
Structural and Electronic Properties of Novel Azothiophene Dyes: A Multilevel Study Incorporating Explicit Solvation Effects

 ,
,
Abstract
:1. Introduction
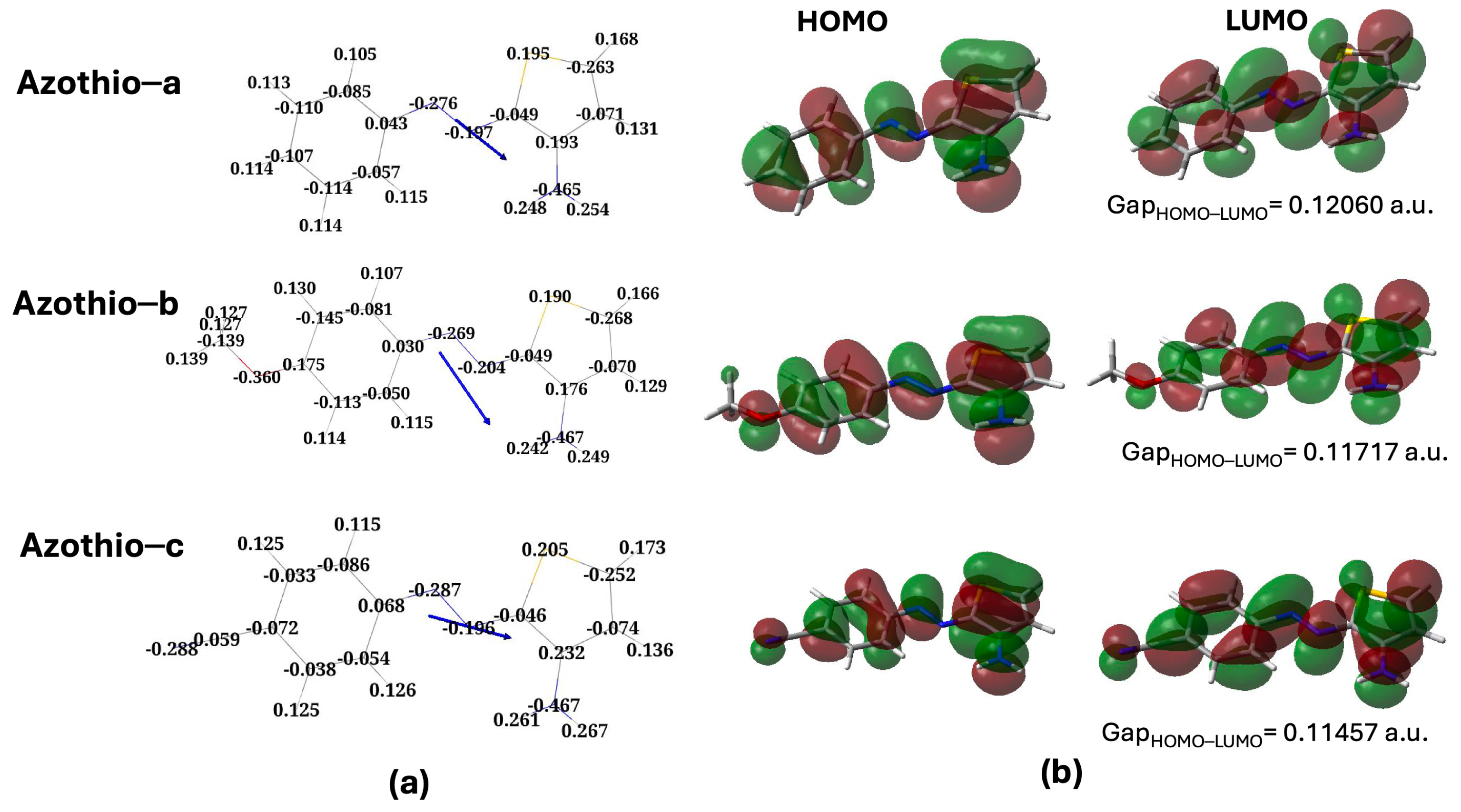
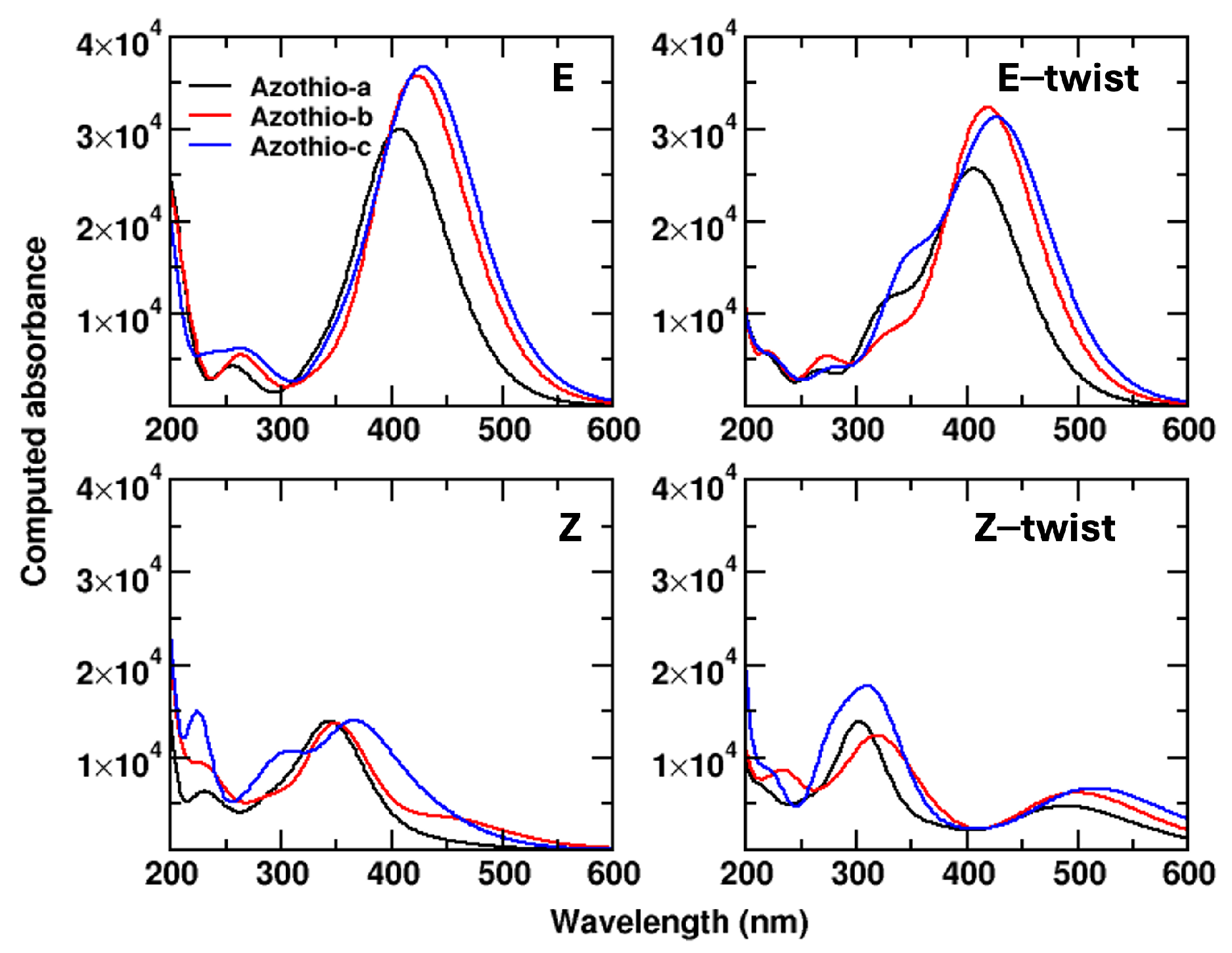
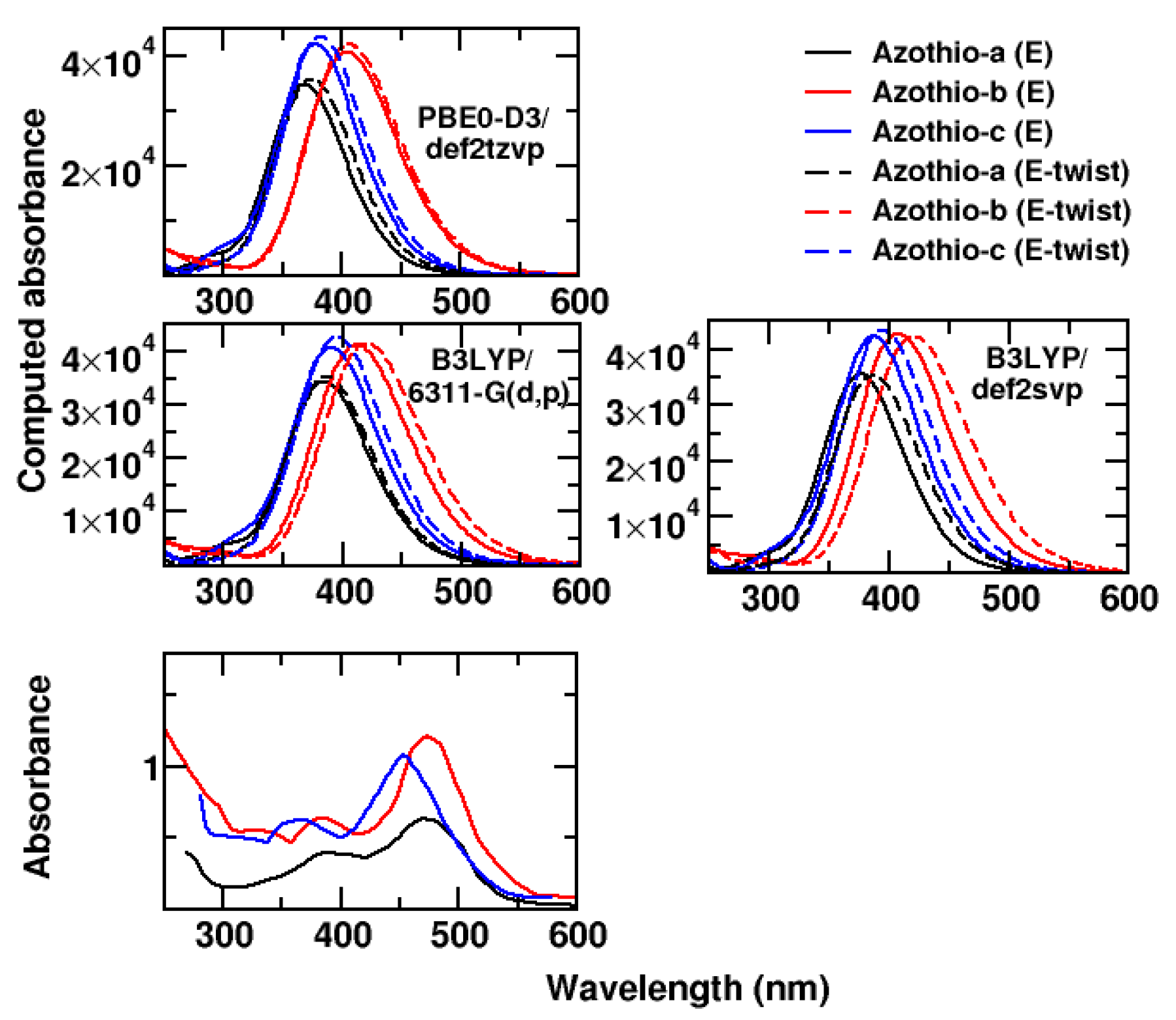
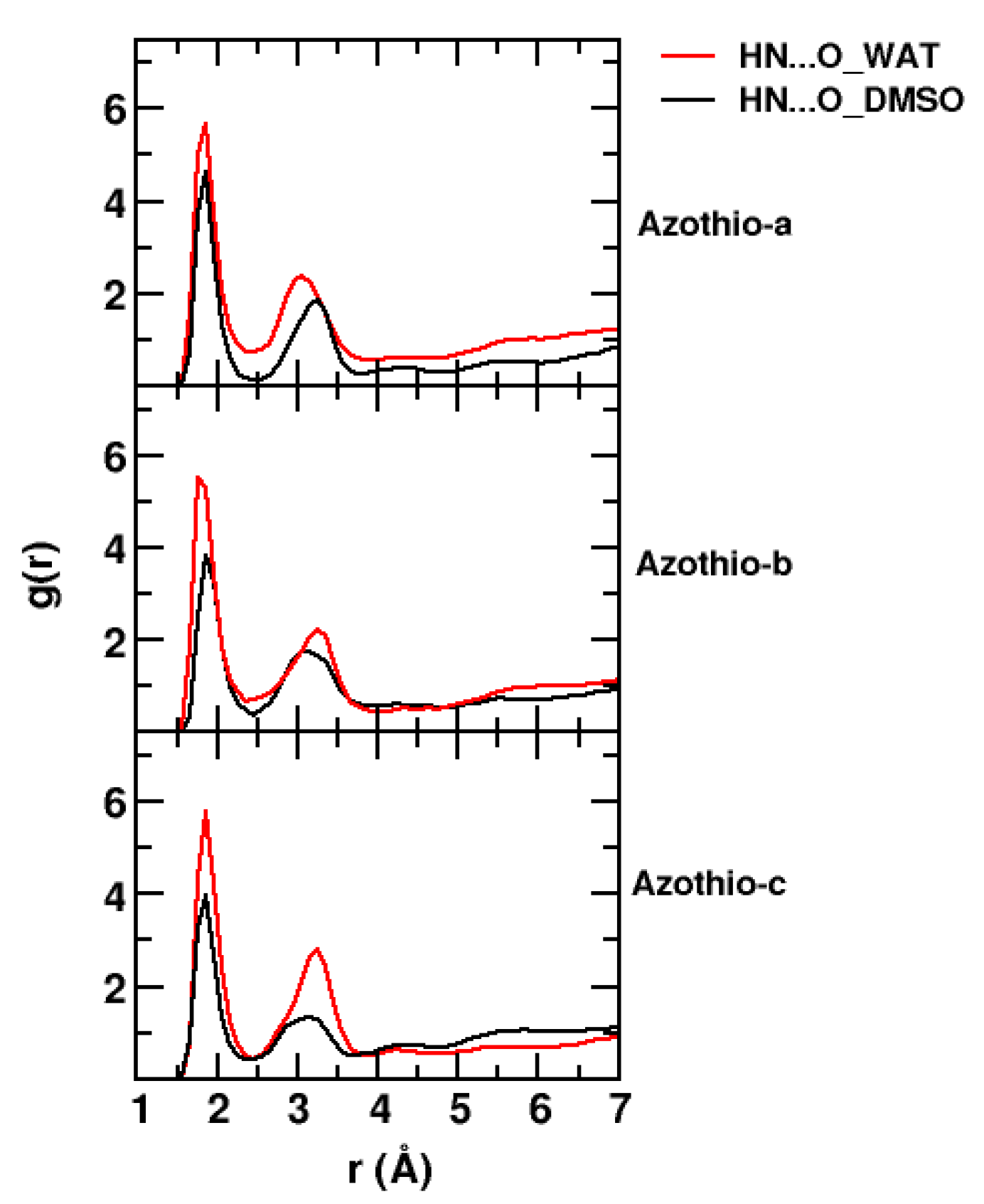
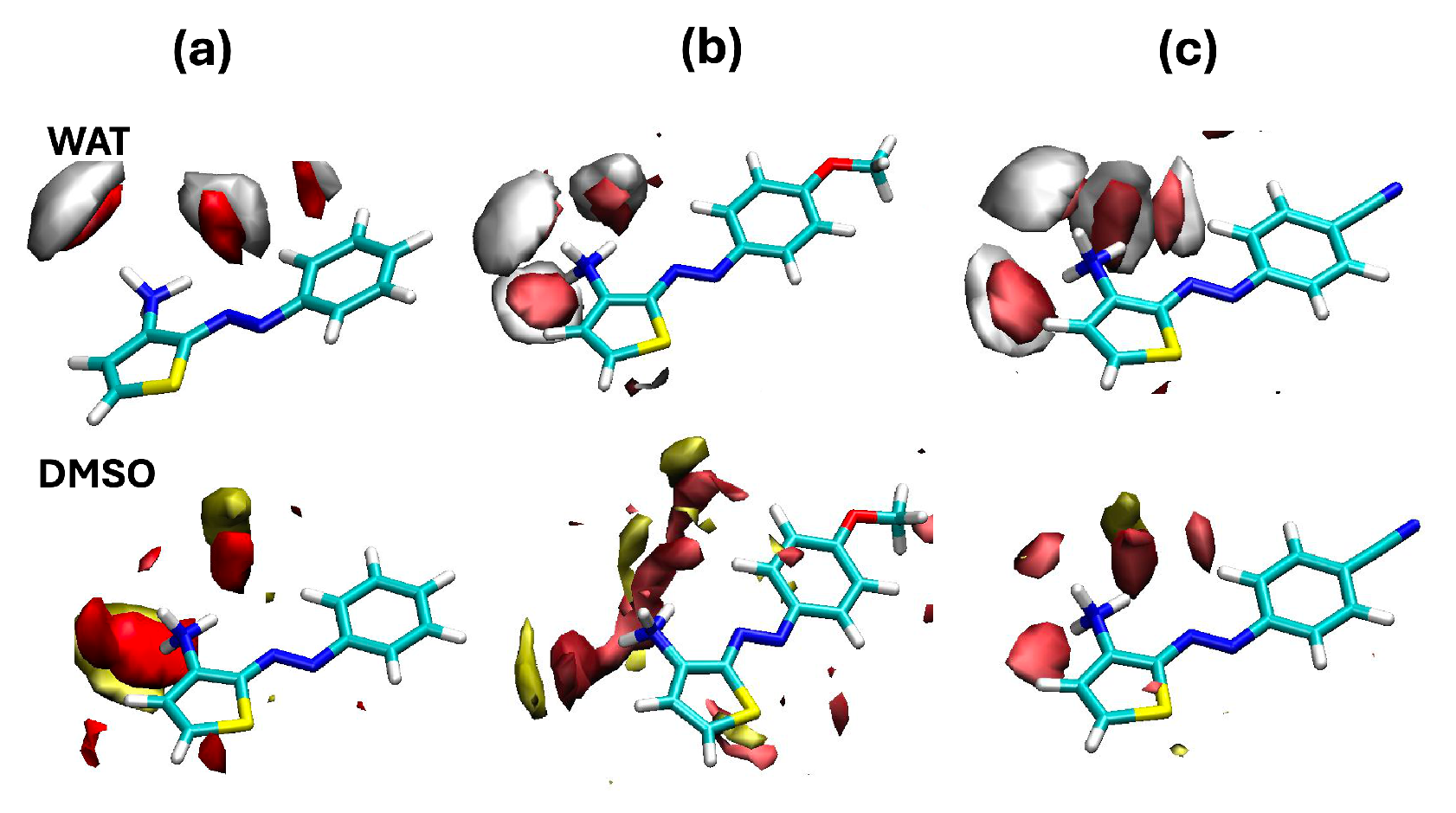
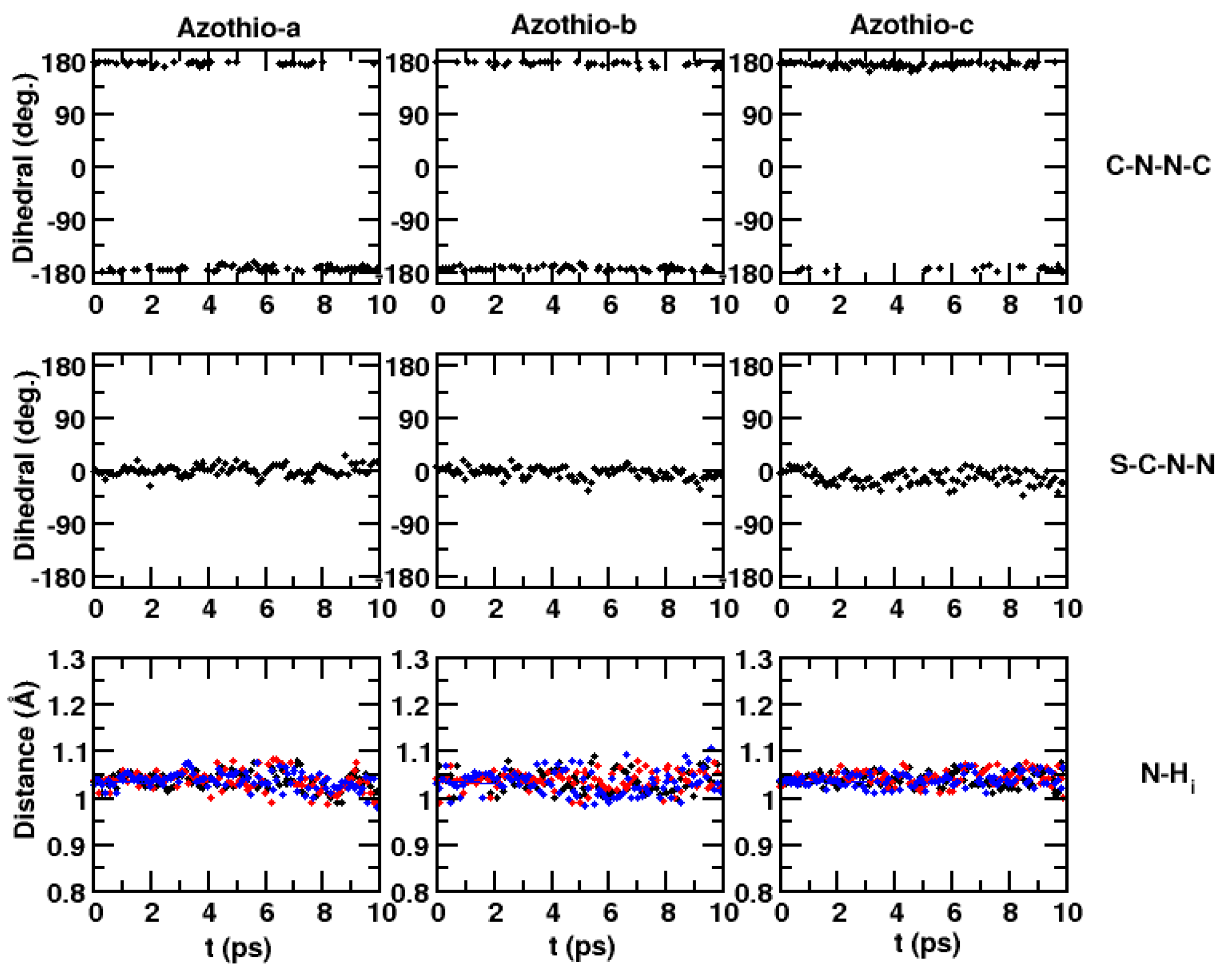
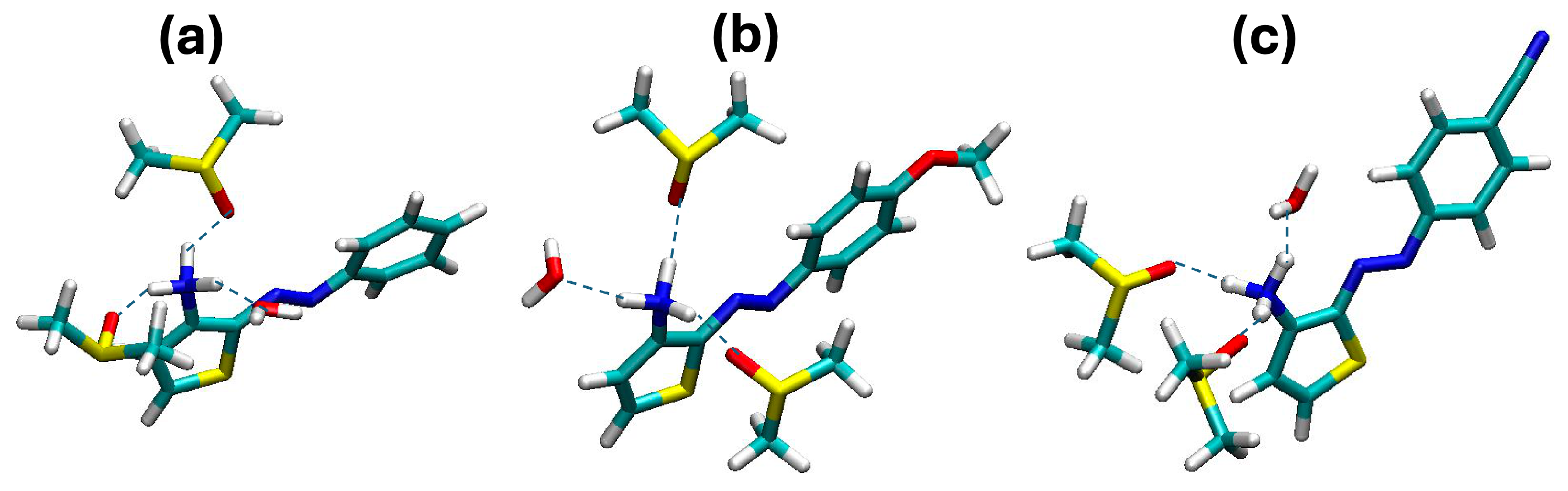
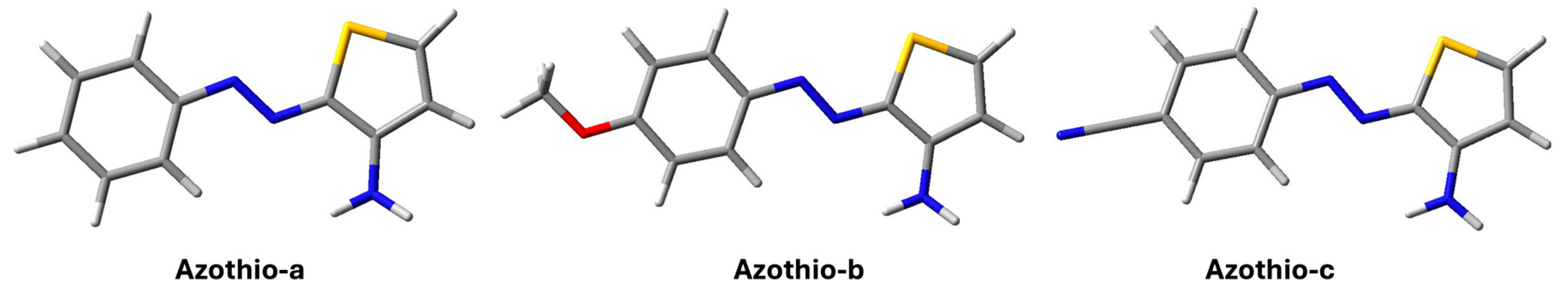
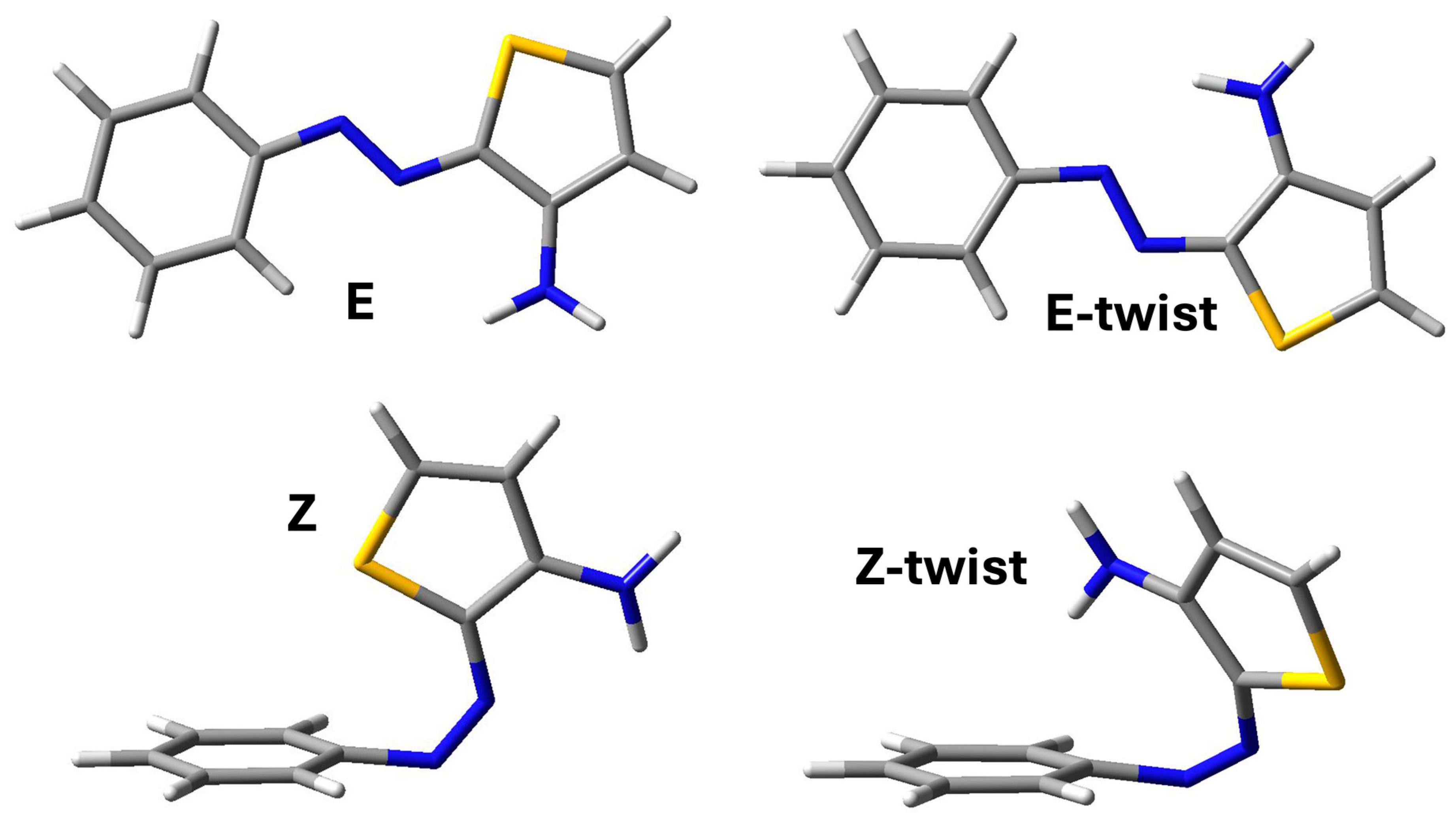
2. Results and Discussion
3. Materials and Methods
3.1. Experimental Procedure
3.2. Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jerca, F.A.; Jerca, V.V.; Hoogenboom, R. Advances and opportunities in the exciting world of azobenzenes. Nat. Rev. Chem. 2022, 6, 51–69. [Google Scholar] [CrossRef] [PubMed]
- Hartley, G.S. The cis-form of azobenzene. Nature 1937, 140, 281. [Google Scholar] [CrossRef]
- Bafana, A.; Devi, S.S.; Chakrabarti, T. Azo dyes: Past, present and the future. Environ. Rev. 2011, 19, 350–371. [Google Scholar] [CrossRef]
- Bronstein, H.; Nielsen, C.B.; Schroeder, B.C.; McCulloch, I. The role of chemical design in the performance of organic semiconductors. Nat. Rev. Chem. 2020, 4, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Towns, A. Developments in azo disperse dyes derived from heterocyclic diazo components. Dyes Pigm. 1999, 42, 3–28. [Google Scholar] [CrossRef]
- Abd-El-Aziz, A.S.; Afifi, T.H. Novel azo disperse dyes derived from aminothiophenes: Synthesis and UV–visible studies. Dyes Pigm. 2006, 70, 8–17. [Google Scholar] [CrossRef]
- Dang, T.; Zhang, Z.Y.; Li, T. Visible-Light-Activated Heteroaryl Azoswitches: Toward a More Colorful Future. J. Am. Chem. Soc. 2024, 146, 19609–19620. [Google Scholar] [CrossRef]
- Christie, R. Colour Chemistry; Royal Society of Chemistry: London, UK, 2015. [Google Scholar]
- Heindl, A.H.; Wegner, H.A. Rational design of azothiophenes—Substitution effects on the switching properties. Chem. Eur. J. 2020, 26, 13730–13737. [Google Scholar] [CrossRef]
- Franz, E.; Kunz, A.; Oberhof, N.; Heindl, A.H.; Bertram, M.; Fusek, L.; Taccardi, N.; Wasserscheid, P.; Dreuw, A.; Wegner, H.A.; et al. Electrochemically Triggered Energy Release from an Azothiophene-Based Molecular Solar Thermal System. ChemSusChem 2022, 15, e202200958. [Google Scholar] [CrossRef]
- Goulet-Hanssens, A.; Utecht, M.; Mutruc, D.; Titov, E.; Schwarz, J.; Grubert, L.; Bléger, D.; Saalfrank, P.; Hecht, S. Electrocatalytic Z→E Isomerization of Azobenzenes. J. Am. Chem. Soc. 2017, 139, 335–341. [Google Scholar] [CrossRef]
- Goulet-Hanssens, A.; Rietze, C.; Titov, E.; Abdullahu, L.; Grubert, L.; Saalfrank, P.; Hecht, S. Hole catalysis as a general mechanism for efficient and wavelength-independent Z→E azobenzene isomerization. Chem 2018, 4, 1740–1755. [Google Scholar] [CrossRef]
- Calbo, J.; Weston, C.E.; White, A.J.; Rzepa, H.S.; Contreras-García, J.; Fuchter, M.J. Tuning azoheteroarene photoswitch performance through heteroaryl design. J. Am. Chem. Soc 2017, 139, 1261–1274. [Google Scholar] [CrossRef] [PubMed]
- Bienfait, B.; Ertl, P. JSME: A free molecule editor in JavaScript. J. Cheminform. 2013, 5, 24. [Google Scholar] [CrossRef] [PubMed]
- Gabsi, W.; Boubaker, T.; Goumont, R. Azo-Coupling Reactions of Para-X-Benzenediazonium Cations with 3-Ethoxythiophene in Acetonitrile. Int. J. Chem. Kin. 2016, 48, 266–273. [Google Scholar] [CrossRef]
- Hartmann, H.; Zug, I. On the coupling of aryldiazonium salts with N,N-disubstituted 2-aminothiophenes and some of their carbocyclic and heterocyclic analogues. J. Chem. Soc. Perkin Trans. 2000, 16, 4316–4320. [Google Scholar] [CrossRef]
- Pan, X.; Wang, H.; Li, C.; Zhang, J.Z.; Ji, C. MolGpka: A web server for small molecule p K a prediction using a graph-convolutional neural network. J. Chem. Inf. Model. 2021, 61, 3159–3165. [Google Scholar] [CrossRef]
- Diana, R.; Sessa, L.; Concilio, S.; Piotto, S.; Di Costanzo, L.; Carella, A.; Panunzi, B. Experimental and Theoretical Insights into a Novel Lightfast Thiophene Azo Dye. Crystals 2023, 14, 31. [Google Scholar] [CrossRef]
- Barone, V.; Polimeno, A. Integrated computational strategies for UV/vis spectra of large molecules in solution. Chem. Soc. Rev. 2007, 36, 1724–1731. [Google Scholar] [CrossRef]
- Laurent, A.D.; Adamo, C.; Jacquemin, D. Dye chemistry with time-dependent density functional theory. Phys. Chem. Chem. Phys. 2014, 16, 14334–14356. [Google Scholar] [CrossRef]
- Huddleston, P.R.; Volkov, V.V.; Perry, C.C. The structural and electronic properties of 3,3′-azothiophene photo-switching systems. Phys. Chem. Chem. Phys. 2019, 21, 1344–1353. [Google Scholar] [CrossRef]
- Fehér, P.P.; Madarász, Á.; Stirling, A. A Practice-Oriented Benchmark Strategy to Predict the UV-Vis Spectra of Organic Photocatalysts. Chem. Methods 2023, 3, e202200069. [Google Scholar] [CrossRef]
- Prampolini, G.; Porwal, V.K.; Carof, A.; Ingrosso, F. Tautomeric contributions to the absorption spectrum of [2,2′-bipyridyl]-3,3′-diol in water unveiled by molecular dynamics with accurate quantum mechanically derived force-fields. J. Mol. Liq. 2024, 396, 123898. [Google Scholar] [CrossRef]
- Loco, D.; Polack, É.; Caprasecca, S.; Lagardere, L.; Lipparini, F.; Piquemal, J.P.; Mennucci, B. A QM/MM Approach Using the AMOEBA Polarizable Embedding: From Ground State Energies to Electronic Excitations. J. Chem. Theor. Comput. 2016, 12, 3654–3661. [Google Scholar] [CrossRef]
- Giovannini, T.; Egidi, F.; Cappelli, C. Molecular spectroscopy of aqueous solutions: A theoretical perspective. Chem. Soc. Rev. 2020, 49, 5664–5677. [Google Scholar] [CrossRef]
- Cerezo, J.; Gao, S.; Armaroli, N.; Ingrosso, F.; Prampolini, G.; Santoro, F.; Ventura, B.; Pastore, M. Non-Phenomenological Description of the Time-Resolved Emission in Solution with Quantum-Classical Vibronic Approaches—Application to Coumarin C153 in Methanol. Molecules 2023, 28, 3910. [Google Scholar] [CrossRef] [PubMed]
- Cerezo, J.; García-Iriepa, C.; Santoro, F.; Navizet, I.; Prampolini, G. Unraveling the contributions to the spectral shape of flexible dyes in solution: Insights on the absorption spectrum of an oxyluciferin analogue. Phys. Chem. Chem. Phys. 2023, 25, 5007–5020. [Google Scholar] [CrossRef]
- Losantos, R.; Prampolini, G.; Monari, A. A Portrait of the Chromophore as a Young System—Quantum-Derived Force Field Unraveling Solvent Reorganization upon Optical Excitation of Cyclocurcumin Derivatives. Molecules 2024, 29, 1752. [Google Scholar] [CrossRef]
- Keegstra, M.A.; Peters, T.H.; Brandsma, L. Copper(I) halide catalysed synthesis of alkyl aryl and alkyl heteroaryl ethers. Tetrahedron 1992, 48, 3633–3652. [Google Scholar] [CrossRef]
- Terrier, F.; Pouet, M.J.; Gzouli, K.; Hallé, J.C.; Outurquin, F.; Paulmier, C. Carbon-carbon coupling of 4,6-dinitrobenzofuroxan with 3-aminothiophenes: A kinetic and structural study. Can. J. Chem. 1998, 76, 937–945. [Google Scholar] [CrossRef]
- Reinhoudt, D.N.; Geevers, J.; Trompenaars, W.P.; Harkema, S.; Van Hummel, G.J. Solvent effects in thermal (2 + 2) cycloaddition reactions. Intramolecular capture of 1,4-dipolar intermediates vs. (2 + 2) cycloaddition in reactions of 3-(1-pyrrolidinyl)thiophenes with electron-deficient acetylenes. J. Org. Chem. 1981, 46, 424–434. [Google Scholar] [CrossRef]
- Terrier, F.; Pouet, M.J.; Kizilian, E.; Halle, J.C.; Outurquin, F.; Paulmier, C. Evidence for a strong enaminic character of 3,4-diaminothiophene: A fast carbon-carbon coupling with 4,6-dinitrobenzofuroxan. J. Org. Chem. 1993, 58, 4696–4702. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView, Version 6; Semichem Inc.: Shawnee Mission, KS, USA, 2019. [Google Scholar]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- McLean, A.; Chandler, G. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z = 11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Zhu, Z.; Wang, Y.; Lu, Y. Time-Dependent Density Functional Theory Study on Polyazopyrrole and Polyazothiophene. Macromolecules 2003, 36, 9585–9593. [Google Scholar] [CrossRef]
- Adrion, D.M.; Lopez, S.A. Design rules for optimization of photophysical and kinetic properties of azoarene photoswitches. Org. Biomol. Chem. 2023, 21, 7351–7357. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Płłowaś, I.; Świergiel, J.; Jadzyn, J. Relative static permittivity of dimethyl sulfoxide + water mixtures. J. Chem. Eng. Data 2013, 58, 1741–1746. [Google Scholar] [CrossRef]
- Case, D.; Betz, R.; Cerutti, D.; Cheatham, T., III; Darden, T.; Duke, R.; Giese, T.; Gohlke, H.; Goetz, A.; Izadi, S.; et al. AMBER 2016; University of California: San Francisco, CA, USA, 2016. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Idrissi, A.; Marekha, B.; Barj, M.; Jedlovszky, P. Thermodynamics of mixing water with dimethyl sulfoxide, as seen from computer simulations. J. Phys. Chem. B 2014, 118, 8724–8733. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comp. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Berendsen, H.J.; Postma, J.v.; Van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Götz, A.W.; Clark, M.A.; Walker, R.C. An extensible interface for QM/MM molecular dynamics simulations with AMBER. J. Comp. Chem. 2014, 35, 95–108. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | E kJ/mol | H kJ/mol |
|---|---|---|
| Azothio-a (E) | 0.0 | 0.0 |
| Azothio-a (E-twist) | 7.0 | 6.6 |
| Azothio-a (Z) | 58.2 | 57.1 |
| Azothio-a (Z-twist) | 86.6 | 84.8 |
| Azothio-b (E) | 0.0 | 0.0 |
| Azothio-b (E-twist) | 7.9 | 7.1 |
| Azothio-b (Z) | 61.7 | 60.1 |
| Azothio-b (Z-twist) | 87.7 | 85.9 |
| Azothio-c (E) | 0.0 | 0.0 |
| Azothio-c (E-twist) | 6.6 | 6.3 |
| Azothio-c (Z) | 60.4 | 59.1 |
| Azothio-c (Z-twist) | 90.3 | 88.5 |
| Molecule | G ‡ E/Z kJ/mol | G ‡ Z/E kJ/mol |
|---|---|---|
| Azothio-a | 146.1 | 89.3 |
| Azothio-b | 160.9 | 101.2 |
| Azothio-c | 120.2 | 59.8 |
| Molecule | E kJ/mol | H kJ/mol | E kJ/mol | H kJ/mol | E kJ/mol | H kJ/mol |
|---|---|---|---|---|---|---|
| B3LYP/6-311G(d,p) | B3LYP/def2svp | PBE0-D3/def2tzvp | ||||
| Azothio-a (E) | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| Azothio-a (E-twist) | 2.2 | 0.7 | −2.9 | −4.7 | −0.7 | −2.5 |
| Azothio-a (Z) | 56.7 | 54.8 | 57.6 | 56.4 | 54.3 | 51.5 |
| Azothio-a (Z-twist) | 92.5 | 91.4 | 90.5 | 88.6 | 70.1 | 69.4 |
| Azothio-b (E) | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| Azothio-b (E-twist) | 1.5 | 0.1 | −3.2 | −4.8 | −8.0 | −7.3 |
| Azothio-b (Z) | 59.3 | 58.1 | 65.1 | 63.3 | 47.8 | 49.4 |
| Azothio-b (Z-twist) | 88.9 | 86.8 | 93.5 | 92.1 | 66.7 | 67.9 |
| Azothio-c (E) | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| Azothio-c (E-twist) | 1.4 | 0.1 | −2.0 | −4.2 | −0.7 | −2.5 |
| Azothio-c (Z) | 67.3 | 65.5 | 54.6 | 52.8 | 54.3 | 51.5 |
| Azothio-c (Z-twist) | 92.0 | 90.4 | 93.7 | 92.8 | 70.1 | 69.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vautrin, L.; Lambert, A.; Mahdhaoui, F.; El Abed, R.; Boubaker, T.; Ingrosso, F. Structural and Electronic Properties of Novel Azothiophene Dyes: A Multilevel Study Incorporating Explicit Solvation Effects. Molecules 2024, 29, 4053. https://doi.org/10.3390/molecules29174053
Vautrin L, Lambert A, Mahdhaoui F, El Abed R, Boubaker T, Ingrosso F. Structural and Electronic Properties of Novel Azothiophene Dyes: A Multilevel Study Incorporating Explicit Solvation Effects. Molecules. 2024; 29(17):4053. https://doi.org/10.3390/molecules29174053
Chicago/Turabian StyleVautrin, Laura, Alexandrine Lambert, Faouzi Mahdhaoui, Riad El Abed, Taoufik Boubaker, and Francesca Ingrosso. 2024. "Structural and Electronic Properties of Novel Azothiophene Dyes: A Multilevel Study Incorporating Explicit Solvation Effects" Molecules 29, no. 17: 4053. https://doi.org/10.3390/molecules29174053