Abstract
A novel and concise synthetic method for arenastatin A, a cytotoxic cyclic depsipeptide of marine origin, was developed in this study. The convergent assembly of the four segments, including the cross-metathesis reaction, gave a cyclization precursor, and Fmoc deprotection caused simultaneous macrocyclization. The Corey–Chaykovsky reaction using a chiral sulfur ylide afforded arenastatin A with complete stereoselectivity in the longest linear sequence of seven reaction steps from the known compound. Using this synthetic method, some analogs of segment B were prepared through a late-stage diversification strategy. The simple SN2 reaction of the thiolate toward the tosylate precursor, prepared using almost the same synthetic method as described above, provided the desired sulfide analogs.
1. Introduction
Marine natural products are rich and promising sources of drug candidates for anticancer drug discovery [1,2]. In 1994, arenastatin A (1, Figure 1), a cyclic depsipeptide with potent cytotoxicity (IC50 = 5 pg/mL against KB cells), was isolated from the Okinawan marine sponge Dysidea arenaria [3]. The chemical structure of the compound was determined by NMR analysis and total synthesis [4,5]. Cryptophycins, a family of closely related depsipeptides, are found in the terrestrial cyanobacterium Nostoc sp. Some of these compounds, such as cryptophycin 1 (2), exhibit potent cytotoxicity [6]. Since then, a number of total synthesis and medicinal chemistry studies have been conducted, resulting in the discovery of a potent analog, cryptophycin-52 [7,8,9]. Although the clinical phase trial of 3 was discontinued, the high potency of the compound has attracted attention in anticancer drug development as a payload for antibody–drug conjugates (ADC) or other conjugate molecules [10,11,12].
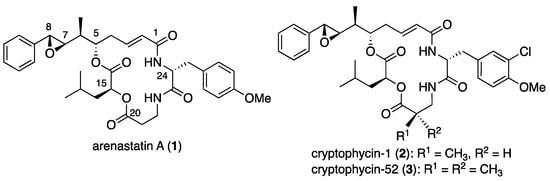
Figure 1.
Chemical structures of arenastatin A (1), cryptophycin-1 (2), and -52 (3).
Biological evaluation of arenastatin A (1) and related compounds or synthetic analogs revealed that the stereochemistry of the compound is important for its potent cytotoxicity. Therefore, stereoselective preparation of the respective asymmetric centers is essential. One of the most important parts is the 7,8-epoxide moiety because its (7S,8S) epimer has no cytotoxicity [13]. Two major approaches have been used in the reported total syntheses of 1, 2, and their analogs: In an earlier general approach, oxidation of the alkene precursor using m-chloroperbenzoic acid (mCPBA) or dioxirane in the final stage of synthesis was applied (Figure 2A) [5,14,15,16,17,18,19,20,21,22,23,24]. It gave a mixture of isomers, up to β:α = 6.5:1 by chiral dioxirane [24], and HPLC is needed for the purification. Another reliable approach is cyclization of the bromohydrin derivative, which was prepared from the corresponding 7,8-diol derivative, although relatively long reaction steps are required (Figure 2B) [25,26]. Previously, we developed a stereoselective synthesis of arenastatin A (1) (Figure 2C) [27]. The Corey–Chaykovsky reaction using chiral sulfonium salt 4, developed by Aggarwal et al. [28], was effective in constructing the natural 7R,8R-epoxide moiety in an almost stereospecific manner (>99:1 d.r.). This avoids HPLC separation of the undesired diastereomer, enabling us to prepare a sufficient amount of 1 and analogs.
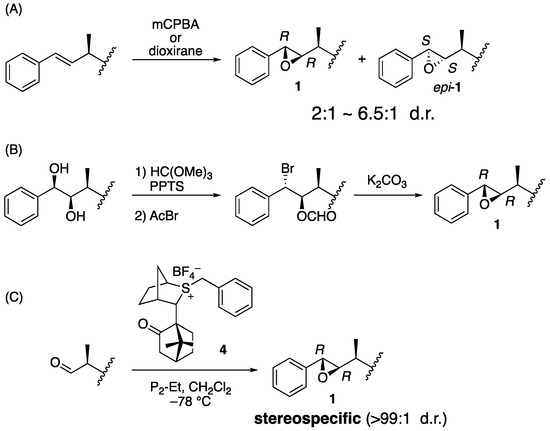
Figure 2.
Reported synthetic method of 7,8-epoxide moiety of 1. (A) Oxidation of the alkene precursor. (B) Cyclization of bromohydrin derivative. (C) Corey–Chaykovsky reaction using chiral sulfonium salt 4.
Based on the above background, a more efficient and scalable synthesis of 1 and its analogs would be interesting for anticancer drug discovery. In this study, we developed a short-step, stereospecific synthetic method for arenastatin A (1). Additionally, some analogs of segment B were prepared using a late-stage diversification approach. The details of this study, together with the bioactivity of the analogs, are described here.
2. Results and Discussions
2.1. Retrosynthetic Analysis of Arenastatin A
The retrosynthetic analysis of arenastatin A (1) is shown in Figure 3. The 7R,8R-epoxide moiety could be constructed by the Corey–Chaykovsky reaction in the final step, and aldehyde 6 could be prepared through the macrolactam formation of linear precursor 7. Assembly of the four segments A–D (10–13) would give 7. As the four segments were linearly and successively connected (A→AB→ABC→ABCD) in our previous synthetic method of 1 [27], the convergent approach by changing the order of assembly would reduce the linear reaction steps, leading to the increased overall yield.
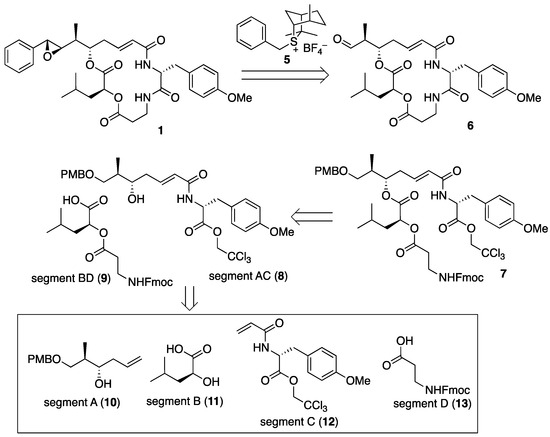
Figure 3.
Retrosynthetic analysis of arenastatin A (1).
A remained difficulty in the scalable synthesis of 1 and its analogs might be the synthesis of sulfonium salt 4, because it requires the hetero Diels–Alder reaction between cyclopentadiene and thioaldehyde, generated by the photoirradiated degradation of phenacyl sulfide [29,30]. Therefore, we decided to use another chiral sulfonium salt, 5 in the reaction. The precursor sulfide isothiocineol can be prepared in only one step from limonene in a scalable manner, and similar stereoselectivities to those using sulfonium salt 5 have been reported [31].
2.2. Total Synthesis of Arenastatin A
First, the four segments were assembled, as shown in Scheme 1. The 2,2,2-trichloroethyl ester of (R)-O-methyltyrosine (14) [14] was converted into the corresponding acrylamide 12 (segment C). Cross-metathesis reaction with segment A (10), separately prepared through Keck’s stereoselective allylation [32,33,34], yielded segment AC (8) in excellent yield using a Grubbs II catalyst (M-204, Umicore: Brussels, Belgium) [35]. A large excess of either segment is not required in this case. By contrast, commercially available Fmoc-β-alanine (13, segment D) was converted to the corresponding acyl chloride using thionyl chloride, and subsequent treatment with l-leucic acid (11, segment B) provided segment BD (9) in quantitative yield. Then, the two fragments were assembled using the Yamaguchi esterification protocol (2,4,6-trichlorobenzoly chloride, NEt3, DMAP) to give cyclization precursor 7. According to a report by Norman et al. [24], the treatment of 7 with piperidine resulted in the removal of the Fmoc group and simultaneous macrocyclization, affording macrolactam 15 in good yield. By changing the order of segment assembly and the choice of coupling reaction, a shortened linear reaction sequence and a higher overall yield than those of our previous method [27] were realized. Finally, the cleavage of the p-methoxybenzyl ether with TFA and subsequent Dess–Martin oxidation gave the desired aldehyde (6), which was then subjected to the Corey–Chaykovsky reaction.
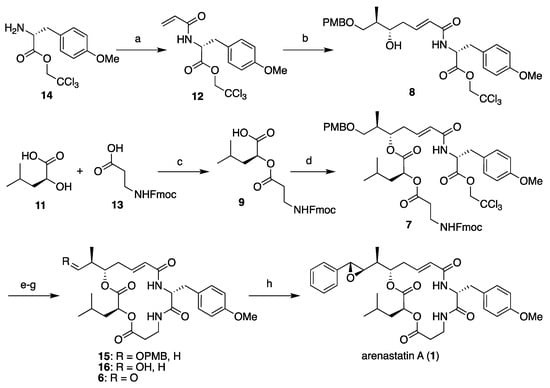
Scheme 1.
Total synthesis of arenastatin A (1). Reagents and conditions: (a) acryloyl chloride, NEt3, CH2Cl2, 0 °C, 77%; (b) 10, Grubbs II catalyst, CH2Cl2, reflux, 95%; (c) SOCl2, DMAP, CH2Cl2, quant.; (d) 2,4,6-trichlorobenzoyl chloride, NEt3, DMAP, THF, 76%; (e) piperidine, DMF, 84%; (f) TFA, (CH3)2S, CH2Cl2, 87%; (g) Dess–Martin periodinane, CH2Cl2; (h) 5, Phosphazene P2-Et, CH2Cl2, −78 °C, 81% (2 steps).
As expected, the reaction with sulfonium salt 5 proceeded smoothly to afford arenastatin A (1) in good yield. Phosphazene P2-Et was found to be the best base, and nearly complete decomposition was observed using LHMDS. Under optimal conditions (1.4 equiv. of 5 and phosphazene P2-Et in CH2Cl2 (0.2 M) at −78 °C), 81% isolated yield and complete stereoselectivity were accomplished. None of the other diastereomers were detected by 1H NMR or HPLC analyses, and nearly pure products were easily obtained by simple silica gel chromatography on a scale of ~100 mg. The same reaction using an achiral sulfonium salt provided a substantial amount of 7,8-epi-1 with a 3:1 diastereomeric ratio, clearly indicating that the chiral, rigid structure of the isothiocineole skeleton was essential for stereoselectivity (Scheme 2).
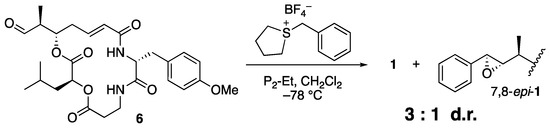
Scheme 2.
Attempted Corey–Chaykovsky reaction using achiral sulfonium salt.
2.3. Diverted Synthesis of Arenastatin A Analogs by Late-Stage Functionalization
Because natural products have complex chemical structures bearing various functional groups, the synthesis of natural product analogs is often difficult. This is mainly because a laborious, separate synthesis of the respective analogs from different starting materials/precursor segments might be required. The late-stage diversification strategy is a useful alternative approach in which only a simple derivatization reaction is conducted on a common late-stage precursor. It provides various analogs in a simple manner, enabling effective analysis of the structure–activity relationship (SAR) [36].
Among the many approaches, we planned to apply a simple SN2 reaction to prepare analogs through late-stage functionalization, as shown in Figure 4. Nucleophilic attack of the thiolate toward the primary sp3-carbon with a leaving group at the terminal of segment B could give substitution products in the presence of labile functionalities such as epoxide, ester, and α,β-unsaturated carbonyl moieties. A similar approach for the derivatization of pleuromutilins has been reported, resulting in the discovery of more potent antibacterial analogs than natural products [37]. Because retained cytotoxicity was observed for the 15-epimer of 1 [23], further derivatization at this position could lead to novel analogs or bioconjugates.
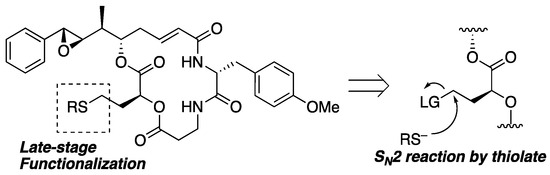
Figure 4.
Synthetic strategy of late-stage functionalization.
The synthesis of segment B analogs was performed as shown in Scheme 3, starting from compound 17 as a surrogate for l-leucic acid. Compound 17 was prepared from l-malic acid according to the literature [38] and coupled with segment D (13) to yield 18. Condensation between 18 and segment AC (8) proceeded smoothly, and subsequent Fmoc deprotection-macrocyclization by the treatment of compound 19 with piperidine provided macrolactam 20 in good yield. Subsequent cleavage of the PMB ether and Dess–Martin oxidation of the resulting primary hydroxyl group of 21 afforded the corresponding aldehyde, which was subjected to the Corey–Chaykovsky reaction. Complete stereoselectivity was observed, and (7R,8R)-22 was obtained as the sole product. Clean removal of the TBDPS group was achieved by treating 22 with tetra-n-butylammonium fluoride (TBAF) in the presence of acetic acid, and subsequent tosylation provided the common derivatization precursor 24 in moderate yield. Objective late-stage functionalization worked effectively by the addition of a thiolate solution to 24, affording the corresponding sulfide analogs 25 and 26. The attempted synthesis of tert-butyl sulfide analog 27 failed, probably because of steric repulsion.
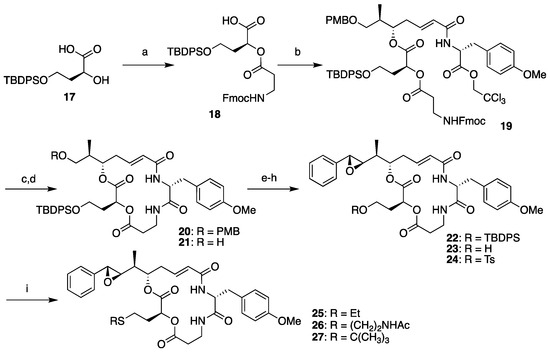
Scheme 3.
Diverted synthesis of arenastatin A analogs. Reagents and conditions: (a) 13, SOCl2, DMAP, CH2Cl2, 69%; (b) 8, 2,4,6-trichlorobenzoyl chloride, NEt3, DMAP, THF, 92%; (c) piperidine, DMF, 85%; (d) TFA, (CH3)2S, CH2Cl2, 83%; (e) Dess–Martin periodinane, CH2Cl2; (f) 5, Phosphazene P2-Et, CH2Cl2, −78 °C, 64% (2 steps); (g) TBAF, CH3COOH, THF, 0 °C; (h) TsCl, pyridine, CH2Cl2, 44% (2 steps); (i) R-SH, NaH, THF or DMF, 0 °C, 49% for 25; 72% for 26; 0% for 27.
The cytotoxicity of analogs 25 and 26 together with arenastatin A (1) against human epidermoid carcinoma KB3-1 cells was evaluated, as summarized in Table 1. Ethyl sulfide analog 25 exhibited approximately one-tenth of the cytotoxicity of 1 (IC50 = 1.8 nM), and less activity was observed for the acetamidoethyl sulfide analog 26 (IC50 = 16.0 nM). These results imply that a longer side chain at this position would lead to diminished cytotoxicity, although more detailed SAR information is required.

Table 1.
Cytotoxicity of arenastatin A (1) and analogs against cancer cells.
3. Materials and Methods
3.1. General
The following instruments were used to obtain physical data: a JASCO DIP-370 digital polarimeter (L = 50 mm) for specific rotations (JASCO: Tokyo, Japan); a JEOL JNM-ECZ500R/S1 (1H-NMR: 500 MHz, 13C-NMR: 125 MHz) spectrometer for 1H and 13C NMR data (JEOL: Tokyo, Japan) using tetramethylsilane as an internal standard; a Waters Xevo G2-XS Q-Tof mass spectrometer for ESI-Q-TOF MS (Waters: Milford, MA, USA). Silica gel (Kanto (Tokyo, Japan) 63–210 μm) and pre-coated thin layer chromatography (TLC) plates (Merck (Darmstadt, Germany) 60F254) were used for column chromatography and TLC. The spots on the TLC plates were detected by spraying with an acidic p-anisaldehyde solution (p-anisaldehyde: 25 mL, c-H2SO4: 25 mL, AcOH: 5 mL, EtOH: 425 mL) with subsequent heating. Unless otherwise noted, all reactions were performed under an N2 atmosphere. Hard copies of 1H and 13C NMR, and HRMS spectra can be found as Supplementary Materials.
3.2. Antiproliferative Activity of the Compounds against Cancer Cells
KB3-1 cells were cultured in RPMI-1640 medium supplemented with 10% heat-inactivated fetal bovine serum and kanamycin (50 μg/mL). Cells were plated into 96-well microplates at 2 × 104 cells/100 μL assay medium/well, and various concentrations of test compounds were added to each well as a 10% DMSO/EtOH solution (1 µL). The plates were incubated at 37 °C in a humidified atmosphere of 5% CO2 for 72 h, and cell proliferation was determined by MTT colorimetric assay, and the averaged IC50 values of the triplicate experiment were shown in Table 1.
3.3. Synthesis
3.3.1. 2,2,2-Trichloroethyl (R)-2-Acrylamido-3-(4-methoxyphenyl)propanoate (12)
NEt3 (0.42 mL, 3.1 mmol) and acryloyl chloride (0.25 mL, 3.1 mmol) were added to a solution of 14 (0.77 g, 2.3 mmol) in CH2Cl2 (12 mL) under N2 atmosphere at 0 °C, and the whole mixture was stirred for 4 h. Then, 1 M HCl aq. was added to the reaction solution and extracted with ethyl acetate. The organic layer was dried over Na2SO4 and was concentrated in vacuo. The crude material was purified by column chromatography (SiO2; n-hexane/AcOEt = 3:1) to afford 12 (0.69 g, 77%) as a white-yellow solid.
mp: 78.0–79.2 °C. = −54.0° (c = 1.54 in CHCl3). 1H NMR (500 MHz, CDCl3) δ: 7.07 (d, J = 8.5 Hz, 2H), 6.83 (d, J = 8.6 Hz, 2H), 6.30 (dd, J = 17.0, 1.3 Hz, 1H), 6.09 (dd, J = 17.0, 10.4 Hz, 1H), 5.91 (d, J = 7.8 Hz, 1H), 5.69 (dd, J = 10.3, 1.3 Hz, 1H), 5.10–5.06 (m, 1H), 4.78 (d, J = 11.9 Hz, 1H), 4.74 (d, J = 11.9 Hz, 1H), 3.78 (s, 3H), 3.23 (dd, J = 14.2, 5.8 Hz, 1H), 3.17 (dd, J = 14.2, 5.9 Hz, 1H). 13C NMR (125 MHz, CDCl3) δ: 170.3, 165.2, 159.0, 130.4 (2C), 130.1, 127.8, 127.2, 114.3 (2C), 94.3, 74.8, 55.3, 53.2, 36.7. HR-ESI MS: m/z 402.0043, calcd for C15H16Cl3NO4Na. Found: 402.0032.
3.3.2. 2,2,2-Trichloroethyl (R)-2-((5S,6R,E)-5-Hydroxy-7-((4-methoxybenzyl)oxy)-6-methylhept-2-enamido)-3-(4-methoxyphenyl)propanoate (8)
Grubbs II catalyst (M204, Umicore: 36 mg, 42 μmol) was added to a solution of 10 (0.39 g, 1.0 mmol) and 12 (0.21 g, 0.85 mmol) in CH2Cl2 (10 mL) under N2 atmosphere at rt, and the reaction solution was heated under reflux at 50 °C for 17 h. The reaction solution was then cooled to rt, and the solvent was removed under reduced pressure below 30 °C. The crude material was purified by column chromatography (SiO2; n-hexane/AcOEt = 2:1) to afford 8 (0.49 g, 95%) as light-brown waxy oil.
= −68.1° (c = 0.90 in CHCl3). 1H NMR (500 MHz, CDCl3) δ: 7.23 (d, J = 8.7 Hz, 2H), 7.07 (d, J = 8.3 Hz, 2H), 6.94–6.89 (m, 1H), 6.87 (d, J = 8.8 Hz, 2H), 6.83 (d, J = 8.7 Hz, 2H), 5.88–5.84 (m, 2H), 5.08–5.04 (m, 1H), 4.76 (d, J = 11.9 Hz, 1H), 4.72 (d, J = 11.9 Hz, 1H), 4.44 (s, 2H), 3.80 (s, 3H), 3.77 (s, 3H), 3.67–3.62 (m, 1H), 3.58 (dd, J = 9.4, 4.0 Hz, 1H), 3.42 (dd, J = 9.3, 7.8 Hz, 1H), 3.20 (dd, J =14.2, 5.9 Hz, 1H), 3.13 (dd, J =14.2, 5.9 Hz, 1H), 2.44–2.39 (m, 1H), 2.33–2.27 (m, 1H), 1.89–1.83 (m, 1H), 0.88 (d, J = 6.9 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ: 170.4, 165.4, 159.4, 158.9, 142.5, 130.4 (2C), 129.7, 129.5 (2C), 127.3, 125.1, 114.3 (2C), 114.0 (2C), 94.4, 75.2, 74.8, 74.7, 73.2, 55.4, 55.3, 53.2, 37.8, 37.6, 36.9, 13.9. HR-ESI MS: m/z 602.1479, calcd for C28H35Cl3NO7. Found: 602.1461.
3.3.3. (S)-2-((3-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)propanoyl)oxy)-4-methylpentanoic acid (9)
SOCl2 (54.3 μL, 0.76 mmol) was added to a solution of N-Fmoc-β-alanine (13, 0.24 g, 0.76 mmol) in CH2Cl2 (0.7 mL) under N2 atmosphere at rt. After the reaction solution was sonicated for 30 min at rt, DMAP (92 mg, 0.76 mmol) was added and sonicated for another 15 min. Then, this reaction solution was slowly added, over 2 min, to a cocktail containing L-leucic acid (11, 50 mg, 0.38 mmol) and DMAP (92 mg, 0.76 mmol) in CH2Cl2 (1.8 mL) at 0 °C. The mixture was stirred at rt for 2 h, then sat. NaHCO3 aq. and n-hexane were added to stop the reaction, and the aqueous layer was collected. This aqueous layer was acidified with 1 M HCl aq. and extracted with CH2Cl2. The organic layer was dried over Na2SO4 and was concentrated in vacuo. The crude material was purified by column chromatography (SiO2; n-hexane/AcOEt/AcOH = 60:10:1) to afford 9 (0.17 g, quant.) as colorless oil.
= −9.2° (c = 1.11 in CHCl3). 1H NMR (500 MHz, DMSO-d6) δ: 7.89 (d, J = 7.6 Hz, 2H), 7.67 (d, J = 7.6 Hz, 2H), 7.41 (t, J = 7.4 Hz, 2H), 7.37 (d, J = 5.5 Hz, 1H), 7.32 (t, J = 7.4 Hz, 2H), 4.83 (dd, J = 8.3, 3.1 Hz, 1H), 4.29 (d, J = 6.9 Hz, 2H), 4.20 (t, J = 6.9 Hz, 1H), 3.24 (q, J = 6.9 Hz, 2H), 2.52 (t, J = 7.0 Hz, 2H), 1.71–1.66 (m, 2H), 1.59–1.54 (m, 1H), 0.89 (d, J = 6.5 Hz, 3H), 0.86 (d, J = 6.5 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ: 175.6, 172.1, 156.6, 144.03, 143.99, 141.4, 127.8 (2C), 127.2 (2C), 125.2 (2C), 120.1 (3C), 70.9, 67.1, 47.2, 39.5, 36.8, 34.6, 24.7, 23.1, 21.5. HR-ESI MS: m/z 424.1760, calcd for C24H26NO6. Found: 424.1742.
3.3.4. (2R,3S,E)-1-((4-Methoxybenzyl)oxy)-7-(((R)-3-(4-methoxyphenyl)-1-oxo-1-(2,2,2-trichloroethoxy)propan-2-yl)amino)-2-methyl-7-oxohept-5-en-3-yl (S)-2-((3-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)propanoyl)oxy)-4-methylpentanoate (7)
2,4,6-Trichlorobenzoyl chloride (70 μL, 0.43 mmol) and DMAP (6.6 mg, 0.054 mmol) were sequentially added to a solution of 8 (130 mg, 0.22 mmol), 9 (101 mg, 0.24 mmol), and NEt3 (60 μL, 0.43 mmol) in THF (1.1 mL) under N2 atmosphere at 0 °C, and the reaction solution was brought to rt and stirred overnight. Then, sat. citric acid aq. was added to the reaction solution and extracted with CHCl3. The organic layer was then washed with sat. NaHCO3 aq. The organic layer was dried over Na2SO4 and was concentrated in vacuo. The crude material was purified by column chromatography (SiO2; n-hexane/AcOEt = 4:1) to afford 7 (167 mg, 76%) as light brown oil.
= −13.5° (c = 1.71 in CHCl3). 1H NMR (500 MHz, CDCl3) δ: 7.75 (d, J = 7.5 Hz, 2H), 7.61 (d, J = 7.2 Hz, 2H), 7.38 (t, J = 7.4 Hz, 2H), 7.28 (t, J = 7.5 Hz, 2H), 7.20 (d, J = 8.5 Hz, 2H), 7.05 (d, J = 8.6 Hz, 2H), 6.84 (d, J = 8.7 Hz, 2H), 6.82–6.79 (m, 1H), 6.76 (d, J = 8.7 Hz, 2H), 6.38 (d, J = 8.0 Hz, 1H), 5.87–5.83 (m, 2H), 5.16–5.12 (m, 1H), 5.09–5.05 (m, 1H), 4.94 (dd, J = 9.7, 3.9 Hz, 1H), 4.73 (d, J = 11.9 Hz, 1H), 4.69 (d, J = 11.9 Hz, 1H), 4.41–4.31 (m, 2H), 4.38 (d, J = 8.4 Hz, 2H), 4.22 (t, J = 7.3 Hz, 1H), 3.77 (s, 3H), 3.74 (s, 3H), 3.54–3.41 (m, 2H), 3.35 (dd, J = 9.4, 5.9 Hz, 1H), 3.27 (dd, J = 9.4, 5.8 Hz, 1H), 3.17 (dd, J = 14.2, 6.0 Hz, 1H), 3.10 (dd, J = 14.2, 6.3 Hz, 1H), 2.68–2.63 (m, 1H), 2.58–2.52 (m, 1H), 2.51–2.43 (m, 2H), 2.13–2.08 (m, 1H), 1.78–1.71 (m, 2H), 1.57–1.52 (m, 1H), 0.95–0.92 (m, 9H). 13C NMR (125 MHz, CDCl3) δ: 172.3, 170.5, 165.4, 159.2, 158.8, 156.6, 144.1 (2C), 141.4 (2C), 139.8, 130.5 (2C), 130.3, 129.3 (2C), 127.8 (2C), 127.6, 127.1 (2C), 125.7 (2C), 125.4, 125.3, 120.0 (2C), 114.1 (2C), 113.9 (2C), 94.4, 75.3, 74.7, 72.8, 71.7, 71.2, 66.9, 55.3, 55.3, 53.3, 47.3, 39.5, 37.0, 36.8, 36.7, 34.7, 32.9, 24.8, 23.1, 21.6, 13.3. HR-ESI MS: m/z 1031.3031, calcd for C52H59Cl3N2O12Na. Found: 1031.3069.
3.3.5. (3S,10R,16S,E)-3-Isobutyl-10-(4-methoxybenzyl)-16-((R)-1-((4-methoxybenzyl)oxy)propan-2-yl)-1,4-dioxa-8,11-diazacyclohexadec-13-ene-2,5,9,12-tetraone (15)
Piperidine (34.1 μL) was added to a solution of 7 (70 mg, 68.9 μmol) in DMF (2.3 mL), at rt, and stirred overnight. Then, H2O was added to the reaction solution and extracted with ethyl acetate. The organic layer was dried over Na2SO4 and was concentrated in vacuo. The crude material was purified by column chromatography (SiO2; n-hexane/AcOEt = 2:1) to afford 15 (37 mg, 84%) as a colorless amorphous solid.
= +49.2° (c = 1.12 in CHCl3). 1H NMR (500 MHz, CDCl3) δ: 7.21 (d, J = 8.6 Hz, 2H), 7.12 (d, J = 8.6 Hz, 2H), 7.04 (t, J = 5.7 Hz, 1H), 6.86 (d, J = 9.1 Hz, 2H), 6.80 (d, J = 8.7 Hz, 2H), 6.72–6.66 (m, 1H), 5.81–5.78 (m, 1H), 5.71 (dd, J = 15.1, 1.6 Hz, 1H), 5.19–5.15 (m, 1H), 4.91 (dd, J = 9.4, 4.0 Hz, 1H), 4.76–4.71 (m, 1H), 4.41 (d, J = 11.7 Hz, 1H), 4.37 (d, J = 11.7 Hz, 1H), 3.79 (s, 3H), 3.77 (s, 3H), 3.55–3.49 (m, 1H), 3.48–3.41 (m, 1H), 3.36 (dd, J = 9.4, 6.0 Hz, 1H), 3.28 (dd, J = 9.4, 6.0 Hz, 1H), 3.15 (dd, J = 14.5, 5.7 Hz, 1H), 3.02 (dd, J = 14.5, 7.7 Hz, 1H), 2.56 (dd, J =6.9, 4.7 Hz, 1H), 2.45–2.32 (m, 2H), 2.07–1.98 (m, 1H), 1.72–1.64(m, 2H), 1.42–1.36 (m, 1H), 0.97 (d, J = 6.9 Hz, 3H), 0.90 (d, J = 6.2 Hz, 3H), 0.86 (d, J = 6.3 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ: 172.9, 170.9, 170.6, 165.7, 159.3, 158.6, 142.2, 130.3 (2C), 130.2, 129.3 (2C), 128.7, 124.9, 114.2 (2C), 113.9 (2C), 75.6, 72.9, 71.4, 71.2, 55.4, 55.3, 54.3, 39.8, 37.9, 35.4, 35.3, 34.3, 32.6, 24.6, 22.9, 21.6, 13.7. HR-ESI MS: m/z 639.3282, calcd for C35H47N2O9. Found: 639.3289.
3.3.6. (3S,10R,16S,E)-16-((R)-1-Hydroxypropan-2-yl)-3-isobutyl-10-(4-methoxybenzyl)-1,4-dioxa-8,11-diazacyclohexadec-13-ene-2,5,9,12-tetraone (16)
Trifluoroacetic acid (1.5 mL, 10%) and dimethyl sulfide (21.3 mg, 0.343 mmol) were added to a solution of 15 (73 mg, 0.114 mmol) in CH2Cl2 (22.8 mL) at rt, and the whole mixture was stirred for 15 min. Then, saturated NaHCO3 solution was slowly added to the reaction solution at 0 °C and extracted with CHCl3. The organic layer was dried over Na2SO4 and was concentrated in vacuo. The crude material was purified by column chromatography (SiO2; CHCl3/MeOH = 40:1) to afford 16 (51.3 mg, 87%) as white powder.
mp: 98.2–100.5 °C. 1H NMR (500 MHz, CDCl3) δ: 7.11 (d, J = 8.8 Hz, 2H), 7.02 (t, J = 5.8 Hz, 1H), 6.80 (d, J = 8.7 Hz, 2H), 6.73–6.66 (m, 1H), 5.79 (d, J = 8.2 Hz, 1H), 5.74 (d, J = 15.2 Hz, 1H), 5.16–5.12 (m, 1H), 4.94 (dd, J = 10.0, 4.1 Hz, 1H), 4.73–4.69 (m, 1H), 3.77 (s, 3H), 3.60 (dd, J = 10.9, 5.5 Hz, 1H), 3.54 (dd, J = 11.0, 5.5 Hz, 1H), 3.51–3.42 (m, 2H), 3.15 (dd, J = 14.4, 5.8 Hz, 1H), 3.01 (dd, J = 14.4, 7.7 Hz, 1H), 2.55 (dd, J = 6.8, 4.7 Hz, 2H), 2.53–2.49 (m, 1H), 2.43–2.36 (m, 1H), 1.94–1.89 (m, 1H), 1.76–1.65 (m, 2H), 1.47–1.42 (m, 1H), 0.99 (d, J = 7.1 Hz, 3H), 0.92 (d, J = 6.6 Hz, 3H), 0.88 (d, J = 6.6 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ: 172.8, 170.9, 170.8, 165.7, 158.6, 142.0, 130.3 (2C), 128.7, 125.1, 114.2 (2C), 75.7, 71.4, 64.2, 55.3, 54.4, 39.8 (2C), 35.7, 35.3, 34.3, 32.6, 24.6, 23.0, 21.6, 13.5. HR-ESI MS: m/z 519.2706, calcd for C27H39N2O8. Found: 519.2705.
3.3.7. Arenastatin A (1)
Dess–Martin periodinane (57.2 mg, 0.13 mmol) was added to a solution of 8 (46.6 mg, 0.090 mmol) in CH2Cl2 (0.57 mL) at rt and stirred for 3 h. Then, sat. Na2S2O3 aq. and sat. NaHCO3 aq. were added at 0 °C to stop the reaction and extracted with AcOEt. After drying the organic layer with Na2SO4, the solvent was removed under reduced pressure to obtain the crude aldehyde 6 (46.2 mg, quant.). To a solution of 6 (46.2 mg, 0.089 mmol) and sulfonium salt 5 (50.7 mg, 0.13 mmol) in anhydrous CH2Cl2 (0.4 mL) under N2 atmosphere was slowly added phosphazene P2-Et (41.9 mg, 0.13 mmol) in CH2Cl2 (40 μL) at −78 °C, and the whole mixture was stirred at −78 °C for 3 h. After stirring, brine was added to stop the reaction and extracted with CHCl3. The organic layer was dried over Na2SO4 and was concentrated in vacuo. The crude material was purified by column chromatography (SiO2; CHCl3/MeOH = 90:1) to afford 1 (44.0 mg, 81%) as white powder.
mp: 263.1–263.8 °C. 1H NMR (500 MHz, CDCl3) δ: 7.37–7.32 (m, 3H), 7.23–7.21 (m, 2H), 7.09 (d, J = 8.8 Hz, 2H), 6.98 (t, J = 6.9 Hz, 1H), 6.79 (d, J = 8.8 Hz, 2H), 6.71–6.65 (m, 1H), 5.67 (dd, J = 15.1, 1.7 Hz, 1H), 5.53 (d, J = 7.3 Hz, 1H), 5.21–5.17 (m, 1H), 4.86 (dd, J = 10.1, 3.6 Hz, 1H), 4.73–4.69 (m, 1H), 3.76 (s, 3H), 3.66 (d, J = 2.0 Hz, 1H), 3.56–3.50 (m, 1H), 3.40–3.34 (m, 1H), 3.12 (dd, J = 14.4, 5.7 Hz, 1H), 3.01 (dd, J = 14.5, 7.5 Hz, 1H), 2.90 (dd, J = 7.7, 2.1 Hz, 1H), 2.56–2.50 (m, 3H), 2.45–2.38 (m, 1H), 1.79–1.75 (m, 1H), 1.69–1.63 (m, 2H), 1.28–1.25 (m, 1H), 1.13 (d, J = 7.0 Hz, 3H), 0.82 (d, J = 6.3 Hz, 3H), 0.80 (d, J = 6.3 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ: 173.0, 170.8 (2C), 165.5, 158.7, 141.3, 136.8, 130.3 (2C), 128.9 (2C), 128.7, 128.5, 125.8 (2C), 125.3, 114.2 (2C), 75.9, 71.3, 63.2, 59.2, 55.4, 54.3, 40.8, 39.6, 36.8, 35.3, 34.2, 32.6, 24.5, 22.9, 21.3, 13.7. HR-ESI MS: m/z 607.3019, calcd for C34H43N2O8. Found: 607.3045.
3.3.8. (S)-2-((3-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)propanoyl)oxy)-4-((tert-butyldiphenylsilyl)oxy)butanoic acid (18)
SOCl2 (0.29 mL, 4.0 mmol) was added to a solution of 13 (1.25 g, 4.0 mmol) in CH2Cl2 (6 mL) under N2 atmosphere at rt. After the reaction solution was sonicated for 1 h, DMAP (0.55 g, 4.5 mmol) was added and sonicated for another 15 min. Then, this reaction solution was slowly added to a cocktail containing (S)-4-((tert-butyldiphenylsilyl)oxy)-2-hydroxybutanoic acid (17) (0.75 g, 2.1 mmol) and DMAP (0.55 g, 4.5 mmol) in CH2Cl2 (16.3 mL) over 2 min at 0 °C. The mixture was stirred at rt for 4 h, then sat. NaHCO3 aq. and n-hexane were added to stop the reaction, and the aqueous layer was collected. This aqueous layer was acidified with 1 M HCl aq. and extracted with CHCl3. The organic layer was dried over Na2SO4 and was concentrated in vacuo. The crude material was purified by column chromatography (SiO2; n-hexane/AcOEt/AcOH = 60:10:1) to afford 18 (0.95 g, 69%) as colorless oil.
= −5.2° (c = 1.06 in CHCl3). 1H NMR (500 MHz, CDCl3) δ: 10.5–9.0 (br, 1H), 7.72 (d, J = 7.6 Hz, 2H), 7.65–7.61 (m, 4H), 7.55 (d, J = 7.6 Hz, 2H), 7.42–7.39 (m, 2H), 7.38–7.34 (m, 6H), 7.29–7.25 (m, 2H), 5.52 (t, J = 6.4 Hz, 1H), 5.37 (dd, J = 8.9, 4.0 Hz, 1H), 4.32 (d, J = 7.9 Hz, 2H), 4.17 (t, J = 7.3 Hz, 1H), 3.80–3.76 (m, 1H), 3.71–3.66 (m, 1H), 3.49–3.44 (m, 2H), 2.53–2.50 (m, 2H), 2.19–2.15 (m, 1H), 2.05–2.00 (m, 1H), 1.02 (s, 9H). 13C NMR (125 MHz, CDCl3) δ: 175.5, 171.8, 156.6, 144.0, 144.0, 141.3, 135.7 (5C), 133.3, 133.2, 129.9 (2C), 127.9 (4C), 127.8 (2C), 127.2 (2C), 125.3 (2C), 120.1 (2C), 69.0, 67.1, 59.0, 47.2, 36.7, 34.5, 33.6, 26.9 (3C), 19.2. HR-ESI MS: m/z 650.2574, calcd for C38H40NO7Si. Found: 650.2552.
3.3.9. (2R,3S,E)-1-((4-Methoxybenzyl)oxy)-7-(((R)-3-(4-methoxyphenyl)-1-oxo-1-(2,2,2-trichloroethoxy)propan-2-yl)amino)-2-methyl-7-oxohept-5-en-3-yl (S)-2-((3-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)propanoyl)oxy)-4-((tert-butyldiphenylsilyl)oxy)butanoate (19)
2,4,6-Trichlorobenzoyl chloride (0.41 mL, 2.6 mmol) and DMAP (40 mg, 0.33 mmol) were sequentially added to a solution of 8 (0.53 g, 0.88 mmol), 18 (0.97 g, 1.5 mmol), and NEt3 (0.37 mL, 2.6 mmol) in THF (2.9 mL) under N2 atmosphere at 0 °C, and the reaction solution was brought to rt and stirred overnight. Then, sat. citric acid aq. was added to the reaction solution and extracted with CHCl3. The organic layer was then washed with sat. NaHCO3 aq. and dried with Na2SO4. Removal of the solvent from the organic layer under reduced pressure gave the crude product, which was purified by column chromatography (SiO2; n-hexane/AcOEt = 2:1) to afford 19 (0.99 g, 92%) as colorless oil.
= −11.4° (c = 1.70 in CHCl3). 1H NMR (500 MHz, CDCl3) δ: 7.73 (d, J = 7.0 Hz, 2H), 7.64–7.60 (m, 4H), 7.60–7.54 (m, 2H), 7.42–7.39 (m, 2H), 7.38–7.34 (m, 7H), 7.28–7.25 (m, 2H), 7.18 (d, J = 8.8 Hz, 2H), 7.02 (d, J = 8.7 Hz, 2H), 6.81 (d, J = 8.7 Hz, 2H), 6.79–6.76 (m, 1H), 6.74 (d, J = 8.6 Hz, 2H), 6.35 (d, J = 8.0 Hz, 1H), 5.85–5.81 (m, 2H), 5.18 (dd, J = 10.0, 3.5 Hz, 1H), 5.13–5.11 (m, 1H), 5.08–5.03 (m, 1H), 4.71 (d, J = 11.9 Hz, 1H), 4.65 (d, J = 11.9 Hz, 1H), 4.37 (d, J = 11.6 Hz, 1H), 4.33 (d, J = 11.6 Hz, 1H), 4.28 (t, J = 7.3 Hz, 1H), 4.20 (t, J = 7.5 Hz, 1H), 3.76–3.72 (m, 2H), 3.73 (s, 3H), 3.69 (s, 3H), 3.48–3.38 (m, 2H), 3.34 (dd, J = 9.4, 5.9 Hz, 1H), 3.25 (dd, J = 9.5, 5.9 Hz, 1H), 3.14 (dd, J = 14.2, 6.2 Hz, 1H), 3.08 (dd, J = 14.1, 6.2 Hz, 1H), 2.59–2.55 (m, 1H), 2.49–2.44 (m, 3H), 2.10–2.04 (m, 2H), 1.92–1.86 (m, 1H), 1.04 (s, 9H), 0.92 (d, J = 6.9 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ: 172.2, 170.6, 170.3, 165.4, 159.2, 158.8, 156.6, 144.1, 141.4, 139.8, 135.6 (4C), 133.4, 130.5 (2C), 130.3, 129.9 (2C), 129.3 (3C), 127.9 (4C), 127.7 (2C), 127.5, 127.1 (2C), 125.7, 125.4, 125.3, 120.0 (2C), 114.1 (2C), 113.9 (3C), 94.4, 75.4, 74.7, 72.8, 71.2, 69.8, 66.9, 59.2, 55.3 (2C), 55.2 (2C), 53.3, 47.3, 37.0, 36.8, 34.5, 33.8, 33.0, 26.9 (4C), 19.3, 13.3. HR-ESI MS: m/z 1235.4026, calcd for C66H74Cl3N2O13Si. Found: 1235.4009.
3.3.10. (3S,10R,16S,E)-3-(2-((tert-Butyldiphenylsilyl)oxy)ethyl)-10-(4-methoxybenzyl)-16-((R)-1-((4-methoxybenzyl)oxy)propan-2-yl)-1,4-dioxa-8,11-diazacyclohexadec-13-ene-2,5,9,12-tetraone (20)
Piperidine (40 μL, 0.41 mmol) was added to a solution of 19 (0.10 g, 81.7 μmol) in DMF (2.7 mL) at rt, and the whole mixture was stirred overnight. Then, H2O was added to the reaction solution and extracted with AcOEt. The organic layer was dried over Na2SO4 and was concentrated in vacuo. The crude material was purified by column chromatography (SiO2; n-hexane/AcOEt = 1:2) to afford 20 (59.7 mg, 85%) as colorless amorphous solid.
= +37.3° (c = 1.77 in CHCl3). 1H NMR (500 MHz, CDCl3) δ: 7.61–7.57 (m, 4H), 7.43–7.39 (m, 2H), 7.37–7.33 (m, 4H), 7.20 (d, J = 8.8 Hz, 2H), 7.12 (d, J = 8.8 Hz, 2H), 6.99 (t, J = 6.0 Hz, 1H), 6.85 (d, J = 8.8 Hz, 2H), 6.82 (d, J = 8.8 Hz, 2H), 6.72–6.66 (m, 1H), 5.71 (d, J = 15.2 Hz, 1H), 5.63 (d, J = 8.3 Hz, 1H), 5.15–5.11 (m, 1H), 5.09 (dd, J = 9.9, 3.7 Hz, 1H), 4.79–4.74 (m, 1H), 4.38 (d, J = 11.6 Hz, 1H), 4.35 (d, J = 11.6 Hz, 1H), 3.77 (s, 3H), 3.76 (s, 3H), 3.70–3.66 (m, 2H), 3.52–3.47 (m, 1H), 3.43–3.37 (m, 1H), 3.36 (dd, J = 9.3, 5.9 Hz, 1H), 3.30 (dd, J = 9.3, 5.6 Hz, 1H), 3.11 (dd, J = 14.4, 5.6 Hz, 1H), 3.04 (dd, J = 14.6, 7.5 Hz, 1H), 2.45–2.32 (m, 4H), 2.04–1.99 (m, 1H), 1.88–1.79 (m, 2H), 1.03 (s, 9H), 0.96 (d, J = 7.0 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ: 172.8, 170.9, 170.6, 165.7, 159.3, 158.6, 142.2, 135.7 (2C), 135.6 (2C), 133.4 (3C), 130.5, 130.2 (2C), 130.0 (2C), 129.4, 129.2, 128.7, 127.9 (2C), 124.8, 114.2, 114.1, 114.0 (2C), 113.8, 75.9, 73.0, 71.3, 69.8, 69.6, 59.0, 55.3, 54.1, 37.9, 35.5, 34.4, 34.0, 32.5, 26.9 (3C), 19.3 (2C), 13.8. HR-ESI MS: m/z 865.4095, calcd for C49H61N2O10Si. Found: 865.4089.
3.3.11. (3S,10R,16S,E)-3-(2-((tert-Butyldiphenylsilyl)oxy)ethyl)-16-((R)-1-hydroxypropan-2-yl)-10-(4-methoxybenzyl)-1,4-dioxa-8,11-diazacyclohexadec-13-ene-2,5,9,12-tetraone (21)
Trifluoroacetic acid (1.1 mL, 10%) and dimethyl sulfide (16 mg, 0.25 mmol) were added to a solution of 20 (73 mg, 84.5 μmol) in CH2Cl2 (16.9 mL), and the whole mixture was stirred for 15 min. Then, sat. NaHCO3 aq. was slowly added to the reaction solution at 0 °C and extracted with CHCl3. The organic layer was dried over Na2SO4 and was concentrated in vacuo. The crude material was purified by column chromatography (SiO2; CHCl3/MeOH = 40:1) to afford 21 (52.1 mg, 83%) as white powder.
mp: 83.3–85.6 °C. = +43.4° (c = 0.99 in CHCl3). 1H NMR (500 MHz, CDCl3) δ: 7.61–7.57 (m, 4H), 7.44–7.40 (m, 2H), 7.38–7.33 (m, 4H), 7.12 (d, J = 8.7 Hz, 2H), 7.00 (t, J = 5.5 Hz, 1H), 6.81 (d, J = 8.7 Hz, 2H), 6.73–6.67 (m, 1H), 5.75 (dd, J = 15.2, 1.6 Hz, 1H), 5.68 (d, J = 8.3 Hz, 1H), 5.12–5.07 (m, 2H), 4.76–4.72 (m, 1H), 3.77 (s, 3H), 3.74–3.68 (m, 2H), 3.58 (dd, J = 10.8, 5.4 Hz, 1H), 3.52 (dd, J = 10.8, 5.5 Hz, 1H), 3.50–3.47 (m, 1H), 3.42–3.37 (m, 1H), 3.16 (dd, J = 14.4, 5.7 Hz, 1H), 3.03 (dd, J = 14.5, 7.6 Hz, 1H), 2.53–2.49 (m, 1H), 2.44–2.38 (m, 3H), 1.97–1.88 (m, 3H), 1.03 (s, 9H), 0.98 (d, J = 7.0 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ: 172.7, 170.9, 170.7, 165.7, 158.6, 142.0, 135.6 (4C), 133.4, 130.3 (2C), 129.9 (2C), 128.7, 127.8 (3C), 127.8 (2C), 125.0, 114.2 (2C), 76.0, 69.7, 64.2, 59.0, 55.3, 54.2, 39.8, 35.8, 35.3, 34.4, 34.0, 32.5, 26.9 (3C), 19.3, 13.6. HR-ESI MS: m/z 745.3520, calcd for C41H53N2O9Si. Found: 745.3532.
3.3.12. (3S,10R,16S,E)-3-(2-((tert-Butyldiphenylsilyl)oxy)ethyl)-10-(4-methoxybenzyl)-16-((S)-1-((2R,3R)-3-phenyloxiran-2-yl)ethyl)-1,4-dioxa-8,11-diazacyclohexadec-13-ene-2,5,9,12-tetraone (22)
Dess–Martin periodinane (36 mg, 0.085 mmol) was added to a solution of 21 (57.9 mg, 0.078 mmol) in CH2Cl2 (0.8 mL) at 0 °C, and the whole mixture was stirred at rt for 3 h. Then, Na2S2O3 and NaHCO3 solutions were added at 0 °C to stop the reaction and extracted with CHCl3. The organic layer was dried over Na2SO4 and was concentrated in vacuo to give an intermediate aldehyde, which was used for the next reaction without further purification. Phosphazene P2-Et (31 mg, 0.092 mmol) in CH2Cl2 (0.1 mL) was slowly added to a solution of above aldehyde and sulfonium salt 5 (50 mg, 0.12 mmol) in anhydrous CH2Cl2 (0.51 mL) under N2 atmosphere at −78 °C, and the whole mixture was stirred at −78 °C for 3 h. Brine was added to stop the reaction and extracted with CHCl3. The organic layer was dried over Na2SO4 and was concentrated in vacuo. The crude material was purified by column chromatography (SiO2; CHCl3/MeOH = 100:1) to afford 22 (32.5 mg, 64%) as white powder.
mp: 81.9–83.2 °C. = +37.2° (c = 0.63 in CHCl3). 1H NMR (500 MHz, CDCl3) δ: 7.61–7.59 (m, 2H), 7.57–7.55 (m, 2H), 7.44–7.29 (m, 9H), 7.24–7.22 (m, 2H), 7.10 (d, J = 8.8 Hz, 2H), 6.98 (t, J = 5.8 Hz, 1H), 6.81 (d, J = 8.7 Hz, 2H), 6.72–6.66 (m, 1H), 5.70 (dd, J = 15.1, 1.5 Hz, 1H), 5.55 (d, J = 8.2 Hz, 1H), 5.16–5.13 (m, 1H), 5.04 (dd, J = 10.1, 3.5 Hz, 1H), 4.77–4.73 (m, 1H), 3.77 (s, 3H), 3.67 (d, J = 2.0 Hz, 1H), 3.63–3.60 (m, 2H), 3.53–3.48 (m, 1H), 3.35–3.31 (m, 1H), 3.14 (dd, J = 14.4, 5.7 Hz, 1H), 3.03 (dd, J = 14.4, 7.6 Hz, 1H), 2.90 (dd, J = 7.8, 2.1 Hz, 1H), 2.57–2.53 (m, 1H), 2.47–2.33 (m, 3H), 1.79–1.73 (m, 3H), 1.14 (d, J = 6.9 Hz, 3H), 1.03 (s, 9H). 13C NMR (125 MHz, CDCl3) δ: 172.9, 170.8, 165.4, 158.7, 141.2, 136.8, 135.6 (4C), 133.3, 130.3 (2C), 130.0, 129.9, 128.8 (2C), 128.6, 128.5, 127.9 (3C), 127.8 (3C), 125.7 (2C), 125.2, 114.2 (2C), 76.2, 69.6, 63.4, 59.4, 58.8, 55.3, 54.1, 40.9, 36.8, 35.3, 34.3, 33.8, 32.4, 27.0 (3C), 19.3, 13.9. HR-ESI MS: m/z 833.3833, calcd for C48H57N2O9Si. Found: 833.3831.
3.3.13. 2-((3S,10R,16S,E)-10-(4-Methoxybenzyl)-2,5,9,12-tetraoxo-16-((S)-1-((2R,3R)-3-phenyloxiran-2-yl)ethyl)-1,4-dioxa-8,11-diazacyclohexadec-13-en-3-yl)ethyl 4-methylbenzenesulfonate (24)
Tetra-n-butylammonium fluoride (ca. 1 mol/L in THF, 0.22 mL, 0.22 mmol) and acetic acid (13.4 mg, 0.22 mmol) were added to a solution of 22 (61.9 mg, 0.074 mmol) in THF (7.4 mL) under N2 atmosphere at 0 °C, and the whole mixture was stirred for 5 h. Then, sat. NH4Cl aq. was added to the reaction solution at 0 °C and extracted with CHCl3. The organic layer was dried over Na2SO4 and was concentrated in vacuo. The crude material containing 23 was dissolved in CH2Cl2 (1.4 mL) under N2 atmosphere, and p-toluenesulfonyl chloride (15.4 mg, 0.081 mmol) and pyridine (12.8 mg, 0.16 mmol) were added at 0 °C. The whole mixture was stirred overnight at rt, then sat. NaHCO3 aq. was added to the reaction solution at 0 °C and extracted with CHCl3. The organic layer was dried over Na2SO4 and was concentrated in vacuo. The crude material was purified by column chromatography (SiO2; CHCl3/MeOH = 90:1) to afford 24 (24.1 mg, 44% in 2 steps) as a colorless amorphous solid.
= +45.2° (c = 0.32 in CHCl3). 1H NMR (500 MHz, CDCl3) δ: 7.73 (d, J = 8.5 Hz, 2H), 7.34–7.30 (m, 5H), 7.24–7.22 (m, 2H), 7.10 (d, J = 8.8 Hz, 2H), 6.93 (t, J = 5.8 Hz, 1H), 6.81 (d, J = 8.7 Hz, 2H), 6.69–6.63 (m, 1H), 5.68 (dd, J = 15.1, 1.6 Hz, 1H), 5.55 (d, J = 8.2 Hz, 1H), 5.12–5.08 (m, 1H), 4.90 (t, J = 6.7 Hz, 1H), 4.74–4.70 (m, 1H), 3.93–3.86 (m, 2H), 3.77 (s, 3H), 3.66 (d, J = 2.0 Hz, 1H), 3.51–3.45 (m, 1H), 3.36–3.30 (m, 1H), 3.14 (dd, J = 14.6, 5.7 Hz, 1H), 3.01 (dd, J = 14.5, 7.6 Hz, 1H), 2.87 (dd, J = 7.9, 2.0 Hz, 1H), 2.61–2.57 (m, 1H), 2.44 (s, 3H), 2.41–2.34 (m, 3H), 1.79–1.74 (m, 3H), 1.13 (d, J = 6.9 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ: 172.3, 170.8, 169.6, 165.4, 158.7, 145.3, 141.1, 136.7, 132.5, 130.3 (2C), 130.0 (2C), 128.8 (2C), 128.7, 128.5, 128.1 (2C), 125.8 (2C), 125.3, 114.2 (2C), 76.7, 68.5, 64.9, 63.6, 59.4, 55.3, 54.3, 40.8, 36.9, 35.3, 34.1, 32.3, 29.9, 21.8, 13.9. HR-ESI MS: m/z 749.2744, calcd for C39H45N2O11S. Found: 749.2746.
3.3.14. (3S,10R,16S,E)-3-(2-(Ethylthio)ethyl)-10-(4-methoxybenzyl)-16-((S)-1-((2R,3R)-3-phenyloxiran-2-yl)ethyl)-1,4-dioxa-8,11-diazacyclohexadec-13-ene-2,5,9,12-tetraone (25)
Sodium hydride (144.5 mg, 6.02 mmol) was added to a solution of ethanethiol (0.67 mL, 9.03 mmol) in THF (10 mL) under N2 atmosphere, and the whole mixture was stirred for 1 h to prepare sodium ethanethiolate solution. An aliquot of the thiolate solution (73.6 μL) was added to a solution of 24 (11.9 mg, 0.016 mmol) in anhydrous DMF (0.54 mL) at 0 °C, and the whole mixture was stirred for 4 h. Then, H2O was added to the reaction mixture at 0 °C and extracted with CHCl3/MeOH = 4/1. The organic layer was dried over Na2SO4 and was concentrated in vacuo. The crude material was purified by reversed-phase HPLC (Cholester; CH3CN/H2O = 6:4) to afford 25 (4.95 mg, 49%) as white powder.
mp: 235.1–237.0 °C. = +40.7° (c = 0.38 in CHCl3). 1H NMR (500 MHz, CDCl3) δ: 7.38–7.33 (m, 3H), 7.24–7.23 (m, 2H), 7.09 (d, J = 8.8 Hz, 2H), 6.97 (t, J = 5.5 Hz, 1H), 6.80 (d, J = 8.7 Hz, 2H), 6.72–6.66 (m, 1H), 5.69 (d, J = 15.2 Hz, 1H), 5.56 (d, J = 8.0 Hz, 1H), 5.17 (dd, J = 11.4, 5.0 Hz, 1H), 4.99 (dd, J = 9.6, 2.6 Hz, 1H), 4.75–4.71 (m, 1H), 3.77 (s, 3H), 3.68 (s, 1H), 3.56–3.51 (m, 1H), 3.42–3.37 (m, 1H), 3.13 (dd, J = 14.2, 6.1 Hz, 1H), 3.02 (dd, J = 14.2, 7.4 Hz, 1H), 2.90 (d, J = 7.5 Hz, 1H), 2.58–2.52 (m, 4H), 2.49–2.41 (m, 4H), 1.99–1.92 (m, 1H), 1.83–1.77 (m, 2H), 1.22 (t, J = 7.4 Hz, 3H), 1.13 (d, J = 6.9 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ: 172.8, 170.8, 170.0, 165.4, 158.7, 141.2, 136.8, 130.3 (2C), 128.8 (2C), 128.7, 128.5, 125.6 (2C), 125.3, 114.2 (2C), 76.3, 71.3, 63.3, 59.0, 55.3, 54.1, 40.6, 36.8, 35.3, 34.3, 32.5, 30.9, 27.1, 26.0, 14.8, 13.7. HR-ESI MS: m/z 639.2740, calcd for C34H43N2O8S. Found: 639.2719.
3.3.15. N-(2-((2-((3S,10R,16S,E)-10-(4-Methoxybenzyl)-2,5,9,12-tetraoxo-16-((S)-1-((2R,3R)-3-phenyloxiran-2-yl)ethyl)-1,4-dioxa-8,11-diazacyclohexadec-13-en-3-yl)ethyl)thio)ethyl)acetamide (26)
Sodium hydride (17.3 mg, 0.72 mmol) was added to a solution of N-acetylcysteamine (111.5 mg, 0.94 mmol) in DMF (3.6 mL) under N2 atmosphere, and the whole mixture was stirred for 1 h to prepare the corresponding thiolate solution. An aliquot of the thiolate solution (0.14 mL) was added to a solution of 24 (10.2 mg, 0.014 mmol) in anhydrous DMF (0.38 mL) at 0 °C, and the whole mixture was stirred for 4 h. Then, H2O was added to the reaction solution at 0 °C and extracted with CHCl3/MeOH = 4:1. The organic layer was dried over Na2SO4 and was concentrated in vacuo. The crude material was purified by reversed-phase HPLC (Cholester; CH3CN/H2O = 4:6) to afford 26 (6.82 mg, 72%) as white powder.
mp: 203.0–204.5 °C. = +42.7° (c = 0.58 in CHCl3/MeOH = 5:1). 1H NMR (500 MHz, CDCl3) δ: 7.38–7.32 (m, 3H), 7.24–7.23 (m, 2H), 7.09 (d, J = 8.8 Hz, 2H), 6.95 (t, J = 5.9 Hz, 1H), 6.80 (d, J = 8.7 Hz, 2H), 6.72–6.66 (m, 1H), 5.81 (s, 1H), 5.69 (d, J = 15.2 Hz, 1H), 5.61 (d, J = 8.2 Hz, 1H), 5.18–5.14 (m, 1H), 4.97 (dd, J = 9.2, 3.6 Hz, 1H), 4.74–4.70 (m, 1H), 3.76 (s, 3H), 3.68 (d, J = 2.2 Hz, 1H), 3.55–3.50 (m, 1H), 3.44–3.40 (m, 1H), 3.37 (dd, J = 12.7, 6.5 Hz, 2H), 3.13 (dd, J = 14.5, 5.8 Hz, 1H), 3.01 (dd, J = 14.5, 7.6 Hz, 1H), 2.92 (dd, J = 7.2, 2.1 Hz, 1H), 2.60–2.53 (m, 6H), 2.45–2.38 (m, 2H), 1.95–1.89 (m, 4H), 1.86–1.79 (m, 2H), 1.12 (d, J = 6.9 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ: 172.7, 170.8, 170.3, 169.9, 165.5, 158.6, 141.2, 136.8, 130.3 (2C), 128.9 (2C), 128.7, 128.5, 125.7 (2C), 125.3, 114.2 (2C), 76.4, 71.1, 63.3, 58.8, 55.3, 54.2, 40.4, 38.5, 36.9, 35.2, 34.2, 32.5, 31.9, 30.9, 27.0, 23.4, 13.6. HR-ESI MS: m/z 696.2955, calcd for C36H46N3O9S. Found: 696.2950.
4. Conclusions
In summary, a short-step stereospecific total synthesis of arenastatin A (1) was accomplished in this study. The convergent assembly of four segments, deprotection-triggered macrocyclization, and the stereoselective Corey–Chaykovsky reaction yielded the natural product in the longest linear sequence of seven steps, starting with a known compound. In addition, the preparation of some segment B analogs using a late-stage functionalization approach was successful. As other diversification methods mediated by thiols have been reported [39], further synthesis and evaluation of various analogs will lead to the development of more potent and selective anticancer drug candidates, which will be undertaken in due course.
Supplementary Materials
The following supporting information can be downloaded from https://www.mdpi.com/article/10.3390/molecules29174058/s1. Supplementary Data S1: NMR spectra of the new compounds.
Author Contributions
Conceptualization, N.K.; methodology, N.K.; validation, N.K.; formal analysis, N.K.; investigation, Y.M. and H.K.; data curation, Y.M., H.K. and S.K.; writing—original draft preparation, Y.M.; writing—review and editing, N.K.; supervision, N.K.; project administration, N.K.; funding acquisition, N.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Hoansha Foundation and a Grant-in-Aid for Scientific Research C (Grant No. 18K05363 and 22K05339) from the Japan Society for the Promotion of Science (JSPS) to N.K.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article and Supplementary Materials.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Molinski, T.F.; Dalisay, D.S.; Lievens, S.L.; Saludes, J.P. Drug development from marine natural products. Nat. Rev. Drug Discov. 2009, 8, 69–85. [Google Scholar] [CrossRef]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; International Natural Product Sciences Taskforce; Supuran, C.T. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef]
- Kobayashi, M.; Aoki, S.; Ohyabu, N.; Kurosu, M.; Wang, W.; Kitagawa, I. Arenastatin A, a potent cytotoxic depsipeptide from the Okinawan marine sponge Dysidea arenaria. Tetrahedron Lett. 1994, 35, 7969–7972. [Google Scholar] [CrossRef]
- Kobayashi, M.; Kurosu, M.; Ohyabu, N.; Wang, W.; Fujii, S.; Kitagawa, I. The absolute stereostructure of arenastatin A, a potent cytotoxic depsipeptide from the Okinawan Marine sponge Dysidea arenaria. Chem. Pharm. Bull. 1994, 42, 2196–2198. [Google Scholar] [CrossRef]
- Kobayashi, M.; Kurosu, M.; Wang, W.; Kitagawa, I. A total synthesis of arenastatin A, an extremely potent cytotoxic depsipeptide, from the Okinawan marine sponge Dysidea arenaria. Chem. Pharm. Bull. 1994, 42, 2394–2396. [Google Scholar] [CrossRef][Green Version]
- Trimurtulu, G.; Ohtani, I.; Patterson, G.M.L.; Moore, R.E.; Corbett, T.H.; Valeriote, F.A.; Demchik, L. Total Structures of Cryptophycins, Potent Antitumor Depsipeptides from the Blue-Green Alga Nostoc sp. Strain GSV 224. J. Am. Chem. Soc. 1994, 116, 4729–4737. [Google Scholar] [CrossRef]
- Weiss, C.; Figueras, E.; Borbely, A.N.; Sewald, N. Cryptophycins: Cytotoxic cyclodepsipeptides with potential for tumor targeting. J. Pept. Sci. 2017, 23, 514–531. [Google Scholar] [CrossRef]
- Eggen, M.; Georg, G.I. The Cryptophycins: Their Synthesis and Anticancer Activity. Med. Res. Rev. 2002, 22, 85–101. [Google Scholar] [CrossRef]
- Weiss, C.; Sammet, B.; Sewald, N. Recent approaches for the synthesis of modified cryptophycins. Nat. Prod. Rep. 2013, 30, 924–940. [Google Scholar] [CrossRef] [PubMed]
- Verma, V.A.; Pillow, T.H.; DePalatis, L.; Li, G.; Phillips, G.L.; Polson, A.G.; Raab, H.E.; Spencer, S.; Zheng, B. The cryptophycins as potent payloads for antibody drug conjugates. Bioorg. Med. Chem. Lett. 2015, 25, 864–868. [Google Scholar] [CrossRef] [PubMed]
- Lai, Q.; Wu, M.; Wang, R.; Lai, W.; Tao, Y.; Lu, Y.; Wang, Y.; Yu, L.; Zhang, R.; Peng, Y.; et al. Cryptophycin-55/52 based antibody-drug conjugates: Synthesis, efficacy, and mode of action studies. Eur. J. Med. Chem. 2020, 199, 112364. [Google Scholar] [CrossRef]
- Cazzamalli, S.; Figueras, E.; Pethő, L.; Borbély, A.; Steinkühler, C.; Neri, D.; Sewald, N. In Vivo Antitumor Activity of a Novel Acetazolamide–Cryptophycin Conjugate for the Treatment of Renal Cell Carcinomas. ACS Omega 2018, 3, 14726–14731. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Wang, W.; Ohyabu, N.; Kurosu, M.; Kitagawa, I. Improved total synthesis and structure-activity relationship of arenastatin A, a potent cytotoxic spongean depsipeptide. Chem. Pharm. Bull. 1995, 43, 1598–1600. [Google Scholar] [CrossRef]
- Barrow, R.A.; Hemscheidt, T.; Liang, J.; Paik, S.; Moore, R.E.; Tius, M.A. Total Synthesis of Cryptophycins. Revision of the Structures of Cryptophycins A and C. J. Am. Chem. Soc. 1995, 117, 2479–2490. [Google Scholar] [CrossRef]
- Rej, R.; Nguyen, D.; Go, B.; Fortin, S.; Lavallée, J.-F. Total Synthesis of Cryptophycins and Their 16-(3-Phenylacryloyl) Derivatives. J. Org. Chem. 1996, 61, 6289–6295. [Google Scholar] [CrossRef]
- White, J.D.; Hong, J.; Robarge, L.A. A concise synthesis of the cytotoxic depsipeptide arenastatin A. Tetrahedron Lett. 1998, 39, 8779–8782. [Google Scholar] [CrossRef]
- White, J.D.; Hong, J.; Robarge, L.A. Total Synthesis of Cryptophycins-1, -3, -4, -24 (Arenastatin A), and -29, Cytotoxic Depsipeptides from Cyanobacteria of the Nostocaceae. J. Org. Chem. 1999, 64, 6206–6216. [Google Scholar] [CrossRef]
- Eggen, M.; Mossman, C.J.; Buck, S.B.; Nair, S.K.; Bhat, L.; Ali, S.M.; Reiff, E.A.; Boge, T.C.; Georg, G.I. Total Synthesis of Cryptophycin-24 (Arenastatin A) Amenable to Structural Modifications in the C16 Side Chain. J. Org. Chem. 2000, 65, 7792–7799. [Google Scholar] [CrossRef]
- Murakami, N.; Wang, W.; Ohyabu, N.; Ito, T.; Tamura, S.; Aoki, S.; Kobayashi, M.; Kitagawa, I. Synthesis of Amide Analogs of Arenastatin A. Tetrahedron 2000, 56, 9121–9128. [Google Scholar] [CrossRef]
- Murakami, N.; Tamura, S.; Wang, W.; Takagi, T.; Kobayashi, M. Synthesis of stable analogs in blood and conformational analysis of arenastatin A, a potent cytotoxic spongean depsipeptide. Tetrahedron 2001, 57, 4323–4336. [Google Scholar] [CrossRef]
- Murakami, N.; Tamura, S.; Koyama, K.; Sugimoto, M.; Maekawa, R.; Kobayashi, M. New analogue of arenastatin A, a potent cytotoxic spongean depsipeptide, with anti-tumor activity. Bioorg. Med. Chem. Lett. 2004, 14, 2597–2601. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Bischoff, A. Asymmetric Syntheses of Potent Antitumor Macrolides Cryptophycin B and Arenastatin A. Eur. J. Org. Chem. 2004, 2131–2141. [Google Scholar] [CrossRef]
- Buck, S.B.; Huff, J.K.; Himes, R.H.; Georg, G.I. Total Synthesis and Anti-Tubulin Activity of Epi-C3 Analogues of Cryptophycin-24. J. Med. Chem. 2004, 47, 3697–3699. [Google Scholar] [CrossRef]
- Hoard, D.W.; Moher, E.D.; Martinelli, M.J.; Norman, B.H. Synthesis of Cryptophycin 52 Using the Shi Epoxidation. Org. Lett. 2002, 4, 1813–1815. [Google Scholar] [CrossRef]
- Tripathy, N.K.; Georg, G.I. RCM approach for the total synthesis of cryptophycin-24 (Arenastatin A). Tetrahedron Lett. 2004, 45, 5309–5311. [Google Scholar] [CrossRef]
- Yadav, J.S.; Purnima, K.V.; Subba Reddy, B.V.; Nagaiah, K.; Ghamdi, A.K. Total synthesis of cryptophycin-24 (arenastatin A) via Prins cyclization. Tetrahedron Lett. 2011, 52, 6709–6712. [Google Scholar] [CrossRef]
- Kotoku, N.; Narumi, F.; Kato, T.; Yamaguchi, M.; Kobayashi, M. Stereoselective total synthesis of arenastatin A, a spongean cytotoxic depsipeptide. Tetrahedron Lett. 2007, 48, 7147–7150. [Google Scholar] [CrossRef]
- Aggarwal, V.K.; Charmant, J.; Dudin, L.; Porcelloni, M.; Richardson, J. Effect of sulfide structure on enantioselectivity in catalytic asymmetric epoxidation of aldehydes: Mechanistic insights and implications. Proc. Natl. Acad. Sci. USA 2004, 101, 5467–5471. [Google Scholar] [CrossRef]
- Vedejs, E.; Stults, J.S.; Wilde, R.G. Diastereoselectivity in the Diels-Alder reactions of thioaldehydes. J. Am. Chem. Soc. 1988, 110, 5452–5460. [Google Scholar] [CrossRef]
- Al Mehedi, M.S.; Tepe, J.J. Diastereoselective One-Pot Synthesis of Oxazolines Using Sulfur Ylides and Acyl Imines. J. Org. Chem. 2019, 84, 7219–7226. [Google Scholar] [CrossRef]
- Illa, O.; Namutebi, M.; Saha, C.; Ostovar, M.; Chen, C.C.; Haddow, M.F.; Nocquet-Thibault, S.; Luci, M.; McGarrigle, E.M.; Aggarwal, V.K. Practical and Highly Selective Sulfur Ylide-Mediated Asymmetric Epoxidations and Aziridinations Using a Cheap and Readily Available Chiral Sulfide: Extensive Studies to Map Out Scope, Limitations, and Rationalization of Diastereo- and Enantioselectivities. J. Am. Chem. Soc. 2013, 135, 11951–11966. [Google Scholar] [CrossRef]
- Trygstad, T.M.; Pang, Y.; Forsyth, C.J. Versatile Synthesis of the C3-C14 Domain of 7-Deoxyokadaic Acid. J. Org. Chem. 2009, 74, 910–913. [Google Scholar] [CrossRef]
- Keck, G.E.; Park, M.; Krishnamurthy, D. Synthetic studies on the rhizoxins. 1. Two stereoselective routes to a functionalized C(1)-C(9) subunit. J. Org. Chem. 1993, 58, 3787–3788. [Google Scholar] [CrossRef]
- Linderman, R.J.; Cusack, K.P.; Jaber, M.R. Preparation of enantiomerically enriched α-alkoxystannanes by regioselective acetal exchange or acetal hydrolysis. Tetrahedron Lett. 1996, 37, 6649–6652. [Google Scholar] [CrossRef]
- Grela, K.; Bieniek, M. Highly selective cross-metathesis with phenyl vinyl sulphone using the ‘second generation’ Grubbs’ catalyst. Tetrahedron Lett. 2001, 42, 6425–6428. [Google Scholar] [CrossRef]
- Hong, B.; Luo, T.; Lei, X. Late-Stage Diversification of Natural Products. ACS Cent. Sci. 2020, 6, 622–635. [Google Scholar] [CrossRef]
- Goethe, O.; Heuer, A.; Ma, X.; Wang, Z.; Herzon, S.B. Antibacterial properties and clinical potential of pleuromutilins. Nat. Prod. Rep. 2019, 36, 220–247. [Google Scholar] [CrossRef]
- Olejniczak, J.; Chan, M.; Almutairi, A. Light-Triggered Intramolecular Cyclization in Poly(lactic-co-glycolic acid)-Based Polymers for Controlled Degradation. Macromolecules 2015, 48, 3166–3172. [Google Scholar] [CrossRef]
- Nolan, M.D.; Schüttel, M.; Scanlan, E.M.; Nielsen, L.N. Nanomole-scale photochemical thiol-ene chemistry for high-throughput late-stage diversification of peptide macrocycles. Pept. Sci. 2024, 116, e24310. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).