The Preparation and Crystal Structures of Octaoxoketocalix[8]arene Derivatives: The Ketocalixarene Counterparts of the Largest “Major” Calixarene

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. General Considerations
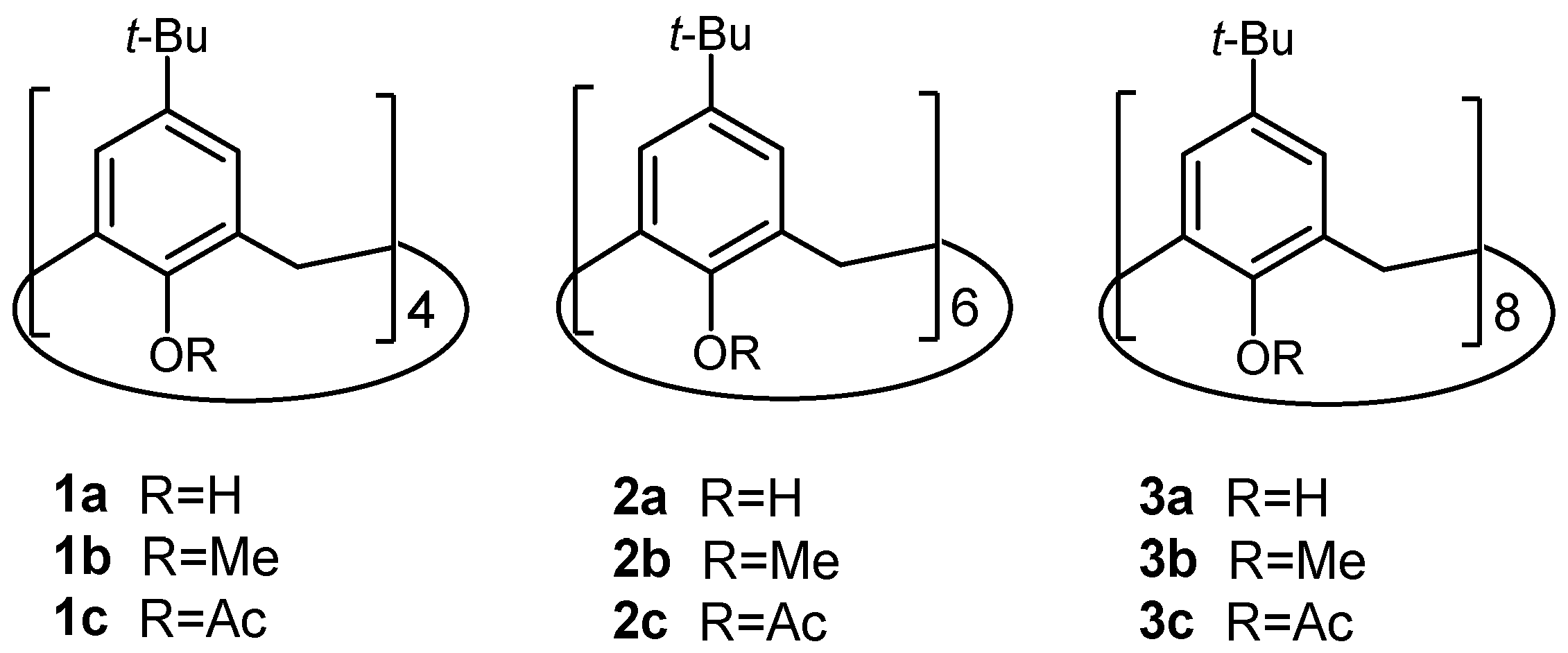
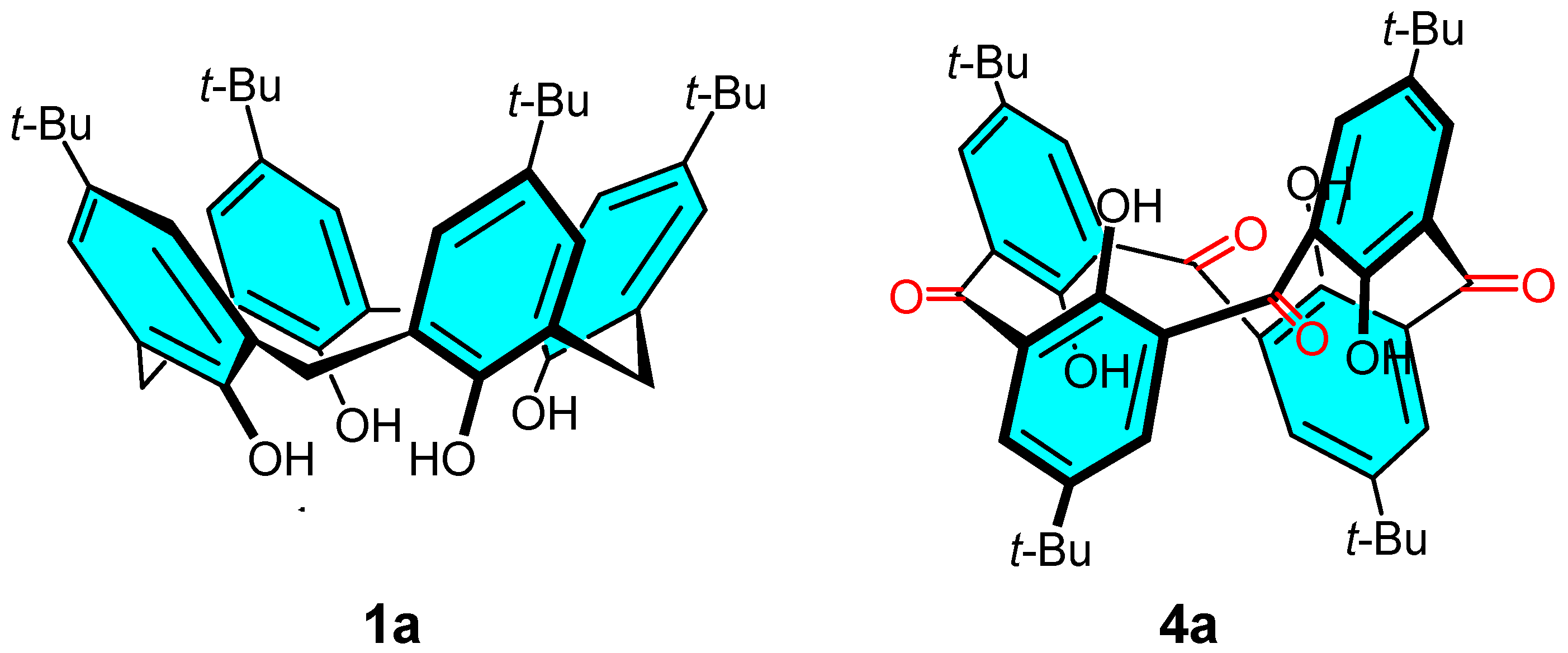
2.2. Oxidation of Octamethylether of Calix[8]arene

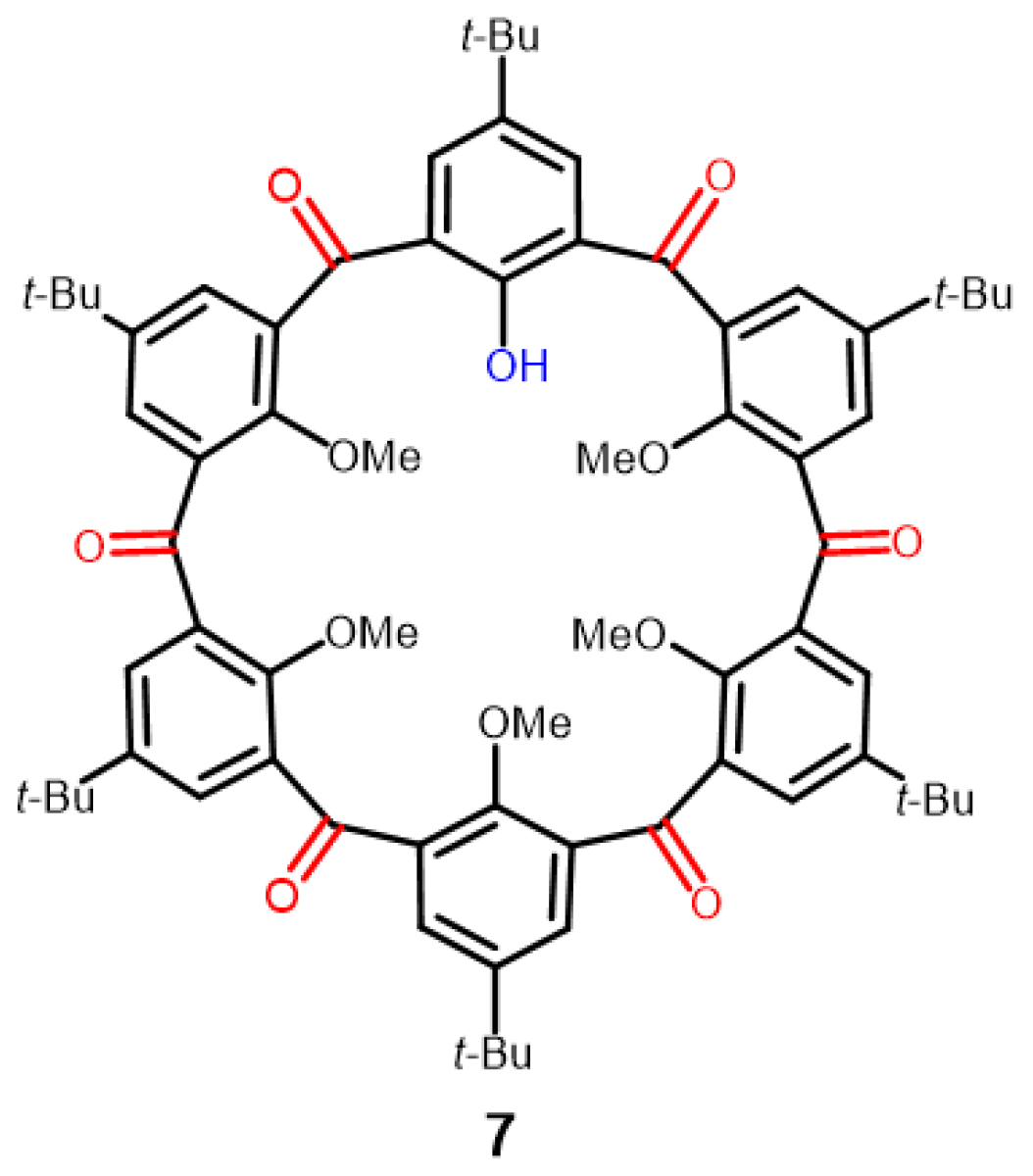
2.3. Crystal Structure of Ketocalix[8]arene 6b
2.4. Demethylation of 6b with Iodocyclohexane/DMF

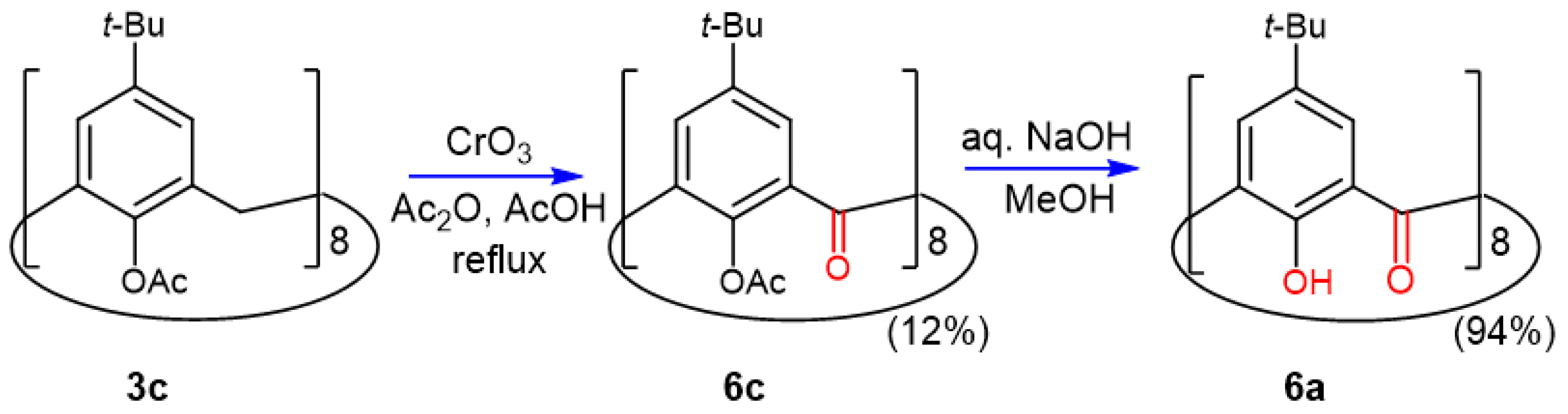
2.5. Oxidation of Octaacetoxy p-tert-Butyl-calix[8]arene 3c
2.6. Crystal Structure of Octaacetoxy Ketocalix[8]arene 6c
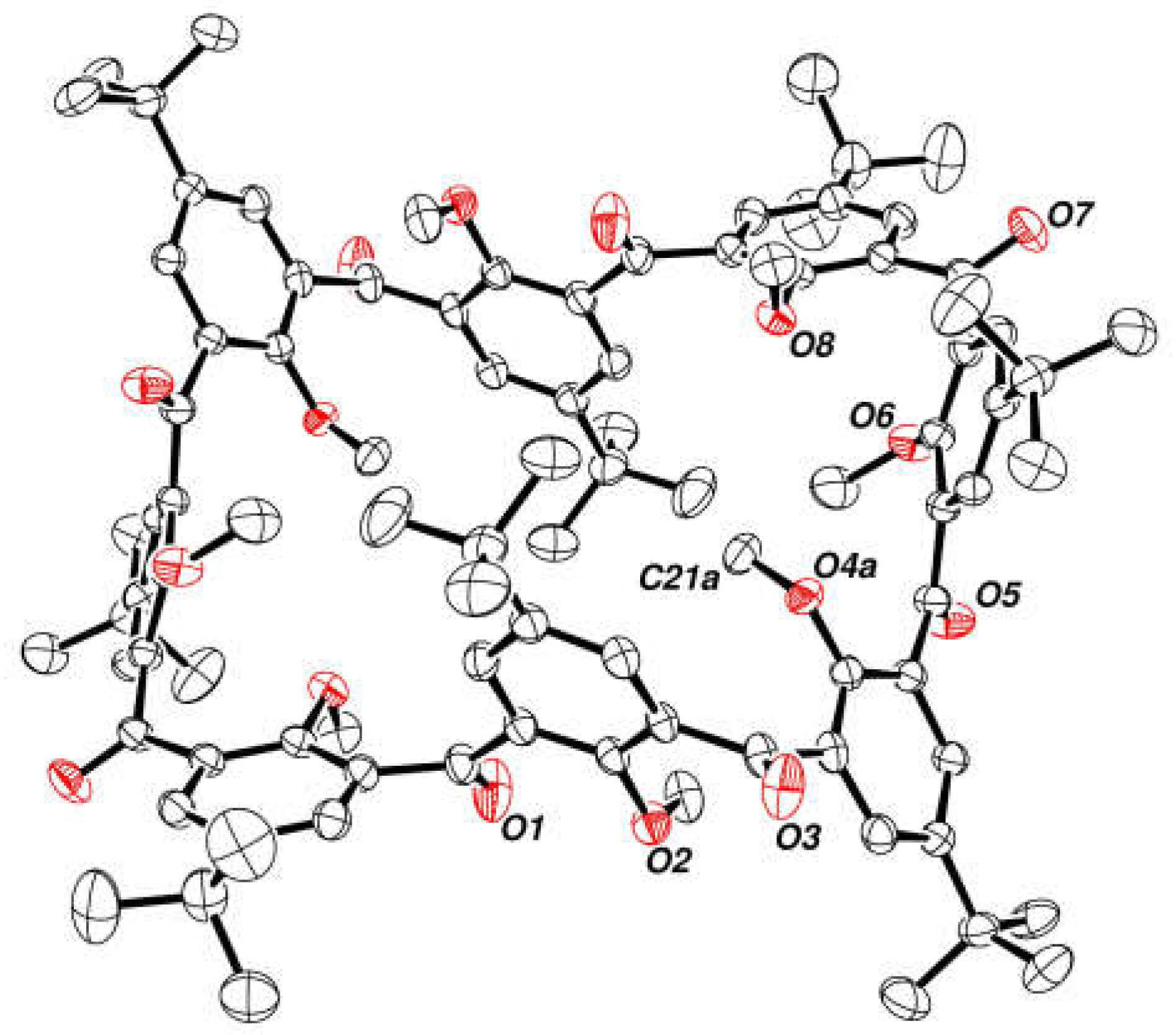
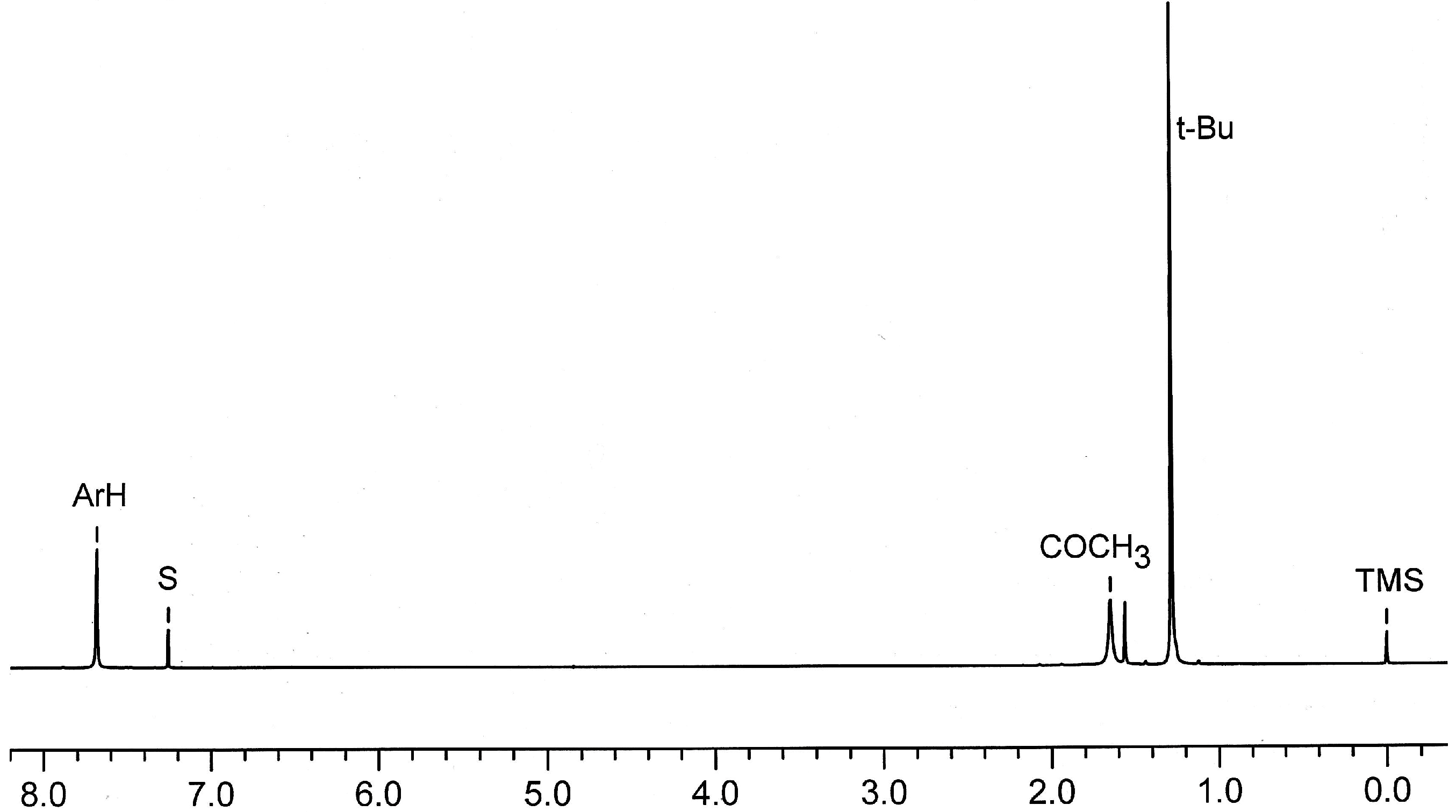
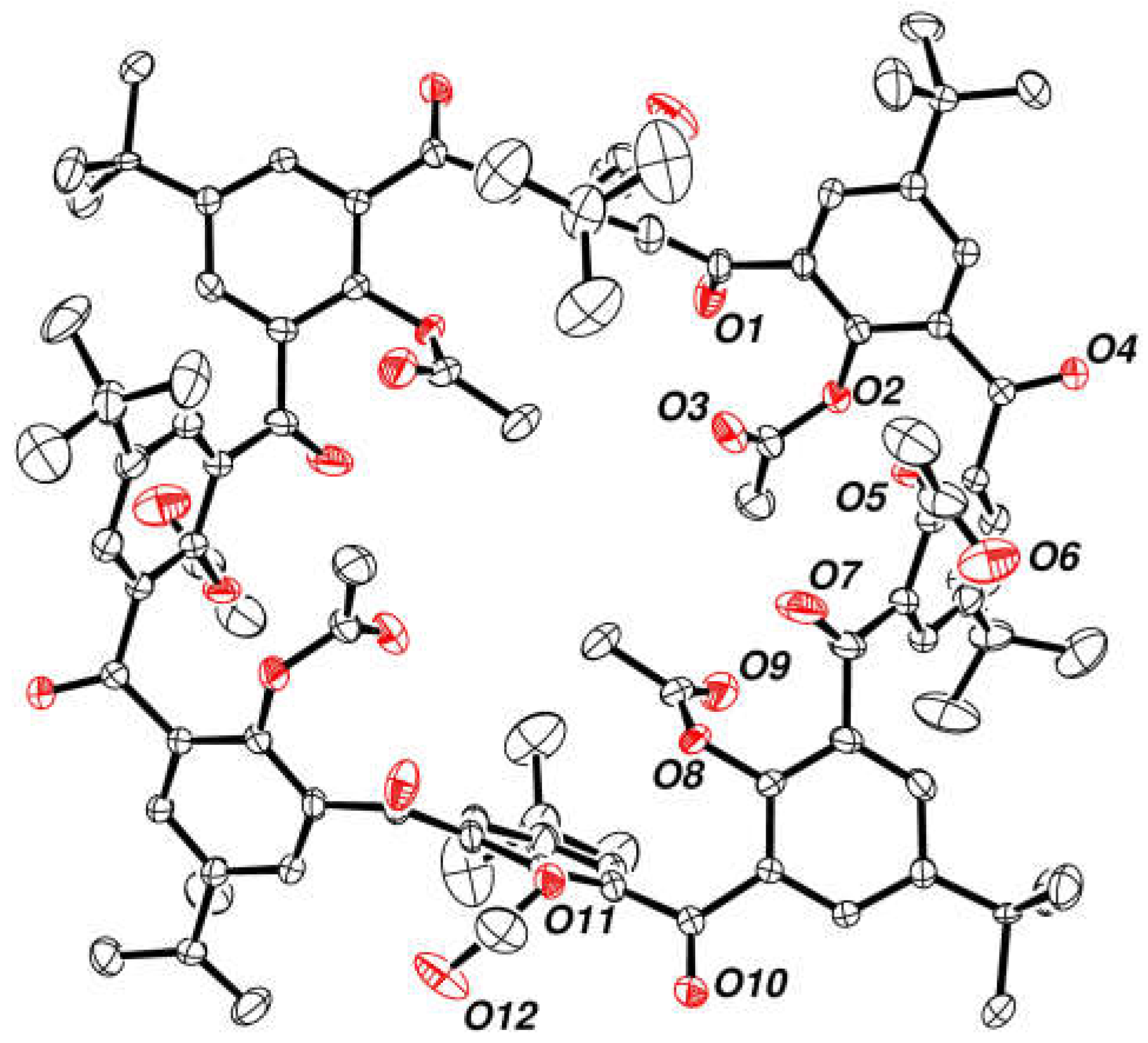
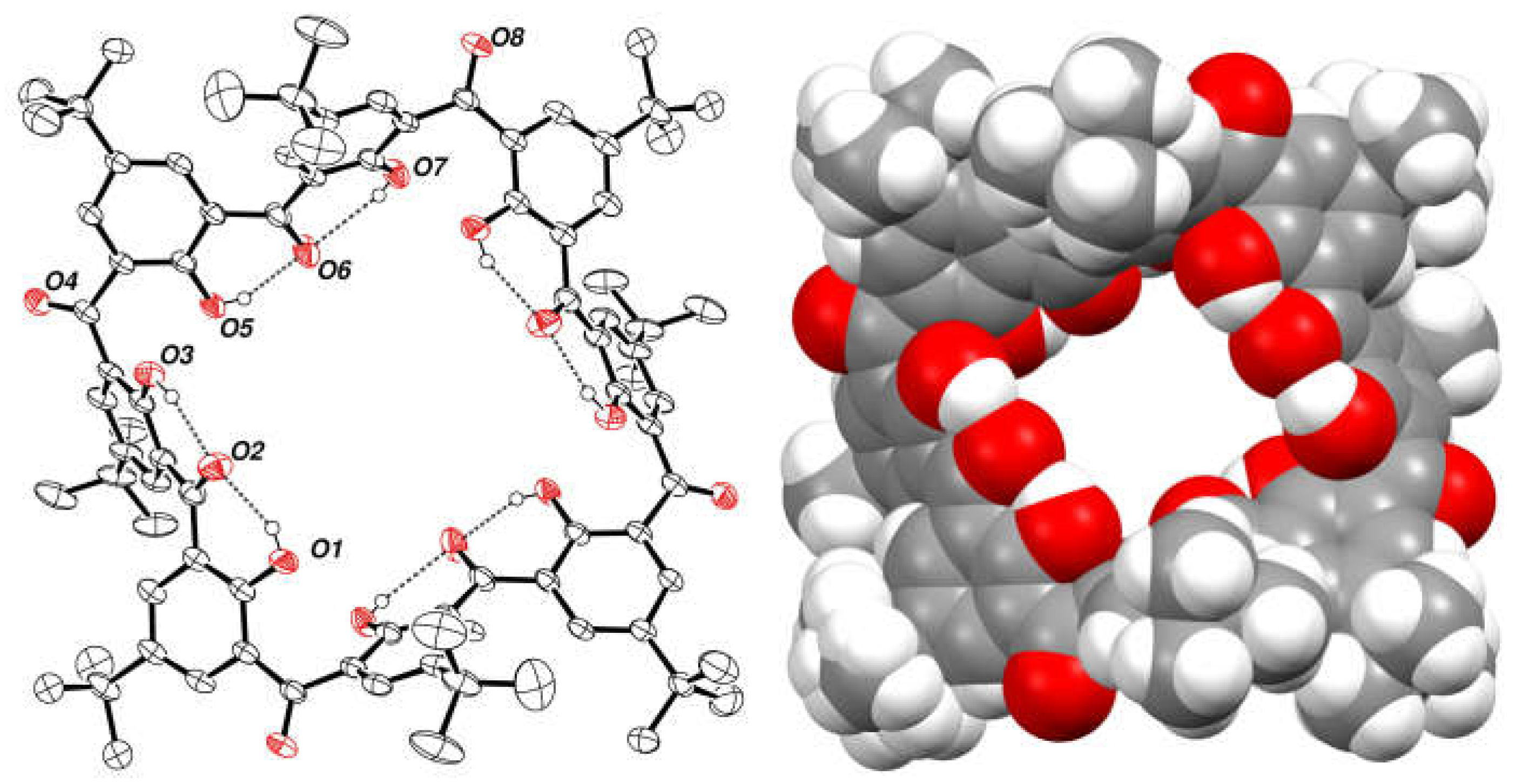
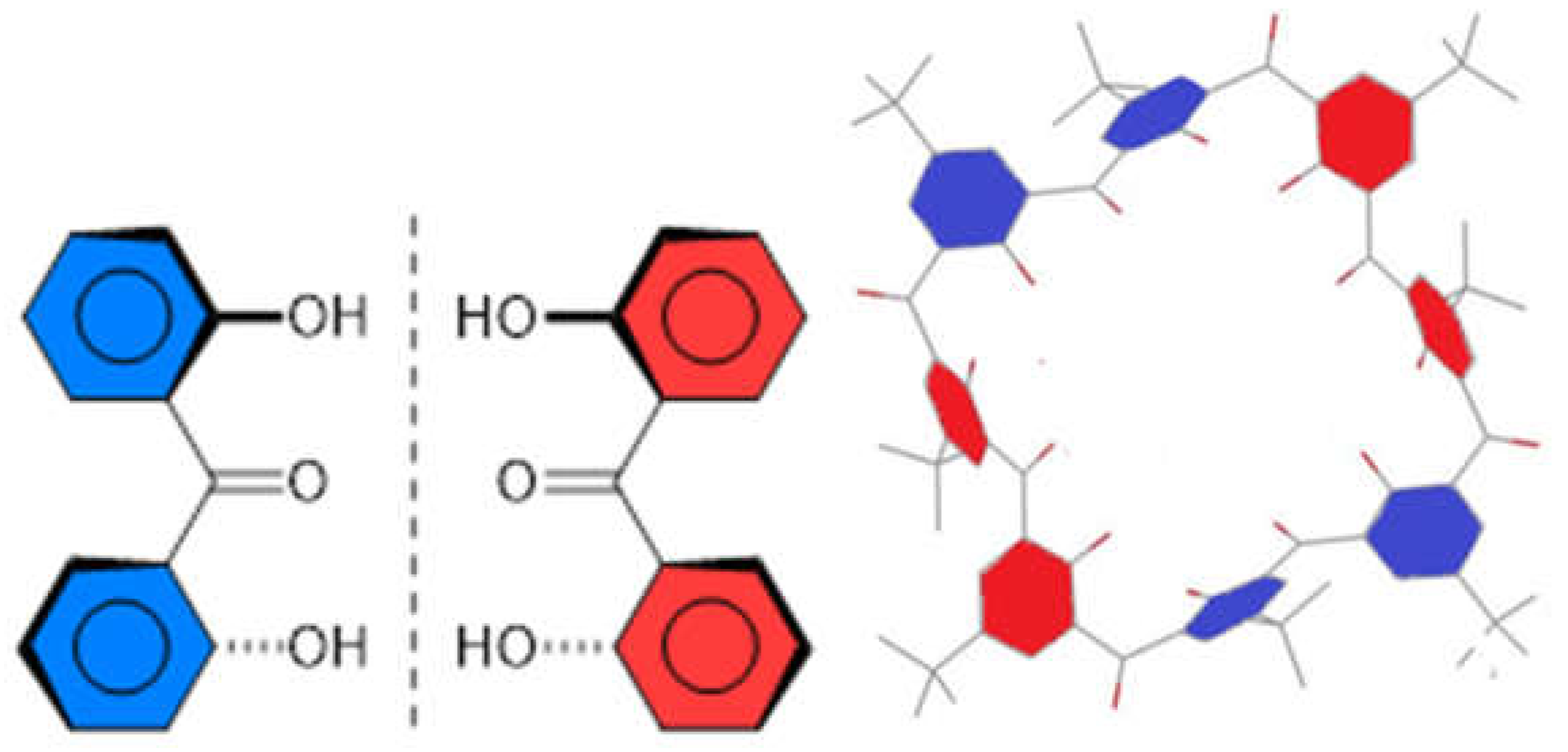
2.7. Preparation and Crystal Structure of Octahydroxyketocalix[8]arene 6a
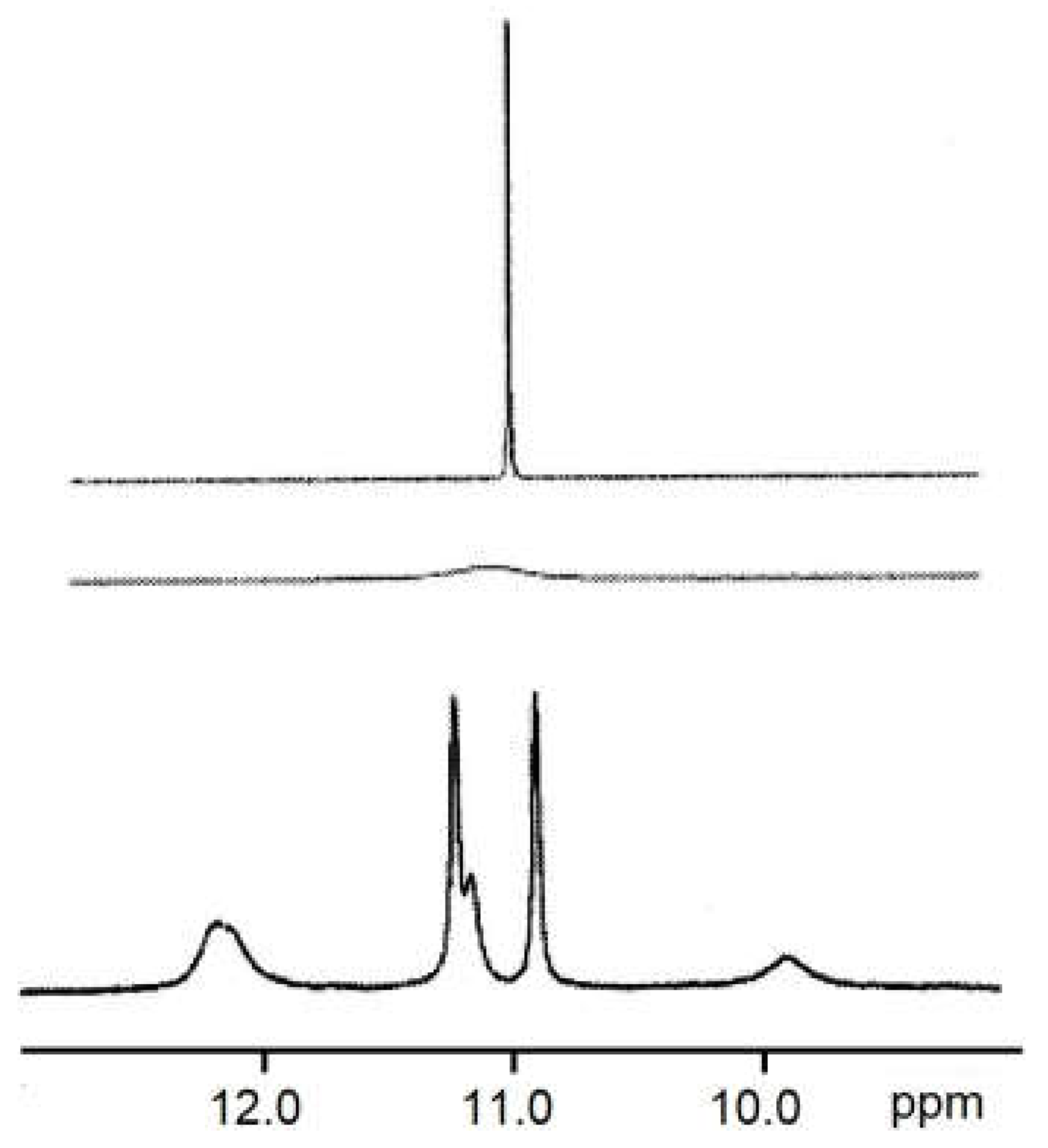
2.8. Low-Temperature 1H NMR Spectra of 6a
3. Experimental Methods
3.1. General Information
3.2. Crystal Data
- Crystal data for 6a: empirical formula: C98.5H108O21.25, formula weight: 1631.84, temperature: 150.0(1) K, crystal system: monoclinic, space group: I2/a, a: 18.4709(5) Å, b: 22.0612(6) Å, c: 23.7204(7) Å, β: 90.484(3)°, V: 9665.5(5) Å3, Z: 4, ρcalc: 1.121 g/cm3, μ: 0.078 mm−1, F(000): 3476.0, crystal size: 0.24 × 0.09 × 0.08 mm3, radiation: Mo Kα (λ = 0.71073), 2θ range for data collection: 4.41 to 64.572°, index ranges: −23 ≤ h ≤ 27, −32 ≤ k ≤ 28, −33 ≤ l ≤ 35, reflections collected: 44,192, independent reflections: 14328 [Rint =0.0435, Rsigma =0.0584], data/restraints/parameters: 14,328/0/606, goodness-of-fit on F2: 1.028, final R indexes [I ≥ 2σ (I)]: R1: 0.0916, wR2: 0. 0.2518, final R indexes [all data]: R1: 0.1404, wR2: 0.2827, largest diff. peak/hole/e Å−3 0.80/−0.34.
- Crystal data for 6b: empirical formula: C97.67H113.33Cl5O16, formula weight: 1720.50, temperature: 200.0(1) K, crystal system: triclinic, space group: P-1, a: 12.4386(1)Å, b: 13.4767(2) Å, c: 14.6784(1) Å, α: 97.887(1)°, β: 93.365(1)°, γ: 96.941(1)°, V: 2412.32(4) Å3, Z: 1, ρcalc: 1.184 g/cm3, μ: 0.212 mm−1, F(000): 912.0, crystal size: 0.43 × 0.11 × 0.07 mm3, radiation: Mo Kα (λ = 0.71073), 2θ range for data collection: 4.174 to 64.604°, index ranges: −17 ≤ h ≤ 18, −19 ≤ k ≤ 19, −21 ≤ l ≤ 21, reflections collected: 104,881, independent reflections: 15145 [Rint = 0.0309, Rsigma = 0.0221], data/restraints/parameters: 15,145/0/631, goodness-of-fit on F2: 1.031, final R indexes [I ≥ 2σ (I)]: R1: 0.0695, wR2: 0.2071, final R indexes [all data]: R1: 0.0871, wR2: 0.2205, largest diff. peak/hole/e Å−3 0.84/−0.66.
- Crystal data for 6c: empirical formula: C116H112Cl2O24, formula weight: 1960.95, temperature: 150.0(1) K, crystal system: triclinic, space group: P-1, a: 10.0905(1) Å, b: 15.8706(2) Å, c: 17.1177(2) Å, α: 84.812(1)°, β: 76.830(1)°, γ: 82.254(1)°, V: 2639.74(5) Å3, Z: 1, ρcalc: 1.234 g/cm3, μ: 0.134 mm−1, F(000): 1034.0, crystal size: 0.26 × 0.19 × 0.09 mm3, radiation: Mo Kα (λ = 0.71073), 2θ range for data collection: 4.174 to 64.798°, index ranges: −14 ≤ h ≤ 14, −22 ≤ k ≤ 23, −23 ≤ l ≤ 24, reflections collected: 94,717, independent reflections: 16,346 [Rint = 0.0412, Rsigma = 0.0312], data/restraints/parameters: 16,346/0/778, goodness-of-fit on F2: 1.027, final R indexes [I ≥ 2σ (I)]: R1: 0.0737, wR2: 0.1987, final R indexes [all data]: R1: 0.0918, wR2: 0.2101, largest diff. peak/hole/e Å−3 0.57/−0.68.
3.3. Synthesis of Compounds 6a–6c
3.3.1. Preparation of 5,11,17,23,29,35,41,47-Octa-tert-butyl-49,50,51,52,53,54,55,56-octahydroxy-2,8,14,20,26,32,38,44-octaoxo-calix[8]arene (6a)
3.3.2. Preparation of 5,11,17,23,29,35,41,47-Octa-tert-butyl-49,50,51,52,53,54,55,56-octamethoxy-2,8,14,20,26,32,38,44-octaoxocalix[8]arene (6b)
3.3.3. Preparation of 5,11,17,23,29,35,41,47-Octa-tert-butyl-49,50,51,52,53,54,55,56-octaacetoxy-2,8,14,20,26,32,38,44-octaoxo-calix[8]arene (6c)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Böhmer, V. Calixarenes, Macrocycles with (Almost) Unlimited Possibilities. Angew. Chem. Int. Ed. Engl. 1995, 34, 713–745. [Google Scholar] [CrossRef]
- Gutsche, C.D. Calixarenes Revisited; Royal Society of Chemistry: Cambridge, UK, 1998. [Google Scholar] [CrossRef]
- Asfari, Z.; Böhmer, V.; Harrowfield, J.; Vicens, J.; Saadioui, M. (Eds.) Calixarenes 2001; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2001. [Google Scholar] [CrossRef]
- Böhmer, V. The Chemistry of Phenols; Rappoport, Z., Ed.; Wiley: Chichester, UK, 2003; pp. 1369–1454. [Google Scholar] [CrossRef]
- Gutsche, C.D. Calixarenes: An Introduction, 2nd ed.; Royal Society of Chemistry: Cambridge, UK, 2008. [Google Scholar] [CrossRef]
- Neri, P.; Sessler, J.L.; Wang, M.-X. (Eds.) Calixarenes and Beyond; Springer: Cham, Switzerland, 2016. [Google Scholar] [CrossRef]
- Gutsche, C.D.; Iqbal, M. p-tert-butylcalix[4]arene. Org. Synth. 1990, 68, 234–237. [Google Scholar] [CrossRef]
- Gutsche, C.D.; Dhawan, B.; Leonis, M.; Stewart, D. p-tert-butylcalix[6]arene . Org. Synth. 1990, 68, 238–242. [Google Scholar] [CrossRef]
- Munch, J.H.; Gutsche, C.D. p-tert-butylcalix[8]arene. Org. Synth. 1990, 68, 243–244. [Google Scholar] [CrossRef]
- For the preparation of “giant” calix[n]arenes derivatives containing up to 90 rings see. Guerineau, V.; Rollet, M.; Viel, S.; Lepoittevin, B.; Costa, L.; Saint-Aguet, P.; Laurent, R.; Roger, P.; Gigmes, D.; Martini, C.; et al. The synthesis and characterization of giant Calixarenes. Nat. Commun. 2019; 10, 113. [Google Scholar] [CrossRef]
- Stewart, D.R.; Gutsche, C.D. Isolation, Characterization, and Conformational Characteristics of p-tert-Butylcalix[9-20]arenes. J. Am. Chem. Soc. 1999, 121, 4136–4146. [Google Scholar] [CrossRef]
- For a recent review on the functionalization at the meta positions of the aromatic rings see. Lhotak, P. Direct meta substitution of calix[4]arenes. Org. Biomol. Chem. 2022; 20, 7377–7390. [Google Scholar] [CrossRef]
- For a review on calixarenes modified at the bridges see. Sliwa, W.; Deska, M. Calixarenes containing modified meso bridges. Arkivoc, 2015; i, 29–47. [Google Scholar] [CrossRef]
- For a review on the intramolecular hydrogen bonding in calixarenes see. Rudkevich, D.M. Intramolecular Hydrogen Bonding in Calixarenes. Chem. Eur. J. 2000; 6, 2679–2686. [Google Scholar] [CrossRef]
- Seri, N.; Simaan, S.; Botoshansky, M.; Kaftory, M.; Biali, S.E. A Conformationally Flexible Tetrahydroxycalix[4]arene Adopting the Unusual 1,3-Alternate Conformation. J. Org. Chem. 2003, 68, 7140–7142. [Google Scholar] [CrossRef]
- A 1,3-alternate conformation was also observed in a tetrahydroxy tetrasulfinylcalix[4]arene. Mislin, G.; Graf, E.; Hosseini, M.W.; De Cian, A.; Fischer, J. Tetrasulfinylcalix[4]arenes: Synthesis and solid state structural analysis. Tetrahedron Lett. 1999; 40, 1129–1132. [Google Scholar] [CrossRef]
- Görmar, G.; Seiffarth, K.; Schulz, M.; Zimmermann, J.; Flämig, G. Synthese und Reduktion des Tetra-tert-butyltetraoxocalix[4]arens. Makromol. Chem. 1990, 191, 81–87. [Google Scholar] [CrossRef]
- Seri, N.; Thondorf, I.; Biali, S.E. Preparation and Conformation of Dioxocalix[4]arene Derivatives. J. Org. Chem. 2004, 69, 4774–4780. [Google Scholar] [CrossRef]
- Kogan, K.; Biali, S.E. Intramolecular SNAr Reactions in a Large-Ring Ketocalix[6]arene. Org. Lett. 2007, 9, 2393–2396. [Google Scholar] [CrossRef]
- Ninagawa, A.; Cho, K.; Matsuda, H. Preparation of 4-tert-butyloxocalix[n]arenes and their properties as UV-absorbers. Makromol. Chem. 1985, 186, 1379–1385. [Google Scholar] [CrossRef]
- Reddy, P.A.; Kashyap, R.P.; Watson, W.H.; Gutsche, C.D. Calixarenes 30. Calixquinones. Isr. J. Chem. 1992, 32, 89–96. [Google Scholar] [CrossRef]
- Neri, P.; Bottino, A.; Cunsolo, F.; Piattelli, M.; Gavuzzo, E. 5,5′-Bicalix[4]arene: The Bridgeless Prototype of Double Calix[4]arenes of the “Head-to-Head” Type. Angew. Chem. Int. Ed. 1998, 37, 166–169. [Google Scholar] [CrossRef]
- Kuno, L.; Seri, N.; Biali, S.E. Tetraaddition of PhLi to a Ketocalixarene Derivative. Org. Lett. 2007, 9, 1577–1580. [Google Scholar] [CrossRef]
- Poms, D.; Itzhak, N.; Kuno, L.; Biali, S.E. Calixradialenes: Calixarene Derivatives with Exocyclic Double Bonds. J. Org. Chem. 2014, 79, 538–545. [Google Scholar] [CrossRef] [PubMed]
- Fischer, C.; Lin, G.; Seichter, W.; Weber, E. Simple two step reaction towards a 2,7,14,21-tetraoxocalix[4]arene via bridge brominated intermediate. Tetrahedron Lett. 2013, 54, 2187–2189. [Google Scholar] [CrossRef]
- Saikia, I.; Borah, A.J.; Phukan, P. Use of Bromine and Bromo-Organic Compounds in Organic Synthesis. Chem. Rev. 2016, 116, 6837–7042. [Google Scholar] [CrossRef]
- Itzhak, N.; Biali, S.E. An Expedient Synthesis of Ketocalix[6]arene Hexamethyl Ether. Synthesis 2018, 50, 3897–3901. [Google Scholar] [CrossRef]
- Zuo, L.; Yao, S.; Wang, W.; Duan, W. An efficient method for demethylation of aryl methyl ethers. Tetrahedron Lett. 2008, 49, 4054–4056. [Google Scholar] [CrossRef]
- Carroll, L.T.; Hill, P.A.; Ngo, C.Q.; Klatt, K.P.; Fantini, J.L. Synthesis and reactions of a 2-chlorocalix[4]arene and a 2,2′-coupled dicalixarene. Tetrahedron 2013, 69, 5002–5007. [Google Scholar] [CrossRef]
- Fong, A.; McCormick, L.; Teat, S.J.; Brechin, E.K.; Dalgarno, S.J. Exploratory studies into 3d/4f cluster formation with fully bridge-substituted calix[4]arenes. Supramol. Chem. 2018, 30, 504–509. [Google Scholar] [CrossRef]
- Itzhak, N.; Biali, S.E. Preparation and Crystal Structure of the 1,3-alternate Atropisomer of a Calix[4]radialene. Eur. J. Org. Chem. 2015, 2015, 3221–3225. [Google Scholar] [CrossRef]
- For a recent study see. Kieliszek, A.; Malinska, M. Conformations of p-tert-Butylcalix[8]arene in Solvated Crystal Structures. Cryst. Growth. Des. 2021; 21, 6862–6871. [Google Scholar] [CrossRef]
- Rappoport, Z.; Biali, S.E.; Kaftory, M. Application of the structural correlation method to ring-flip processes in benzophenones. J. Am. Chem. Soc. 1990, 112, 7742–7748. [Google Scholar] [CrossRef]
- Abraham, R.J.; Haworth, I.S. Conformational analysis. Part 12. A theoretical and lanthanide-induced shift (LIS) investigation of the conformation of benzophenone and 3,4′-dichlorobenzophenone. J. Chem. Soc. Perkin Trans. 1988, 8, 1429–1437. [Google Scholar] [CrossRef]
- Grilli, S.; Lunazzi, L.; Mazzanti, A.; Casarini, D.; Femoni, C. Conformational Studies by Dynamic NMR. 78. Stereomutation of the Helical Enantiomers of Trigonal Carbon Diaryl-Substituted Compounds: Dimesitylketone, Dimesitylthioketone, and Dimesitylethylene. J. Org. Chem. 2001, 66, 488–495. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kogan, K.; Omar, S.; Bogoslavsky, B.; Biali, S.E. The Preparation and Crystal Structures of Octaoxoketocalix[8]arene Derivatives: The Ketocalixarene Counterparts of the Largest “Major” Calixarene. Molecules 2024, 29, 4094. https://doi.org/10.3390/molecules29174094
Kogan K, Omar S, Bogoslavsky B, Biali SE. The Preparation and Crystal Structures of Octaoxoketocalix[8]arene Derivatives: The Ketocalixarene Counterparts of the Largest “Major” Calixarene. Molecules. 2024; 29(17):4094. https://doi.org/10.3390/molecules29174094
Chicago/Turabian StyleKogan, Katerina, Suheir Omar, Benny Bogoslavsky, and Silvio E. Biali. 2024. "The Preparation and Crystal Structures of Octaoxoketocalix[8]arene Derivatives: The Ketocalixarene Counterparts of the Largest “Major” Calixarene" Molecules 29, no. 17: 4094. https://doi.org/10.3390/molecules29174094