
Identification of Novel Bromodomain-Containing Protein 4 (BRD4) Binders through 3D Pharmacophore-Based Repositioning Screening Campaign

 ,
, 


Abstract

1. Introduction
2. Results and Discussion
2.1. Development of 3D Structure-Based Pharmacophore Model of BRD4
2.2. Drug Repositioning Campaign of In-House Library through Developed BRD4 3D Structure-Based Pharmacophore Model
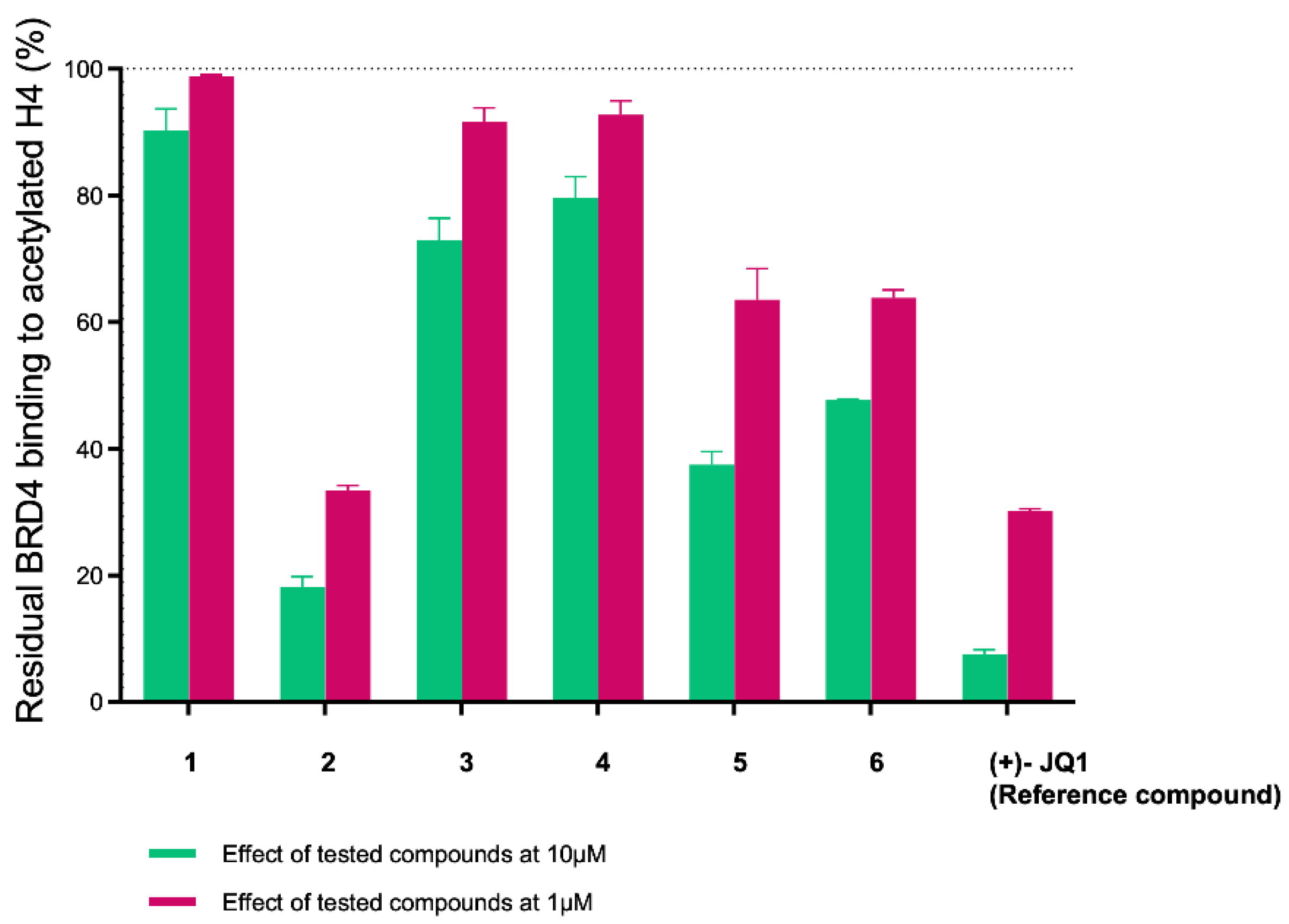
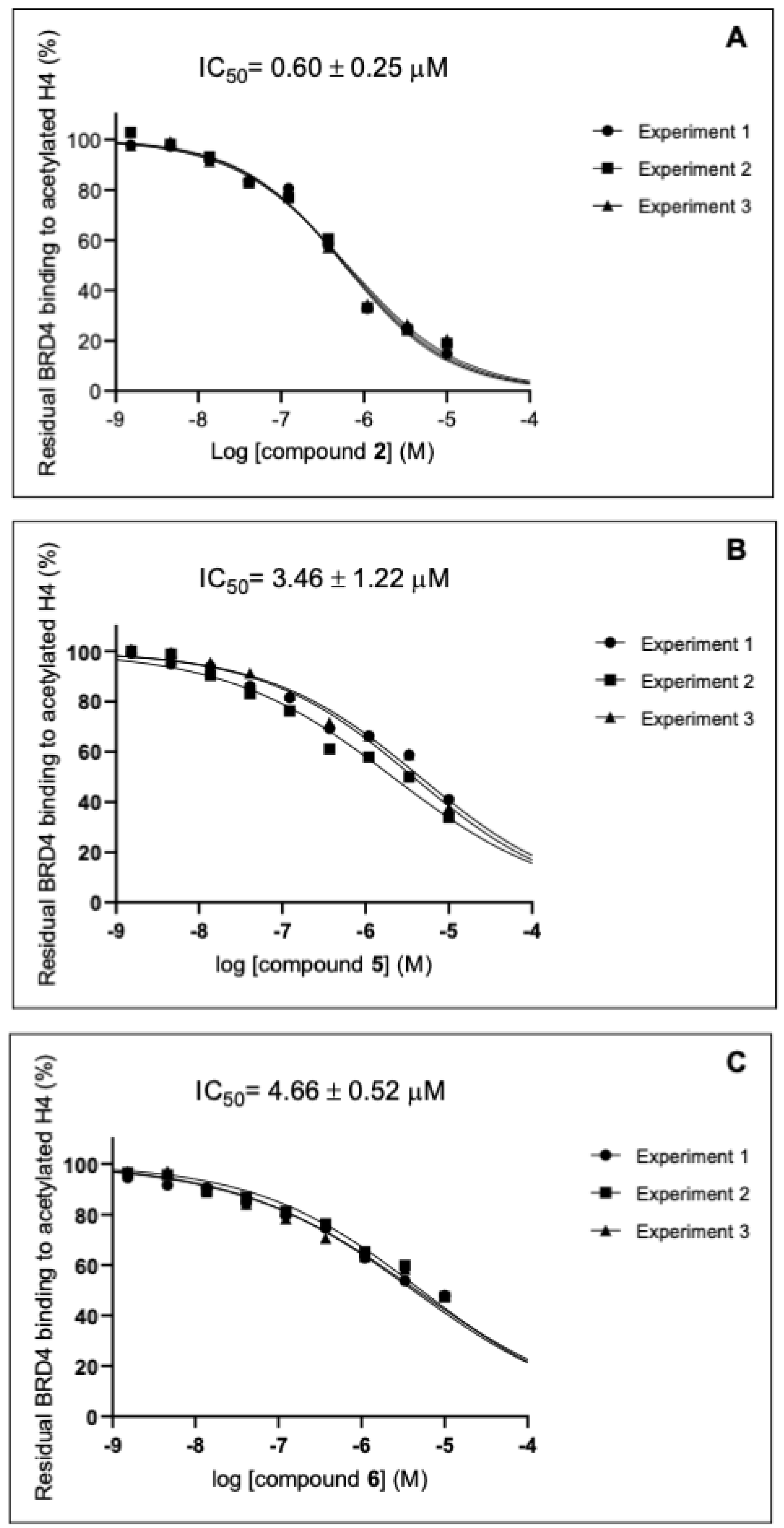
2.3. Evaluation of In Silico Predicted Binding on BRD4 by AlphaScreen Assay
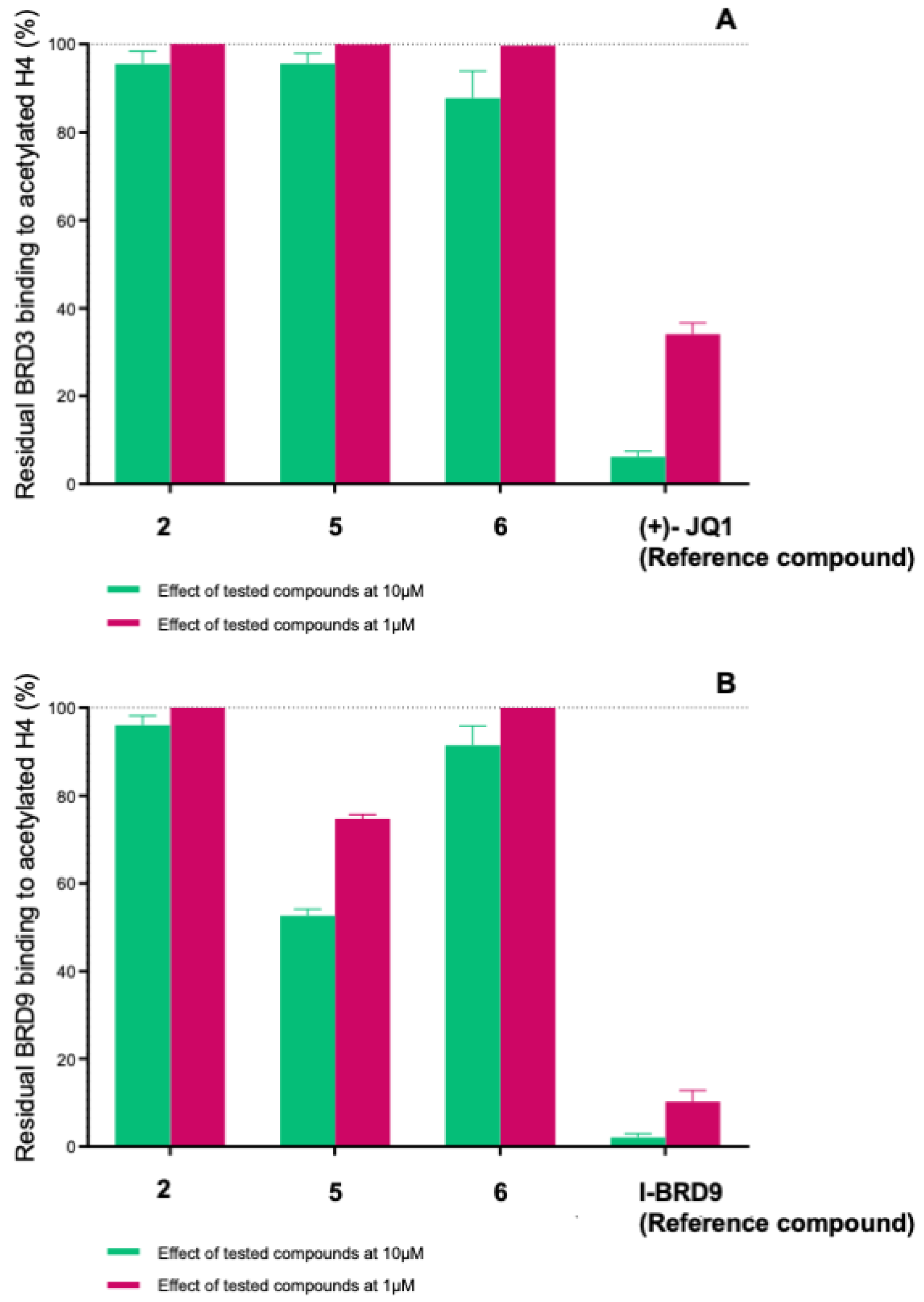
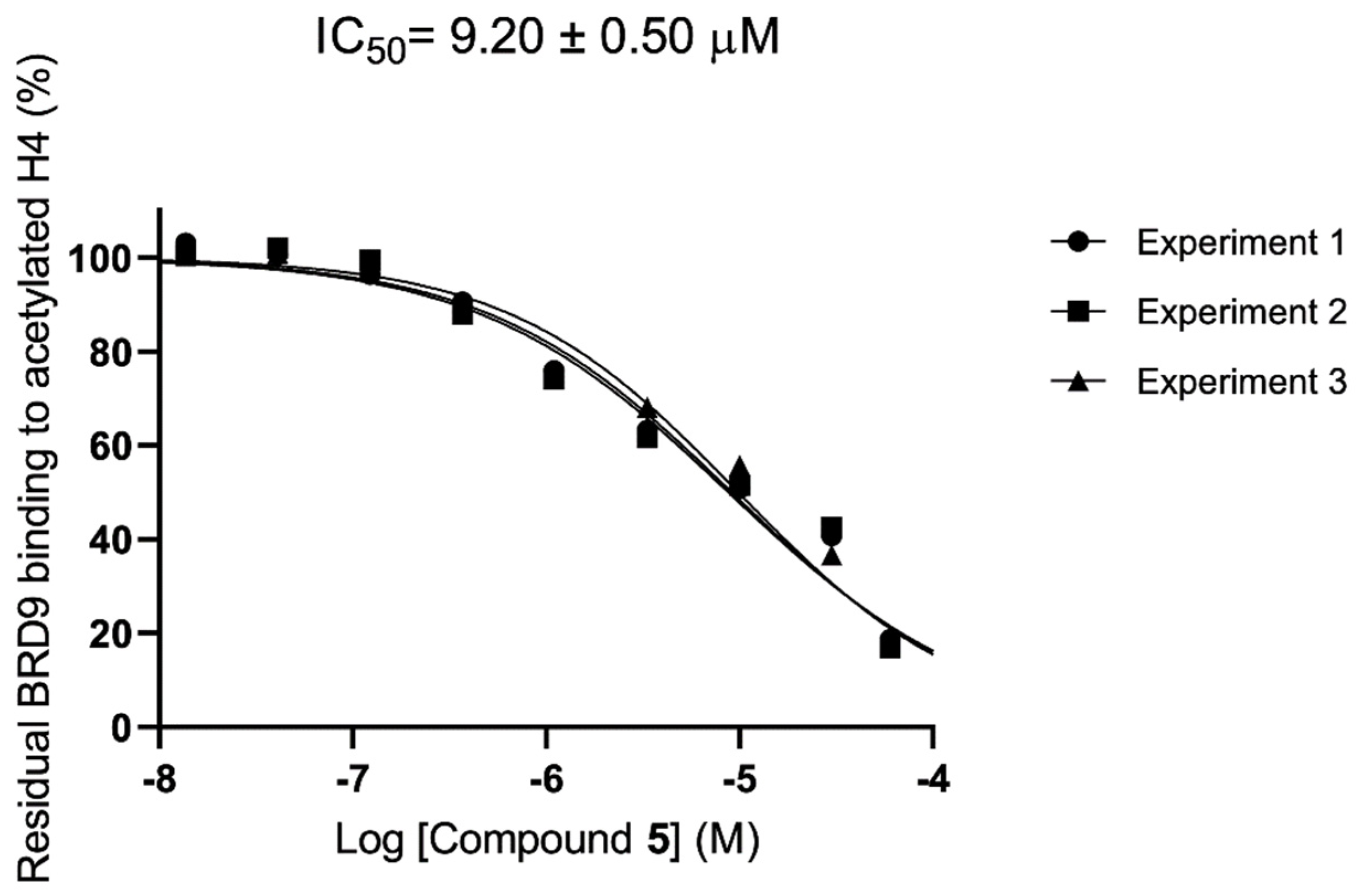
2.4. Evaluation of Selectivity among Other Bromodomains
3. Materials and Methods
3.1. Computational Details
3.1.1. Preparation of Library
3.1.2. Development of 3D Structure-Based Pharmacophore Model for BRD4
3.1.3. Ligand-Based Pharmacophore Screening
3.1.4. Preparation of Protein Structure and Molecular Docking Experiments on BRD4
3.1.5. Structure-Based Pharmacophore Screening
3.2. Biological Evaluation on BRD4(BD1), BRD3(BD1), and BRD9
- BRD4(BD1) and BRD3(BD1): GST-tag-protein concentration = 1.6 ng/μL; buffer type = Hepes solution (BSP-31091); AlphaLISA acceptor beads = 1/250-fold dilution (PerkinElmer #AL109C), streptavidin donor beads = 1/250-fold dilution (PerkinElmer #6760002);
- BRD9: GST-tag-protein concentration = 2 ng/μL; buffer type = tris−HCl solution (BSP-33007); AlphaLISA acceptor beads = 1/250-fold dilution (PerkinElmer #AL109C), streptavidin donor beads = 1/125-fold dilution (PerkinElmer #6760002);
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Huang, Q.-X.; Fan, D.-M.; Zheng, Z.-Z.; Ran, T.; Bai, A.; Xiao, R.-Q.; Hu, G.-S.; Liu, W. Peptide Inhibitor Targeting the Extraterminal Domain in BRD4 Potently Suppresses Breast Cancer Both In Vitro and In Vivo. J. Med. Chem. 2024, 67, 6658–6672. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.V.; Alauddin, S. Review on: BRD4 inhibitors for anticancer research. Human Gene 2023, 37, 201196. [Google Scholar] [CrossRef]
- White, M.E.; Fenger, J.M.; Carson, W.E. Emerging roles of and therapeutic strategies targeting BRD4 in cancer. Cell. Immunol. 2019, 337, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Guan, Y.; Qin, W.; Zhai, X.; Yu, B.; Liu, H. Targeting Brd4 for cancer therapy: Inhibitors and degraders. MedChemComm 2018, 9, 1779–1802. [Google Scholar] [CrossRef] [PubMed]
- Gazzillo, E.; Pierri, M.; Colarusso, E.; Chini, M.G.; Ferraro, M.G.; Piccolo, M.; Irace, C.; Bruno, I.; Bifulco, G.; Terracciano, S.; et al. Exploring the chemical space of functionalized [1,2,4]triazolo[4,3-a]quinoxaline-based compounds targeting the bromodomain of BRD9. Bioorg. Chem. 2023, 139, 106677. [Google Scholar] [CrossRef]
- Colarusso, E.; Gazzillo, E.; Boccia, E.; Giordano, A.; Chini, M.G.; Bifulco, G.; Lauro, G. 6-Methylquinazolin-4(3H)-one Based Compounds as BRD9 Epigenetic Reader Binders: A Rational Combination of in silico Studies and Chemical Synthesis. Eur. J. Org. Chem. 2022, 2022, e202200868. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef]
- Gazzillo, E.; Terracciano, S.; Ruggiero, D.; Potenza, M.; Chini, M.G.; Lauro, G.; Fischer, K.; Hofstetter, R.K.; Giordano, A.; Werz, O.; et al. Repositioning of Quinazolinedione-Based Compounds on Soluble Epoxide Hydrolase (sEH) through 3D Structure-Based Pharmacophore Model-Driven Investigation. Molecules 2022, 27, 3866–3886. [Google Scholar] [CrossRef]
- Gacias, M.; Gerona-Navarro, G.; Plotnikov, A.N.; Zhang, G.; Zeng, L.; Kaur, J.; Moy, G.; Rusinova, E.; Rodriguez, Y.; Matikainen, B.; et al. Selective Chemical Modulation of Gene Transcription Favors Oligodendrocyte Lineage Progression. Chem. Biol. 2014, 21, 841–854. [Google Scholar] [CrossRef]
- Chung, C.-W.; Coste, H.; White, J.H.; Mirguet, O.; Wilde, J.; Gosmini, R.L.; Delves, C.; Magny, S.M.; Woodward, R.; Hughes, S.A.; et al. Discovery and Characterization of Small Molecule Inhibitors of the BET Family Bromodomains. J. Med. Chem. 2011, 54, 3827–3838. [Google Scholar] [CrossRef]
- Nicodeme, E.; Jeffrey, K.L.; Schaefer, U.; Beinke, S.; Dewell, S.; Chung, C.-W.; Chandwani, R.; Marazzi, I.; Wilson, P.; Coste, H.; et al. Suppression of inflammation by a synthetic histone mimic. Nature 2010, 468, 1119–1123. [Google Scholar] [CrossRef]
- Zhang, G.; Liu, R.; Zhong, Y.; Plotnikov, A.N.; Zhang, W.; Zeng, L.; Rusinova, E.; Gerona-Nevarro, G.; Moshkina, N.; Joshua, J.; et al. Down-regulation of NF-κB Transcriptional Activity in HIV-associated Kidney Disease by BRD4 Inhibition. J. Biol. Chem. 2012, 287, 28840–28851. [Google Scholar] [CrossRef]
- Gehling, V.S.; Hewitt, M.C.; Vaswani, R.G.; Leblanc, Y.; Côté, A.; Nasveschuk, C.G.; Taylor, A.M.; Harmange, J.-C.; Audia, J.E.; Pardo, E.; et al. Discovery, Design, and Optimization of Isoxazole Azepine BET Inhibitors. ACS Med. Chem. Lett. 2013, 4, 835–840. [Google Scholar] [CrossRef]
- Albrecht, B.K.; Gehling, V.S.; Hewitt, M.C.; Vaswani, R.G.; Côté, A.; Leblanc, Y.; Nasveschuk, C.G.; Bellon, S.; Bergeron, L.; Campbell, R.; et al. Identification of a Benzoisoxazoloazepine Inhibitor (CPI-0610) of the Bromodomain and Extra-Terminal (BET) Family as a Candidate for Human Clinical Trials. J. Med. Chem. 2016, 59, 1330–1339. [Google Scholar] [CrossRef] [PubMed]
- Ozer, H.G.; El-Gamal, D.; Powell, B.; Hing, Z.A.; Blachly, J.S.; Harrington, B.; Mitchell, S.; Grieselhuber, N.R.; Williams, K.; Lai, T.-H.; et al. BRD4 Profiling Identifies Critical Chronic Lymphocytic Leukemia Oncogenic Circuits and Reveals Sensitivity to PLX51107, a Novel Structurally Distinct BET Inhibitor. Cancer Discov. 2018, 8, 458–477. [Google Scholar] [CrossRef] [PubMed]
- Picaud, S.; Leonards, K.; Lambert, J.-P.; Dovey, O.; Wells, C.; Fedorov, O.; Monteiro, O.; Fujisawa, T.; Wang, C.-Y.; Lingard, H.; et al. Promiscuous targeting of bromodomains by bromosporine identifies BET proteins as master regulators of primary transcription response in leukemia. Sci. Adv. 2016, 2, e1600760. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhang, Y.; Song, M.; Xue, X.; Wang, J.; Wang, C.; Zhang, C.; Li, C.; Xiang, Q.; Zou, L.; et al. Structure-Based Discovery and Optimization of Benzo[d]isoxazole Derivatives as Potent and Selective BET Inhibitors for Potential Treatment of Castration-Resistant Prostate Cancer (CRPC). J. Med. Chem. 2018, 61, 3037–3058. [Google Scholar] [CrossRef]
- Colarusso, E.; Chini, M.G.; Bifulco, G.; Lauro, G.; Giordano, A. Identification and Development of BRD9 Chemical Probes. Pharmaceuticals 2024, 17, 392–423. [Google Scholar] [CrossRef]
- Gajjela, B.K.; Zhou, M.-M. Bromodomain inhibitors and therapeutic applications. Curr. Opin. Chem. Biol. 2023, 75, 102323. [Google Scholar] [CrossRef]
- Schrödinger Release 2021-1: LigPrep; Schrödinger, LLC: New York, NY, USA, 2021.
- Laoui, A.; Polyakov, V.R. Web services as applications’ integration tool: QikProp case study. J. Comput. Chem. 2011, 32, 1944–1951. [Google Scholar] [CrossRef]
- Lauber, D.T.; Fülöp, A.; Kovács, T.; Szigeti, K.; Máthé, D.; Szijártó, A. State of the art in vivo imaging techniques for laboratory animals. Lab. Anim. 2017, 51, 465–478. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2021-1: QikProp; Schrödinger, LLC: New York, NY, USA, 2021.
- Dixon, S.L.; Smondyrev, A.M.; Knoll, E.H.; Rao, S.N.; Shaw, D.E.; Friesner, R.A. PHASE: A new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1. Methodology and preliminary results. J. Comput. Aided Mol. Des. 2006, 20, 647–671. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.L.; Smondyrev, A.M.; Rao, S.N. PHASE: A novel approach to pharmacophore modeling and 3D database searching. Chem. Biol. Drug Des. 2006, 67, 370–372. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2021-1: Phase; Schrödinger, LLC: New York, NY, USA, 2021.
- Bradbury, R.H.; Callis, R.; Carr, G.R.; Chen, H.; Clark, E.; Feron, L.; Glossop, S.; Graham, M.A.; Hattersley, M.; Jones, C.; et al. Optimization of a Series of Bivalent Triazolopyridazine Based Bromodomain and Extraterminal Inhibitors: The Discovery of (3R)-4-[2-[4-[1-(3-Methoxy-[1,2,4]triazolo[4,3-b]pyridazin-6-yl)-4-piperidyl]phenoxy]ethyl]-1,3-dimethyl-piperazin-2-one (AZD5153). J. Med. Chem. 2016, 59, 7801–7817. [Google Scholar] [CrossRef]
- Crawford, T.D.; Tsui, V.; Flynn, E.M.; Wang, S.; Taylor, A.M.; Cote, A.; Audia, J.E.; Beresini, M.H.; Burdick, D.J.; Cummings, R.; et al. Diving into the Water: Inducible Binding Conformations for BRD4, TAF1(2), BRD9, and CECR2 Bromodomains. J. Med. Chem. 2016, 59, 5391–5402. [Google Scholar] [CrossRef]
- Dawson, M.A.; Prinjha, R.K.; Dittmann, A.; Giotopoulos, G.; Bantscheff, M.; Chan, W.I.; Robson, S.C.; Chung, C.W.; Hopf, C.; Savitski, M.M.; et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 2011, 478, 529–533. [Google Scholar] [CrossRef]
- Ember, S.W.; Lambert, Q.T.; Berndt, N.; Gunawan, S.; Ayaz, M.; Tauro, M.; Zhu, J.Y.; Cranfill, P.J.; Greninger, P.; Lynch, C.C.; et al. Potent Dual BET Bromodomain-Kinase Inhibitors as Value-Added Multitargeted Chemical Probes and Cancer Therapeutics. Mol. Cancer Ther. 2017, 16, 1054–1067. [Google Scholar] [CrossRef]
- Ember, S.W.; Zhu, J.Y.; Olesen, S.H.; Martin, M.P.; Becker, A.; Berndt, N.; Georg, G.I.; Schonbrunn, E. Acetyl-lysine binding site of bromodomain-containing protein 4 (BRD4) interacts with diverse kinase inhibitors. ACS Chem. Biol. 2014, 9, 1160–1171. [Google Scholar] [CrossRef]
- Karim, R.M.; Bikowitz, M.J.; Chan, A.; Zhu, J.Y.; Grassie, D.; Becker, A.; Berndt, N.; Gunawan, S.; Lawrence, N.J.; Schonbrunn, E. Differential BET Bromodomain Inhibition by Dihydropteridinone and Pyrimidodiazepinone Kinase Inhibitors. J. Med. Chem. 2021, 64, 15772–15786. [Google Scholar] [CrossRef]
- McLure, K.G.; Gesner, E.M.; Tsujikawa, L.; Kharenko, O.A.; Attwell, S.; Campeau, E.; Wasiak, S.; Stein, A.; White, A.; Fontano, E.; et al. RVX-208, an inducer of ApoA-I in humans, is a BET bromodomain antagonist. PLoS ONE 2013, 8, e83190. [Google Scholar] [CrossRef]
- Picaud, S.; Wells, C.; Felletar, I.; Brotherton, D.; Martin, S.; Savitsky, P.; Diez-Dacal, B.; Philpott, M.; Bountra, C.; Lingard, H.; et al. RVX-208, an inhibitor of BET transcriptional regulators with selectivity for the second bromodomain. Proc. Natl. Acad. Sci. USA 2013, 110, 19754–19759. [Google Scholar] [CrossRef]
- Ran, X.; Zhao, Y.; Liu, L.; Bai, L.; Yang, C.Y.; Zhou, B.; Meagher, J.L.; Chinnaswamy, K.; Stuckey, J.A.; Wang, S. Structure-Based Design of gamma-Carboline Analogues as Potent and Specific BET Bromodomain Inhibitors. J. Med. Chem. 2015, 58, 4927–4939. [Google Scholar] [CrossRef]
- Sullivan, J.M.; Badimon, A.; Schaefer, U.; Ayata, P.; Gray, J.; Chung, C.W.; von Schimmelmann, M.; Zhang, F.; Garton, N.; Smithers, N.; et al. Autism-like syndrome is induced by pharmacological suppression of BET proteins in young mice. J. Exp. Med. 2015, 212, 1771–1781. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Roberts, J.M.; Seo, H.S.; Souza, A.; Paulk, J.; Scott, T.G.; DeAngelo, S.L.; Dhe-Paganon, S.; Bradner, J.E. Design and characterization of bivalent BET inhibitors. Nat. Chem. Biol. 2016, 12, 1089–1096. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.M.; Vaswani, R.G.; Gehling, V.S.; Hewitt, M.C.; Leblanc, Y.; Audia, J.E.; Bellon, S.; Cummings, R.T.; Cote, A.; Harmange, J.C.; et al. Discovery of Benzotriazolo[4,3-d][1,4]diazepines as Orally Active Inhibitors of BET Bromodomains. ACS Med. Chem. Lett. 2016, 7, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Theodoulou, N.H.; Bamborough, P.; Bannister, A.J.; Becher, I.; Bit, R.A.; Che, K.H.; Chung, C.W.; Dittmann, A.; Drewes, G.; Drewry, D.H.; et al. Discovery of I-BRD9, a Selective Cell Active Chemical Probe for Bromodomain Containing Protein 9 Inhibition. J. Med. Chem. 2016, 59, 1425–1439. [Google Scholar] [CrossRef]
- Waring, M.J.; Chen, H.; Rabow, A.A.; Walker, G.; Bobby, R.; Boiko, S.; Bradbury, R.H.; Callis, R.; Clark, E.; Dale, I.; et al. Potent and selective bivalent inhibitors of BET bromodomains. Nat. Chem. Biol. 2016, 12, 1097–1104. [Google Scholar] [CrossRef]
- Watts, E.; Heidenreich, D.; Tucker, E.; Raab, M.; Strebhardt, K.; Chesler, L.; Knapp, S.; Bellenie, B.; Hoelder, S. Designing Dual Inhibitors of Anaplastic Lymphoma Kinase (ALK) and Bromodomain-4 (BRD4) by Tuning Kinase Selectivity. J. Med. Chem. 2019, 62, 2618–2637. [Google Scholar] [CrossRef]
- Yang, S.M.; Urban, D.J.; Yoshioka, M.; Strovel, J.W.; Fletcher, S.; Wang, A.Q.; Xu, X.; Shah, P.; Hu, X.; Hall, M.D.; et al. Discovery and lead identification of quinazoline-based BRD4 inhibitors. Bioorg. Med. Chem. Lett. 2018, 28, 3483–3488. [Google Scholar] [CrossRef]
- Zhao, Y.; Bai, L.; Liu, L.; McEachern, D.; Stuckey, J.A.; Meagher, J.L.; Yang, C.Y.; Ran, X.; Zhou, B.; Hu, Y.; et al. Structure-Based Discovery of 4-(6-Methoxy-2-methyl-4-(quinolin-4-yl)-9H-pyrimido[4,5-b]indol-7-yl)-3,5-dimethylisoxazole (CD161) as a Potent and Orally Bioavailable BET Bromodomain Inhibitor. J. Med. Chem. 2017, 60, 3887–3901. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhou, B.; Bai, L.; Liu, L.; Yang, C.Y.; Meagher, J.L.; Stuckey, J.A.; McEachern, D.; Przybranowski, S.; Wang, M.; et al. Structure-Based Discovery of CF53 as a Potent and Orally Bioavailable Bromodomain and Extra-Terminal (BET) Bromodomain Inhibitor. J. Med. Chem. 2018, 61, 6110–6120. [Google Scholar] [CrossRef] [PubMed]
- Luescher, M.U.; Bode, J.W. Catalytic Synthesis of N-Unprotected Piperazines, Morpholines, and Thiomorpholines from Aldehydes and SnAP Reagents. Angew. Chem. Int. Ed. Engl. 2015, 54, 10884–10888. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein−ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Schrödinger Release 2021-1: Glide; Schrödinger, LLC: New York, NY, USA, 2021.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Structure | Docking Score | PhaseScreen Score | Number of Matched Features | Interactions |
|---|---|---|---|---|---|
| 1 |  | −6.6 | 1.1 | 5/7 | H bond (Tyr97, Tyr139, Asn140); π-π interaction (Tyr139) |
| 2 |  | −3.7 | 1.2 | 5/7 | H bond (Tyr97, Asn140) |
| 3 |  | −5.7 | 0.5 | 5/7 | H bond (Tyr97, Asn140, Ile146); π-π interaction (Trp81) |
| 4 |  | −5.1 | 1.2 | 5/7 | H bond (Asn93, Tyr97, Asn140) |
| 5 |  | −5.2 | 1.0 | 5/7 | H bond (Asn140, Lys141) |
| 6 |  | −5.9 | 1.3 | 5/7 | H bond (Tyr97, Asn140, Asp144) |
| Compound | Residual Binding of Histone H4Ac to BRD4(BD1) ± SD (%) [Compound] = 10 μM r | Residual Binding of Histone H4Ac to BRD4(BD1) ± SD (%) [Compound] = 1 μM r | IC50 ± SD (µM) | Residual Binding of Histone H4Ac to BRD3(BD1) ± SD (%) [Compound] = 10 μM r | Residual Binding of Histone H4Ac to BRD3(BD1) ± SD (%) [Compound] = 1 μM | Residual Binding of Histone H4Ac to BRD9 ± SD (%) [Compound] = 10 μM | Residual Binding of Histone H4Ac to BRD9 ± SD (%) [Compound] = 1 μM | IC50 ± SD (µM) |
|---|---|---|---|---|---|---|---|---|
| 1 | 90.3 ± 4.8 | 98.8 ± 0.2 | \ | \ | \ | \ | \ | \ |
| 2 | 18.2 ± 2.9 | 33.5 ± 0.7 | 0.60 ± 0.25 | 94.8 ± 3.3 | ≥100 | 96.0 ± 3.7 | ≥100 | \ |
| 3 | 72.9 ± 4.8 | 91.6 ± 2.3 | \ | \ | \ | \ | \ | \ |
| 4 | 79.6 ± 4.7 | 92.7 ± 2.2 | \ | \ | \ | \ | \ | \ |
| 5 | 37.5 ± 3.6 | 63.5 ± 4.9 | 3.46 ± 1.22 | 95.6 ± 4.1 | ≥100 | 52.2 ± 3.1 | 77.7 ± 4.7 | 9.20 ± 0.50 |
| 6 | 47.7 ± 0.3 | 63.8 ±1.3 | 4.66 ± 0.52 | 87.8 ± 8.6 | ≥100 | 91.4 ± 7.4 | ≥100 | \ |
| (+)-JQ1 (Reference compound) | 7.6 ± 1.2 | 30.2 ± 0.3 | 0.23 ± 0.03 | 6.1 ± 2.2 | 34.1 ± 2.5 | \ | \ | \ |
| I-BRD9 (Reference compound) | \ | \ | \ | \ | \ | 8.6 ± 1.3 | 13.6 ± 0.9 | 2.5 ± 0.9 × 10−2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Colarusso, E.; Gazzillo, E.; Boccia, E.; Terracciano, S.; Bruno, I.; Bifulco, G.; Chini, M.G.; Lauro, G. Identification of Novel Bromodomain-Containing Protein 4 (BRD4) Binders through 3D Pharmacophore-Based Repositioning Screening Campaign. Molecules 2024, 29, 4025. https://doi.org/10.3390/molecules29174025
Colarusso E, Gazzillo E, Boccia E, Terracciano S, Bruno I, Bifulco G, Chini MG, Lauro G. Identification of Novel Bromodomain-Containing Protein 4 (BRD4) Binders through 3D Pharmacophore-Based Repositioning Screening Campaign. Molecules. 2024; 29(17):4025. https://doi.org/10.3390/molecules29174025
Chicago/Turabian StyleColarusso, Ester, Erica Gazzillo, Eleonora Boccia, Stefania Terracciano, Ines Bruno, Giuseppe Bifulco, Maria Giovanna Chini, and Gianluigi Lauro. 2024. "Identification of Novel Bromodomain-Containing Protein 4 (BRD4) Binders through 3D Pharmacophore-Based Repositioning Screening Campaign" Molecules 29, no. 17: 4025. https://doi.org/10.3390/molecules29174025
APA StyleColarusso, E., Gazzillo, E., Boccia, E., Terracciano, S., Bruno, I., Bifulco, G., Chini, M. G., & Lauro, G. (2024). Identification of Novel Bromodomain-Containing Protein 4 (BRD4) Binders through 3D Pharmacophore-Based Repositioning Screening Campaign. Molecules, 29(17), 4025. https://doi.org/10.3390/molecules29174025