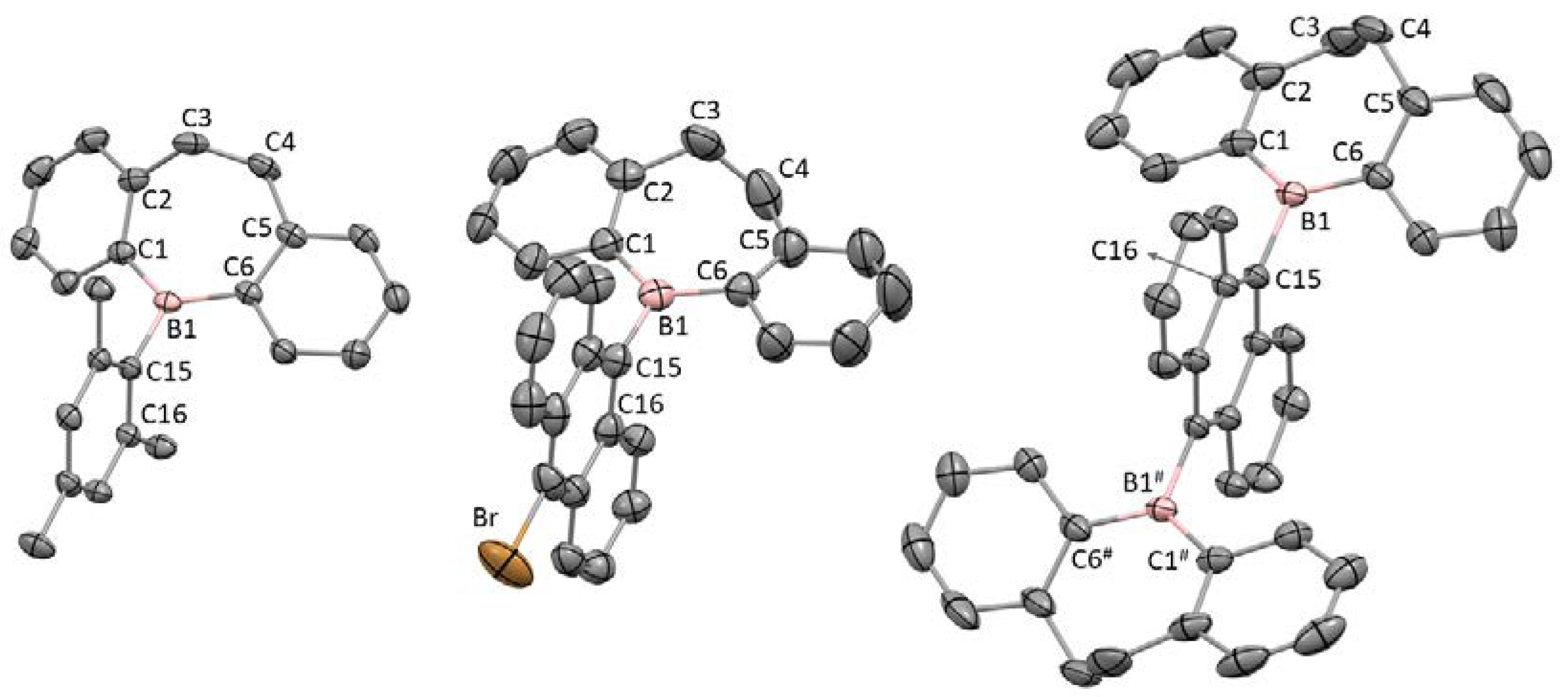
2.2. Single-Crystal X-ray Diffraction Analysis
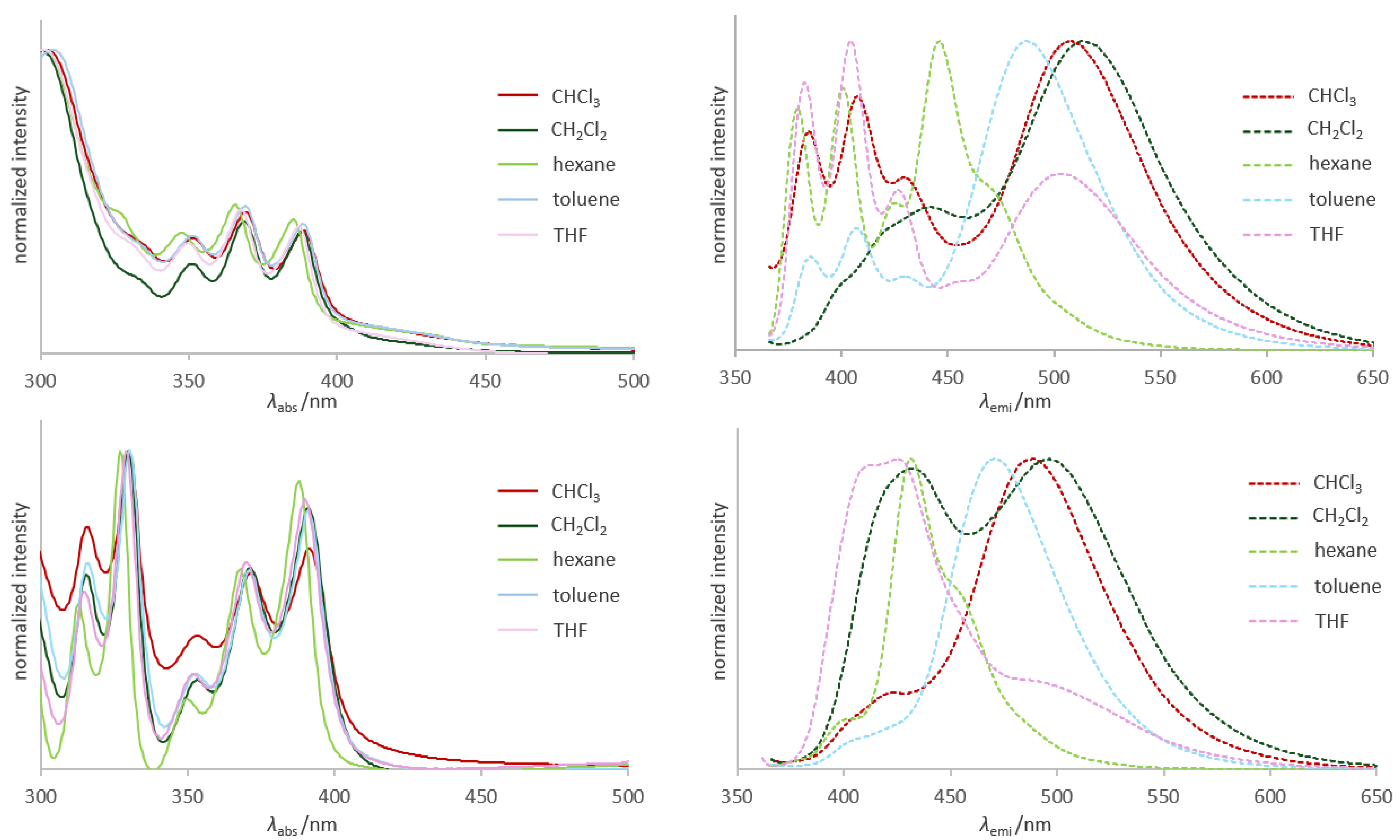
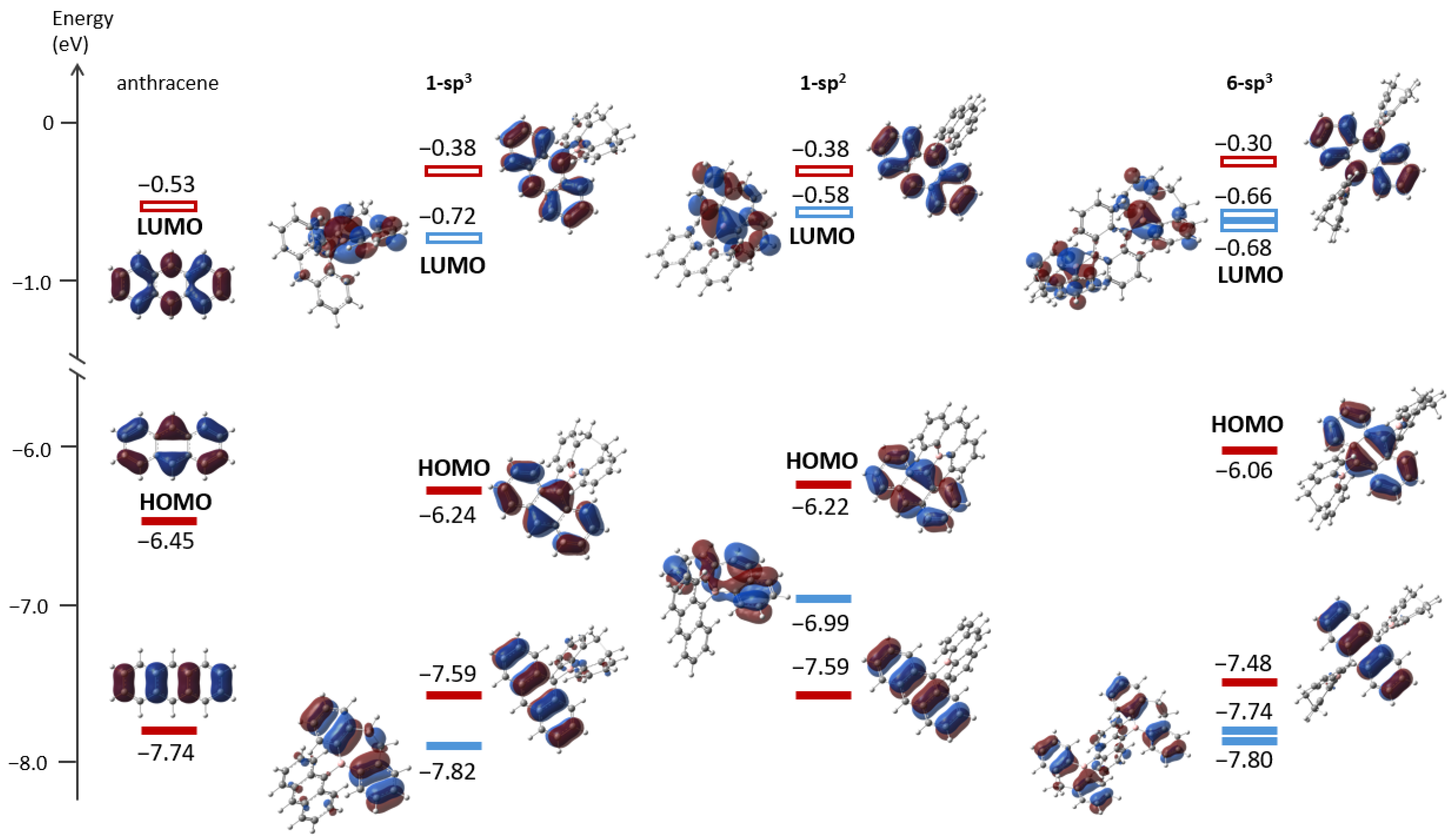
2.3. Photophysical Properties and Theoretical Calculations
3. Materials and Method
3.1. General Considerations
3.2. Preparation of 2Mg-sp3
3.3. Synthesis of 5-(9-Anthryl)-5H-9,10-dihydrodibenzo[b,f]borepin (1-sp3) Using AnthBpin
3.4. Synthesis of 5-(2,4,6-Trimethylphenyl)-5H-9,10-dihydrodibenzo[b,f]borepin (3)
3.5. Synthesis of 5-(2,4,6-Triisopropylphenyl)-5H-9,10-dihydrodibenzo[b,f]borepin (4)
3.6. Synthesis of 5-(10-Bromo-9-anthryl)-5H-9,10-dihydrodibenzo[b,f]borepin (5-sp3)
3.7. Synthesis of 9,10-Bis(5H-9,10-Dihydrodibenzo[b,f]borepin-5-yl)anthracene (6-sp3)
3.8. Synthesis of 5-sp2 from 5-sp3
3.9. Synthesis of 1-sp2 from 5-sp2
3.10. Synthesis of 7
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ji, L.; Griesbeck, S.; Marder, T.B. Recent developments in and perspectives on three-coordinate boron materials: A bright future. Chem. Sci. 2017, 8, 846–863. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, S.; Wakamiya, A. Boron as a key component for new π-electron materials. Pure Appl. Chem. 2006, 78, 1413–1424. [Google Scholar] [CrossRef]
- Hirai, M.; Tanaka, N.; Sakai, M.; Yamaguchi, S. Structurally Constrained Boron-, Nitrogen-, Silicon-, and Phosphorus-Centered Polycyclic π-Conjugated Systems. Chem. Rev. 2019, 119, 8291–8331. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Wang, S.; Dewhurst, R.D.; Ignat’ev, N.V.; Finze, M.; Braunschweig, H. Boron: Its Role in Energy-Related Processes and Applications. Angew. Chem. Int. Ed. 2020, 59, 8800–8816. [Google Scholar] [CrossRef]
- Braunschweig, H.; Kupfer, T. Recent developments in the chemistry of antiaromatic boroles. Chem. Commun. 2011, 47, 10903–10914. [Google Scholar] [CrossRef]
- Messersmith, R.E.; Tovar, J.D. Assessment of the aromaticity of borepin rings by spectroscopic, crystallographic and computational methods: A historical overview. J. Phys. Org. Chem. 2015, 28, 378–387. [Google Scholar] [CrossRef]
- Iida, A.; Yamaguchi, S. Thiophene-Fused Ladder Boroles with High Antiaromaticity. J. Am. Chem. Soc. 2011, 133, 6952–6955. [Google Scholar] [CrossRef]
- Mercier, L.G.; Piers, W.E.; Parvez, M. Benzo- and Naphthoborepins: Blue-Emitting Boron Analogues of Higher Acenes. Angew. Chem. Int. Ed. 2009, 48, 6108–6111. [Google Scholar] [CrossRef]
- Messersmith, R.E.; Siegler, M.A.; Tovar, J.D. Aromaticity Competition in Differentially Fused Borepin-Containing Polycyclic Aromatics. J. Org. Chem. 2016, 81, 5595–5605. [Google Scholar] [CrossRef]
- Ando, N.; Kushida, T.; Yamaguchi, S. A planarized B-phenyldibenzoborepin: Impact of structural constraint on its electronic properties and Lewis acidity. Chem. Commun. 2018, 54, 5213–5216. [Google Scholar] [CrossRef]
- Iqbal, S.A.; Pahl, J.; Yuan, K.; Ingleson, M.J. Intramolecular (directed) electrophilic C–H borylation. Chem. Soc. Rev. 2020, 49, 4564–4591. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Bartholome, T.A.; Tidwell, J.R.; Pujol, A.; Yruegas, S.; Martinez, J.J.; Martin, C.D. 9-Borafluorenes: Synthesis, Properties, and Reactivity. Chem. Rev. 2021, 121, 4147–4192. [Google Scholar] [CrossRef] [PubMed]
- Oda, S.; Hatakeyama, T. Development of One-Shot/One-Pot Borylation Reactions toward Organoboron-Based Materials. Bull. Chem. Soc. Jpn. 2021, 94, 950–960. [Google Scholar] [CrossRef]
- He, J.; Rauch, F.; Finze, M.; Marder, T.B. (Hetero)arene-fused boroles: A broad spectrum of applications. Chem. Sci. 2021, 12, 128–147. [Google Scholar] [CrossRef]
- Eisch, J.J.; Hota, N.K.; Kozima, S. Synthesis of pentaphenylborole, a potentially antiaromatic system. J. Am. Chem. Soc. 1969, 91, 4575–4577. [Google Scholar] [CrossRef]
- Eisch, J.J.; Galle, J.E.; Kozima, S. Bora-aromatic systems. Part 8. The physical and chemical consequences of cyclic conjugation in boracyclopolyenes. The antiaromatic character of pentaarylboroles. J. Am. Chem. Soc. 1986, 108, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Chase, P.A.; Piers, W.E.; Patrick, B.O. New Fluorinated 9-Borafluorene Lewis Acids. J. Am. Chem. Soc. 2000, 122, 12911–12912. [Google Scholar] [CrossRef]
- Wakamiya, A.; Mishima, K.; Ekawa, K.; Yamaguchi, S. Kinetically stabilized dibenzoborole as an electron-accepting building unit. Chem. Commun. 2008, 579–581. [Google Scholar] [CrossRef] [PubMed]
- Su, B.; Kinjo, R. Construction of Boron-Containing Aromatic Heterocycles. Synthesis 2017, 49, 2985–3034. [Google Scholar]
- Kuwabara, T.; Saito, M. 3.17—Siloles, Germoles, Stannoles, and Plumboles. In Comprehensive Heterocyclic Chemistry IV; Black, D.S., Cossy, J., Stevens, C.V., Eds.; Elsevier: Oxford, UK, 2022; pp. 798–832. [Google Scholar]
- Hatakeyama, T.; Hashimoto, S.; Seki, S.; Nakamura, M. Synthesis of BN-Fused Polycyclic Aromatics via Tandem Intramolecular Electrophilic Arene Borylation. J. Am. Chem. Soc. 2011, 133, 18614–18617. [Google Scholar] [CrossRef]
- Hirai, H.; Nakajima, K.; Nakatsuka, S.; Shiren, K.; Ni, J.; Nomura, S.; Ikuta, T.; Hatakeyama, T. One-Step Borylation of 1,3-Diaryloxybenzenes Towards Efficient Materials for Organic Light-Emitting Diodes. Angew. Chem. Int. Ed. 2015, 54, 13581–13585. [Google Scholar] [CrossRef] [PubMed]
- Nakatsuka, S.; Yasuda, N.; Hatakeyama, T. Four-Step Synthesis of B2N2-Embedded Corannulene. J. Am. Chem. Soc. 2018, 140, 13562–13565. [Google Scholar] [CrossRef]
- Matsui, K.; Oda, S.; Yoshiura, K.; Nakajima, K.; Yasuda, N.; Hatakeyama, T. One-Shot Multiple Borylation toward BN-Doped Nanographenes. J. Am. Chem. Soc. 2018, 140, 1195–1198. [Google Scholar] [CrossRef]
- Yamaguchi, S.; Shirasaka, T.; Akiyama, S.; Tamao, K. Dibenzoborole-Containing π-Electron Systems: Remarkable Fluorescence Change Based on the “On/Off” Control of the pπ–π* Conjugation. J. Am. Chem. Soc. 2002, 124, 8816–8817. [Google Scholar] [CrossRef] [PubMed]
- Iida, A.; Sekioka, A.; Yamaguchi, S. Heteroarene-fused boroles: What governs the antiaromaticity and Lewis acidity of the borole skeleton? Chem. Sci. 2012, 3, 1461–1466. [Google Scholar] [CrossRef]
- Brend’amour, S.; Gilmer, J.; Bolte, M.; Lerner, H.-W.; Wagner, M. C-Halogenated 9,10-Diboraanthracenes: How the Halogen Load and Distribution Influences Key Optoelectronic Properties. Chem. Eur. J. 2018, 24, 16910–16918. [Google Scholar] [CrossRef]
- Urban, M.; Durka, K.; Górka, P.; Wiosna-Sałyga, G.; Nawara, K.; Jankowski, P.; Luliński, S. The effect of locking π-conjugation in organoboron moieties in the structures of luminescent tetracoordinate boron complexes. Dalton Trans. 2019, 48, 8642–8663. [Google Scholar] [CrossRef]
- Lennox, A.J.J.; Lloyd-Jones, G.C. Selection of boron reagents for Suzuki–Miyaura coupling. Chem. Soc. Rev. 2014, 43, 412–443. [Google Scholar] [CrossRef]
- Asakawa, H.; Lee, K.-H.; Lin, Z.; Yamashita, M. Facile scission of isonitrile carbon–nitrogen triple bond using a diborane(4) reagent. Nat. Commun. 2014, 5, 4245. [Google Scholar] [CrossRef]
- Ishiyama, T.; Murata, M.; Miyaura, N. Palladium(0)-Catalyzed Cross-Coupling Reaction of Alkoxydiboron with Haloarenes: A Direct Procedure for Arylboronic Esters. J. Org. Chem. 1995, 60, 7508–7510. [Google Scholar] [CrossRef]
- Ishiyama, T.; Miyaura, N. Metal-catalyzed reactions of diborons for synthesis of organoboron compounds. Chem. Rec. 2004, 3, 271–280. [Google Scholar] [CrossRef]
- Hartwig, J.F. Borylation and Silylation of C–H Bonds: A Platform for Diverse C–H Bond Functionalizations. Acc. Chem. Res. 2012, 45, 864–873. [Google Scholar] [CrossRef]
- Mkhalid, I.A.I.; Barnard, J.H.; Marder, T.B.; Murphy, J.M.; Hartwig, J.F. C–H Activation for the Construction of C–B Bonds. Chem. Rev. 2010, 110, 890–931. [Google Scholar] [CrossRef] [PubMed]
- Levin, G.; Ward, T.A.; Szwarc, M. Electron-transfer catalyzed cis-trans isomerization of stilbene. Stability of sodium cis-stilbenide and the existence of sodium salts of cis- and of trans-stilbene dianions. J. Am. Chem. Soc. 1974, 96, 270–272. [Google Scholar] [CrossRef]
- Ito, S.; Kuwabara, T.; Ishii, Y. A Tin Analogue of the Cycloheptatrienyl Anion: Synthesis, Structure, and Further Reduction to Form a Dianionic Species. Organometallics 2020, 39, 640–644. [Google Scholar] [CrossRef]
- Sieg, G.; Müller, I.; Weißer, K.; Werncke, C.G. Taming the stilbene radical anion. Chem. Sci. 2022, 13, 13872–13878. [Google Scholar] [CrossRef] [PubMed]
- Grenz, D.C.; Schmidt, M.; Kratzert, D.; Esser, B. Dibenzo[a,e]pentalenes with Low-Lying LUMO Energy Levels as Potential n-Type Materials. J. Org. Chem. 2018, 83, 656–663. [Google Scholar] [CrossRef] [PubMed]
- Yuan, K.; Suzuki, N.; Mellerup, S.K.; Wang, X.; Yamaguchi, S.; Wang, S. Pyridyl Directed Catalyst-Free trans-Hydroboration of Internal Alkynes. Org. Lett. 2016, 18, 720–723. [Google Scholar] [CrossRef]
- Pospiech, S.; Bolte, M.; Lerner, H.-W.; Wagner, M. Diborylated Magnesium Anthracene as Precursor for B2H5−-Bridged 9,10-Dihydroanthracene. Chem. Eur. J. 2015, 21, 8229–8236. [Google Scholar] [CrossRef]
- van Tamelen, E.E.; Brieger, G.; Untch, K.G. Synthesis of a borepin. Tetrahedron Lett. 1960, 1, 14–15. [Google Scholar] [CrossRef]
- Matsuoka, W.; Ito, H.; Sarlah, D.; Itami, K. Diversity-oriented synthesis of nanographenes enabled by dearomative annulative π-extension. Nat. Commun. 2021, 12, 3940. [Google Scholar] [CrossRef] [PubMed]
- Pedireddi, V.R.; Reddy, D.S.; Goud, B.S.; Craig, D.C.; Rae, A.D.; Desiraju, G.R. The nature of halogen···halogen interactions and the crystal structure of 1,3,5,7-tetraiodoadamantane. J. Chem. Soc. Perkin Trans. 2 1994, 2353–2360. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef]
- Matter, H.; Nazaré, M.; Güssregen, S.; Will, D.W.; Schreuder, H.; Bauer, A.; Urmann, M.; Ritter, K.; Wagner, M.; Wehner, V. Evidence for C−Cl/C−Br⋅⋅⋅π Interactions as an Important Contribution to Protein–Ligand Binding Affinity. Angew. Chem. Int. Ed. 2009, 48, 2911–2916. [Google Scholar] [CrossRef]
- Förster, C.; Seichter, W.; Schwarzer, A.; Weber, E. Supramolecular behaviour of bulky arylboranes in the crystalline state. Supramol. Chem. 2010, 22, 571–581. [Google Scholar] [CrossRef]
- Griesbeck, S.; Ferger, M.; Czernetzi, C.; Wang, C.; Bertermann, R.; Friedrich, A.; Haehnel, M.; Sieh, D.; Taki, M.; Yamaguchi, S.; et al. Optimization of Aqueous Stability versus π-Conjugation in Tetracationic Bis(triarylborane) Chromophores: Applications in Live-Cell Fluorescence Imaging. Chem. Eur. J. 2019, 25, 7679–7688. [Google Scholar] [CrossRef] [PubMed]
- Iida, A.; Saito, S.; Sasamori, T.; Yamaguchi, S. Borylated Dibenzoborepin: Synthesis by Skeletal Rearrangement and Photochromism Based on Bora-Nazarov Cyclization. Angew. Chem. Int. Ed. 2013, 52, 3760–3764. [Google Scholar] [CrossRef]
- Chen, C.; Harhausen, M.; Liedtke, R.; Bussmann, K.; Fukazawa, A.; Yamaguchi, S.; Petersen, J.L.; Daniliuc, C.G.; Fröhlich, R.; Kehr, G.; et al. Dibenzopentalenes from B(C6F5)3-Induced Cyclization Reactions of 1,2-Bis(phenylethynyl)benzenes. Angew. Chem. Int. Ed. 2013, 52, 5992–5996. [Google Scholar] [CrossRef] [PubMed]
- Ashe, A.J.; Klein, W.; Rousseau, R. Evaluation of the aromaticity of borepin: Synthesis and properties of 1-substituted borepins. Organometallics 1993, 12, 3225–3231. [Google Scholar] [CrossRef]
- Jones, R.N. The Ultraviolet Absorption Spectra of Anthracene Derivatives. Chem. Rev. 1947, 41, 353–371. [Google Scholar] [CrossRef]
- Yamaguchi, S.; Akiyama, S.; Tamao, K. Tri-9-anthrylborane and Its Derivatives: New Boron-Containing π-Electron Systems with Divergently Extended π-Conjugation through Boron. J. Am. Chem. Soc. 2000, 122, 6335–6336. [Google Scholar] [CrossRef]
- Charlot, M.; Porrès, L.; Entwistle, C.D.; Beeby, A.; Marder, T.B.; Blanchard-Desce, M. Investigation of two-photon absorption behavior in symmetrical acceptor–π–acceptor derivatives with dimesitylboryl end-groups. Evidence of new engineering routes for TPA/transparency trade-off optimization. Phys. Chem. Chem. Phys. 2005, 7, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Kosower, E.M. Intramolecular donor-acceptor systems. 9. Photophysics of (phenylamino)naphthalenesulfonates: A paradigm for excited-state intramolecular charge transfer. Acc. Chem. Res. 1982, 15, 259–266. [Google Scholar] [CrossRef]
- Kuwabara, T.; Orii, J.; Segawa, Y.; Itami, K. Curved Oligophenylenes as Donors in Shape-Persistent Donor–Acceptor Macrocycles with Solvatofluorochromic Properties. Angew. Chem. Int. Ed. 2015, 54, 9646–9649. [Google Scholar] [CrossRef] [PubMed]
- Hermann, M.; Wassy, D.; Esser, B. Conjugated Nanohoops Incorporating Donor, Acceptor, Hetero- or Polycyclic Aromatics. Angew. Chem. Int. Ed. 2021, 60, 15743–15766. [Google Scholar] [CrossRef] [PubMed]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Francl, M.M.; Pietro, W.J.; Hehre, W.J.; Binkley, J.S.; Gordon, M.S.; DeFrees, D.J.; Pople, J.A. Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. J. Chem. Phys. 1982, 77, 3654–3665. [Google Scholar] [CrossRef]
- Zhu, J.; Lin, Z.; Marder, T.B. Trans Influence of Boryl Ligands and Comparison with C, Si, and Sn Ligands. Inorg. Chem. 2005, 44, 9384–9390. [Google Scholar] [CrossRef]
- Kupracz, L.; Kirschning, A. Multiple Organolithium Generation in the Continuous Flow Synthesis of Amitriptyline. Adv. Synth. Catal. 2013, 355, 3375–3380. [Google Scholar] [CrossRef]
- Zhang, H.; Yu, T.; Zhao, Y.; Fan, D.; Xia, Y.; Zhang, P. Synthesis, crystal structure, photo- and electro-luminescence of 3-(4-(anthracen-10-yl)phenyl)-7-(N,N′-diethylamino)coumarin. Synth. Met. 2010, 160, 1642–1647. [Google Scholar] [CrossRef]
- Zhu, Y.; Rabindranath, A.R.; Beyerlein, T.; Tieke, B. Highly Luminescent 1,4-Diketo-3,6-diphenylpyrrolo[3,4-c]pyrrole- (DPP-) Based Conjugated Polymers Prepared Upon Suzuki Coupling. Macromolecules 2007, 40, 6981–6989. [Google Scholar] [CrossRef]
- Filthaus, M.; Oppel, I.M.; Bettinger, H.F. Supramolecular structures and spontaneous resolution: The case of ortho-substituted phenylboronic acids. Org. Biomol. Chem. 2008, 6, 1201–1207. [Google Scholar] [CrossRef] [PubMed]
- Yuan, K.; Wang, X.; Wang, S. Cascade Dehydrogenative Hydroboration for the Synthesis of Azaborabenzofulvenes. Org. Lett. 2018, 20, 1617–1620. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev.A.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
| Entry | Boron Source | Nucleophile | Conditions | Yield |
| 1 | AnthBpin | 2Li-sp2 | C6H6/Et2O, 5 °C to r.t., overnight | 1-sp2 (–) |
| 2 | AnthBpin | 2Li-sp3 | C6H6/Et2O, 5 °C to r.t., overnight | 1-sp3 (–) |
| 3 | AnthBpin | 2Mg-sp3 | THF, reflux, overnight | 1-sp3 (62%) |
| 4 | AnthB(OMe)2 | 2Mg-sp3 | THF, reflux, overnight | 1-sp3 (31%) |
| 5 | AnthBpin | 2Mg-sp2 a | THF, reflux, overnight | 1-sp2 (–) |
| 3 | 5-sp3 | 6-sp3 | |
|---|---|---|---|
| B1–C1/C6 | 1.5682(18)/ 1.5706(19) | 1.562(5)/ 1.567(5) | 1.572(3)/ 1.569(4) |
| B1–C15 | 1.5866(17) | 1.592(5) | 1.594(3) |
| C1–B1–C15–C16 | 89.80(14) | 89.2(4) | 80.2(2) |
| C1–C2–C5–C6 | 19.1(1) | 20.2(6) | 18.9(2) |
| Solvent | λabs/nm (ε) | λemi/nm | Stokes Shift/cm−1 | |
|---|---|---|---|---|
| 1-sp3 | hexane | 385 (9300), 366 (10,000), 348 (8300) | 380, 400, 425, 446 | 3550 |
| toluene | 388 (9300), 369 (11,000), 351 (8400) | 385, 407, 431, 487 | 5240 | |
| THF | 387 (9700), 367 (11,000), 350 (8400) | 382, 404, 426, 503 | 5960 | |
| CHCl3 | 389 (7200), 369 (8300), 348 (6800) | 384, 407, 430, 508 | 6020 | |
| CH2Cl2 | 388 (9400), 369 (10,000), 351 (6900) | 441, 513 | 6280 | |
| 1-sp2 | hexane | 388 (14000), 368 (10,000), 350 (3900), 327 (16,000), 313 (8600) | 432, 451 | 3600 |
| toluene | 391 (17,000), 371 (13,000), 353 (6400), 330 (21,000), 316 (14,000) | 471 | 4340 | |
| THF | 390 (15,000), 370 (12,000), 352 (5400), 329 (18,000), 315 (10,000) | 410, 425 | 2110 | |
| CHCl3 | 391 (15,000), 372 (13,000), 353 (9100), 330 (22,000), 316 (16,000) | 489 | 5130 | |
| CH2Cl2 | 391 (12,000), 371 (9200), 353 (4000), 330 (14,000), 316 (8700) | 432, 496 | 5410 | |
| 6-sp3 | CH2Cl2 | 398 (9300), 378 (10,000), 360 (7900) | 532 | 6330 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kawaguchi, H.; Fuse, K.; Maeda, N.; Kuwabara, T. Arylboronic Acid Pinacol Esters as Stable Boron Sources for Dihydrodibenzoborepin Derivatives and a Dibenzoborole. Molecules 2024, 29, 4024. https://doi.org/10.3390/molecules29174024
Kawaguchi H, Fuse K, Maeda N, Kuwabara T. Arylboronic Acid Pinacol Esters as Stable Boron Sources for Dihydrodibenzoborepin Derivatives and a Dibenzoborole. Molecules. 2024; 29(17):4024. https://doi.org/10.3390/molecules29174024
Chicago/Turabian StyleKawaguchi, Himeko, Kotomi Fuse, Nanoka Maeda, and Takuya Kuwabara. 2024. "Arylboronic Acid Pinacol Esters as Stable Boron Sources for Dihydrodibenzoborepin Derivatives and a Dibenzoborole" Molecules 29, no. 17: 4024. https://doi.org/10.3390/molecules29174024
APA StyleKawaguchi, H., Fuse, K., Maeda, N., & Kuwabara, T. (2024). Arylboronic Acid Pinacol Esters as Stable Boron Sources for Dihydrodibenzoborepin Derivatives and a Dibenzoborole. Molecules, 29(17), 4024. https://doi.org/10.3390/molecules29174024