
Desulfonylative Functionalization of Organosulfones via Inert (Hetero)Aryl C(sp2)–SO2 Bond Cleavage



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Transition Metal-Catalyzed Desulfonylative Cross-Coupling Reaction
2.1. Kumada-Type Cross-Coupling Reaction
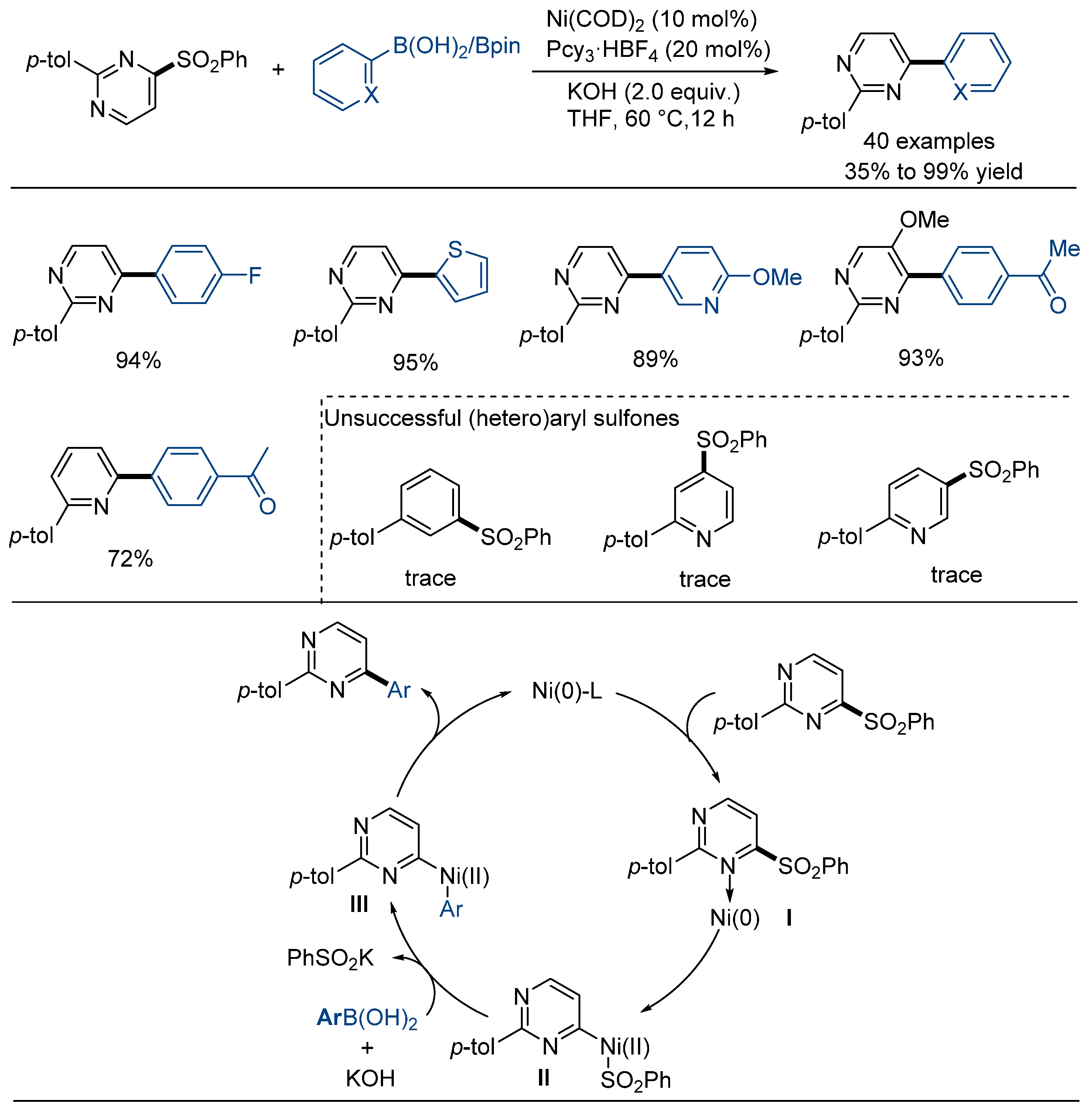
2.2. Suzuki–Miyaura-Type Cross-Coupling Reaction
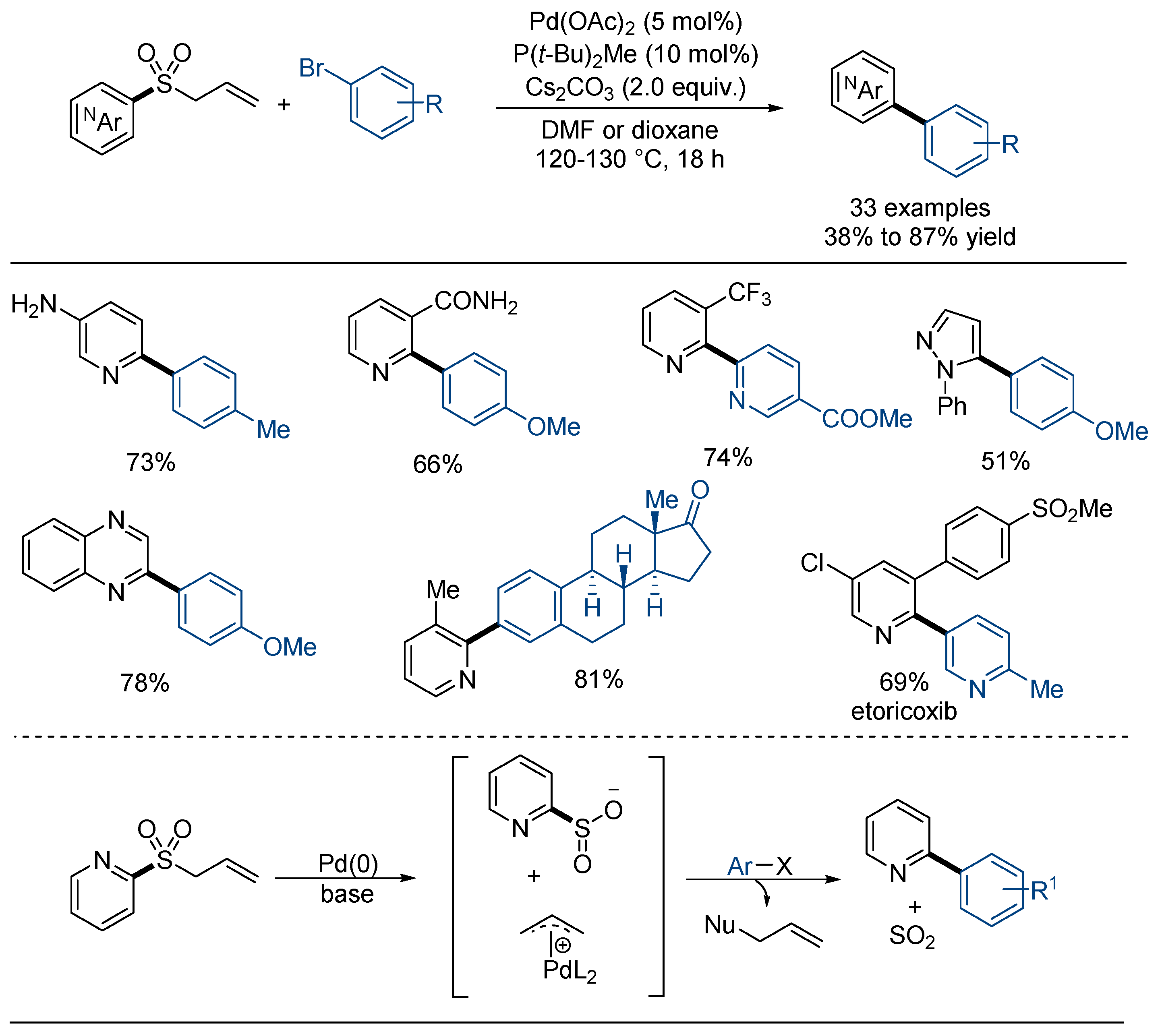
2.3. Desulfonylative Cross-Coupling Reaction with (Hetero)Aryl Halides
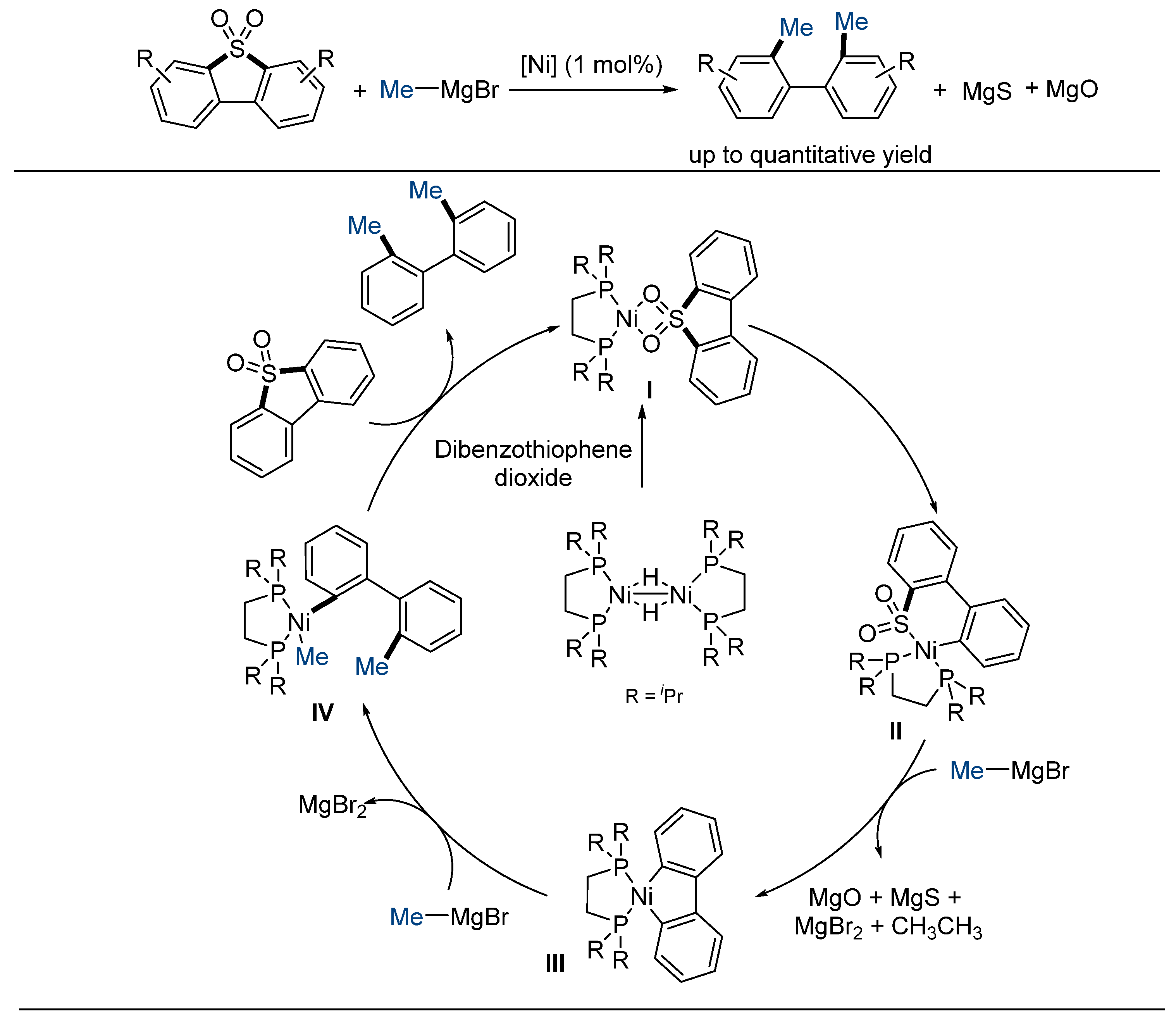
2.4. Intramolecular Desulfonylative Cross-Coupling Reaction
2.5. Desulfonylative Cross-Coupling Reaction with Other Reagents
3. Photo-/Electrocatalytic Desulfonylative Cross-Coupling
3.1. Desulfonylative Cross-Coupling with Alkylating Agents
3.2. Desulfonylative Cross-Coupling with Alkenes
4. Conclusion
Author Contributions
Funding
Conflicts of Interest
References
- Trost, B.M. Chemical Chameleons. Organosulfones as Synthetic Building Blocks. Bull. Chem. Soc. Jpn. 2006, 61, 107–124. [Google Scholar] [CrossRef]
- Elena, N.P. Sulfones and Sulfoxides in the Total Synthesis of Biologically Active Natural Compounds. Russ. Chem. Rev. 2000, 69, 367. [Google Scholar]
- Wang, X.; Chen, L.; Chong, S.Y.; Little, M.A.; Wu, Y.; Zhu, W.-H.; Clowes, R.; Yan, Y.; Zwijnenburg, M.A.; Sprick, R.S.; et al. Sulfone-containing covalent organic frameworks for photocatalytic hydrogen evolution from water. Nat. Chem. 2018, 10, 1180–1189. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.M.; Kalnmals, C.A. Sulfones as Chemical Chameleons: Versatile Synthetic Equivalents of Small-Molecule Synthons. Chem. Eur. J. 2019, 25, 11193–11213. [Google Scholar] [CrossRef] [PubMed]
- Nájera, C.; Yus, M. Desulfonylation Reactions: Recent Developments. Tetrahedron 1999, 55, 10547–10658. [Google Scholar] [CrossRef]
- Chu, X.-Q.; Ge, D.; Cui, Y.-Y.; Shen, Z.-L.; Li, C.-J. Desulfonylation via Radical Process: Recent Developments in Organic Synthesis. Chem. Rev. 2021, 121, 12548–12680. [Google Scholar] [CrossRef]
- Nambo, M.; Crudden, C.M. Transition Metal-Catalyzed Cross-Couplings of Benzylic Sulfone Derivatives. Chem. Rec. 2021, 21, 3978–3989. [Google Scholar] [CrossRef]
- Corpas, J.; Kim-Lee, S.-H.; Mauleón, P.; Arrayás, R.G.; Carretero, J.C. Beyond Classical Sulfone Chemistry: Metal- and Photocatalytic Approaches for C–S Bond Functionalization of Sulfones. Chem. Soc. Rev. 2022, 51, 6774–6823. [Google Scholar] [CrossRef]
- Huang, S.; Wang, M.; Jiang, X. Ni-catalyzed C–S bond construction and cleavage. Chem. Soc. Rev. 2022, 51, 8351–8377. [Google Scholar] [CrossRef]
- Nambo, M.; Maekawa, Y.; Crudden, C.M. Desulfonylative Transformations of Sulfones by Transition-Metal Catalysis, Photocatalysis, and Organocatalysis. ACS Cat. 2022, 12, 3013–3032. [Google Scholar] [CrossRef]
- Paul, B.; Paul, H.; Chatterjee, I. Photoredox-Mediated Desulfonylative Radical Reactions: An Excellent Approach Towards C–C and C–Heteroatom Bond Formation. Synthesis 2022, 54, 5409–5422. [Google Scholar] [CrossRef]
- Wenkert, E.; Ferreira, T.W.; Michelotti, E.L. Nickel-Induced Conversion of Carbon–Sulphur into Carbon–Carbon Bonds. One-Step Transformations of Enol Sulphides into Olefins and Benzenethiol Derivatives into Alkylarenes and Biaryls. J. Chem. Soc. Chem. Commun. 1979, 14, 637–638. [Google Scholar] [CrossRef]
- Clayden, J.; Julia, M. ortho-Substituted Unsymmetrical Biaryls from Aryl tert-Butyl Sulfones. J. Chem. Soc. Chem. Commun. 1993, 1682–1683. [Google Scholar] [CrossRef]
- Clayden, J.; Cooney, J.J.A.; Julia, M. Nickel-Catalysed Substitutions of Aryl tert-Butyl Sulfones with Organometallic Reagents: Synthesis of ortho-Substituted Unsymmetrical Biaryls. J. Chem. Soc. Perkin Trans. 1995, 1, 7–14. [Google Scholar] [CrossRef]
- Oviedo, A.; Torres-Nieto, J.; Arévalo, A.; García, J.J. Deoxydesulfurization of Sulfones Derived from Dibenzothiophene Using Nickel Compounds. J. Mol. Catal. A Chem. 2008, 293, 65–71. [Google Scholar] [CrossRef]
- Li, W.-X.; Huo, B.-J.; Huang, J.-Y.; Rao, W.; Xu, H.; Zhou, X.; Shen, Z.-L. Nickel-Catalyzed Cross-electrophile Coupling of Unactivated (hetero)aryl Sulfone with Aryl Bromide. J. Catal. 2024, 430, 115359. [Google Scholar] [CrossRef]
- Liu, J.; Robins, M.J. Fluoro, Alkylsulfanyl, and Alkylsulfonyl Leaving Groups in Suzuki Cross-Coupling Reactions of Purine 2‘-Deoxynucleosides and Nucleosides. Org. Lett. 2005, 7, 1149–1151. [Google Scholar] [CrossRef]
- Hennessy, E.J.; Cornebise, M.; Gingipalli, L.; Grebe, T.; Hande, S.; Hoesch, V.; Huynh, H.; Throner, S.; Varnes, J.; Wu, Y. Preparation of Highly Functionalized 1,5-Disubstituted Tetrazoles via Palladium-Catalyzed Suzuki Coupling. Tetrahedron Lett. 2017, 58, 1709–1713. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhang, W.; Cui, R.; Zhang, Y.; Niu, D. Synthesis of 2,4-Diarylated Pyrimidines Enabled by Ni-Catalyzed C–Sulfone Bond Activation. Org. Chem. Front. 2023, 10, 645–650. [Google Scholar] [CrossRef]
- Chatelain, P.; Sau, A.; Rowley, C.N.; Moran, J. Suzuki–Miyaura Coupling of (Hetero)Aryl Sulfones: Complementary Reactivity Enables Iterative Polyaryl Synthesis. Angew. Chem. Int. Ed. 2019, 58, 14959–14963. [Google Scholar] [CrossRef]
- Fukuda, J.-I.; Nogi, K.; Yorimitsu, H. Cross-Coupling of Aryl Trifluoromethyl Sulfones with Arylboronates by Cooperative Palladium/Rhodium Catalysis. Org. Lett. 2019, 21, 8987–8991. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Ordaz, R.; García, J.J. Desulfurization of Dibenzothiophene and Dibenzothiophene Sulfone via Suzuki–Miyaura Type Reaction: Direct Access to o-Terphenyls and Polyphenyl Derivatives. Polyhedron 2018, 154, 373–381. [Google Scholar] [CrossRef]
- Markovic, T.; Murray, P.R.D.; Rocke, B.N.; Shavnya, A.; Blakemore, D.C.; Willis, M.C. Heterocyclic Allylsulfones as Latent Heteroaryl Nucleophiles in Palladium-Catalyzed Cross-Coupling Reactions. J. Am. Chem. Soc. 2018, 140, 15916–15923. [Google Scholar] [CrossRef] [PubMed]
- Cook, X.A.F.; Pantaine, L.R.E.; Blakemore, D.C.; Moses, I.B.; Sach, N.W.; Shavnya, A.; Willis, M.C. Base-Activated Latent Heteroaromatic Sulfinates as Nucleophilic Coupling Partners in Palladium-Catalyzed Cross-Coupling Reactions. Angew. Chem. Int. Ed. 2021, 60, 22461–22468. [Google Scholar] [CrossRef]
- Huang, X.; Tang, L.; Song, Z.; Jiang, S.; Liu, X.; Ma, M.; Chen, B.; Ma, Y. Nickel-Catalyzed Desulfonylative Reductive Cross-Coupling of Aryl Sulfones with Aryl Bromides. Org. Lett. 2023, 25, 1198–1203. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, F.; Nogi, K.; Yorimitsu, H. Intramolecular Desulfitative Coupling: Nickel-Catalyzed Transformation of Diaryl Sulfones into Biaryls via Extrusion of SO2. Org. Lett. 2018, 20, 6601–6605. [Google Scholar] [CrossRef]
- Yu, T.-Y.; Zheng, Z.-J.; Bai, J.-H.; Fang, H.; Wei, H. Nickel-Catalyzed Intramolecular Coupling of Sulfones via the Extrusion of Sulfur Dioxide. Adv. Synth. Catal. 2019, 361, 2020–2024. [Google Scholar] [CrossRef]
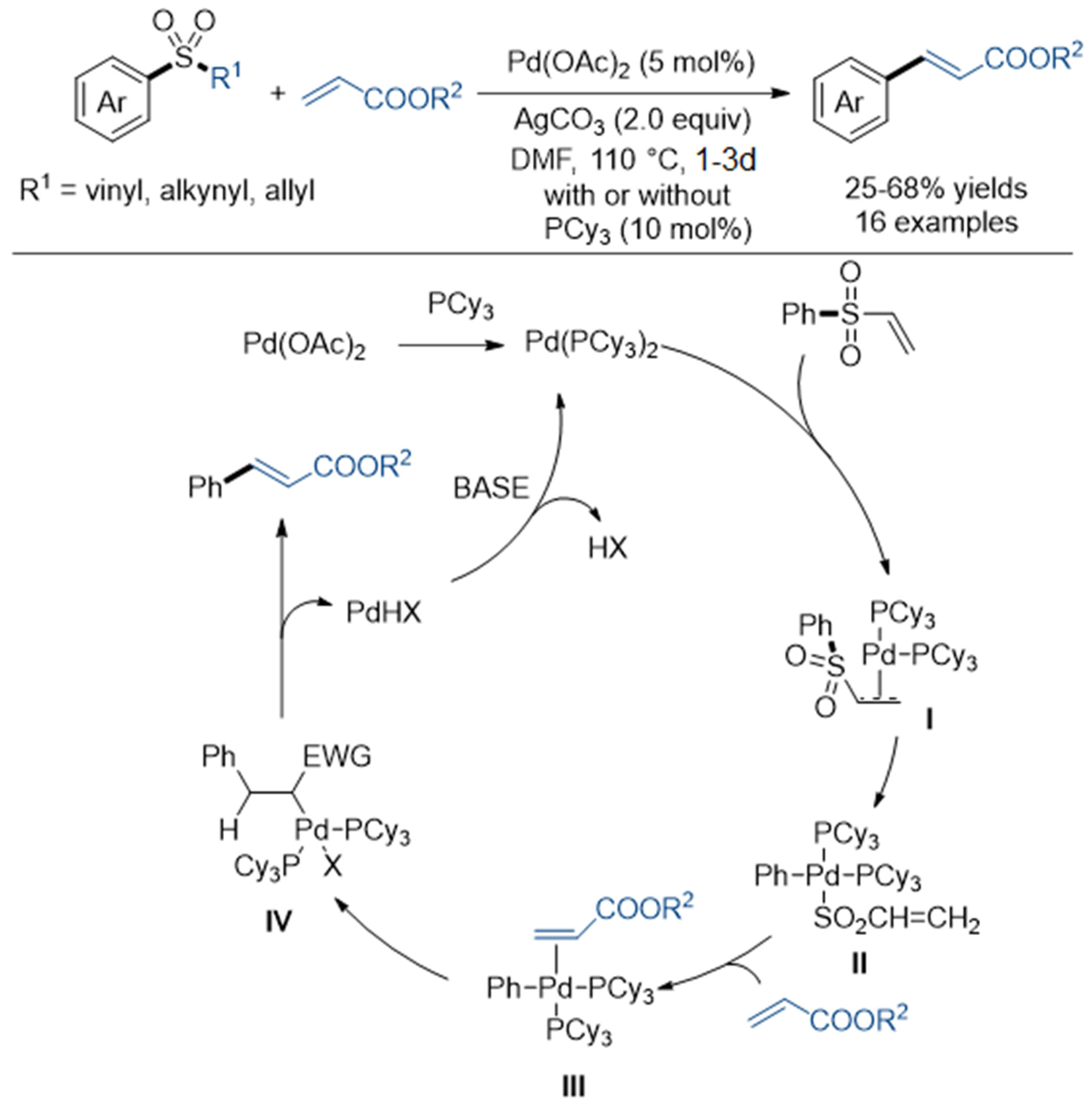
- García Ruano, J.L.; Alemán, J.; Paredes, C.G. Oxidative Addition of Pd(0) to Ar−SO2R Bonds: Heck-Type Reactions of Sulfones. Org. Lett. 2006, 8, 2683–2686. [Google Scholar] [CrossRef]
- Matsubara, R.; Kim, H.; Sakaguchi, T.; Xie, W.; Zhao, X.; Nagoshi, Y.; Wang, C.; Tateiwa, M.; Ando, A.; Hayashi, M.; et al. Modular Synthesis of Carbon-Substituted Furoxans via Radical Addition Pathway. Useful Tool for Transformation of Aliphatic Carboxylic Acids Based on “Build-and-Scrap” Strategy. Org. Lett. 2020, 22, 1182–1187. [Google Scholar] [CrossRef]
- Yang, J.; Xiao, J.; Chen, T.; Yin, S.-F.; Han, L.-B. Efficient nickel-catalyzed phosphinylation of C–S bonds forming C–P bonds. Chem. Commun. 2016, 52, 12233–12236. [Google Scholar] [CrossRef]
- Saito, H.; Nogi, K.; Yorimitsu, H. Palladium-Catalyzed Double Borylation of Diaryl Sulfoxides with Diboron. Synthesis 2017, 49, 4769–4774. [Google Scholar]
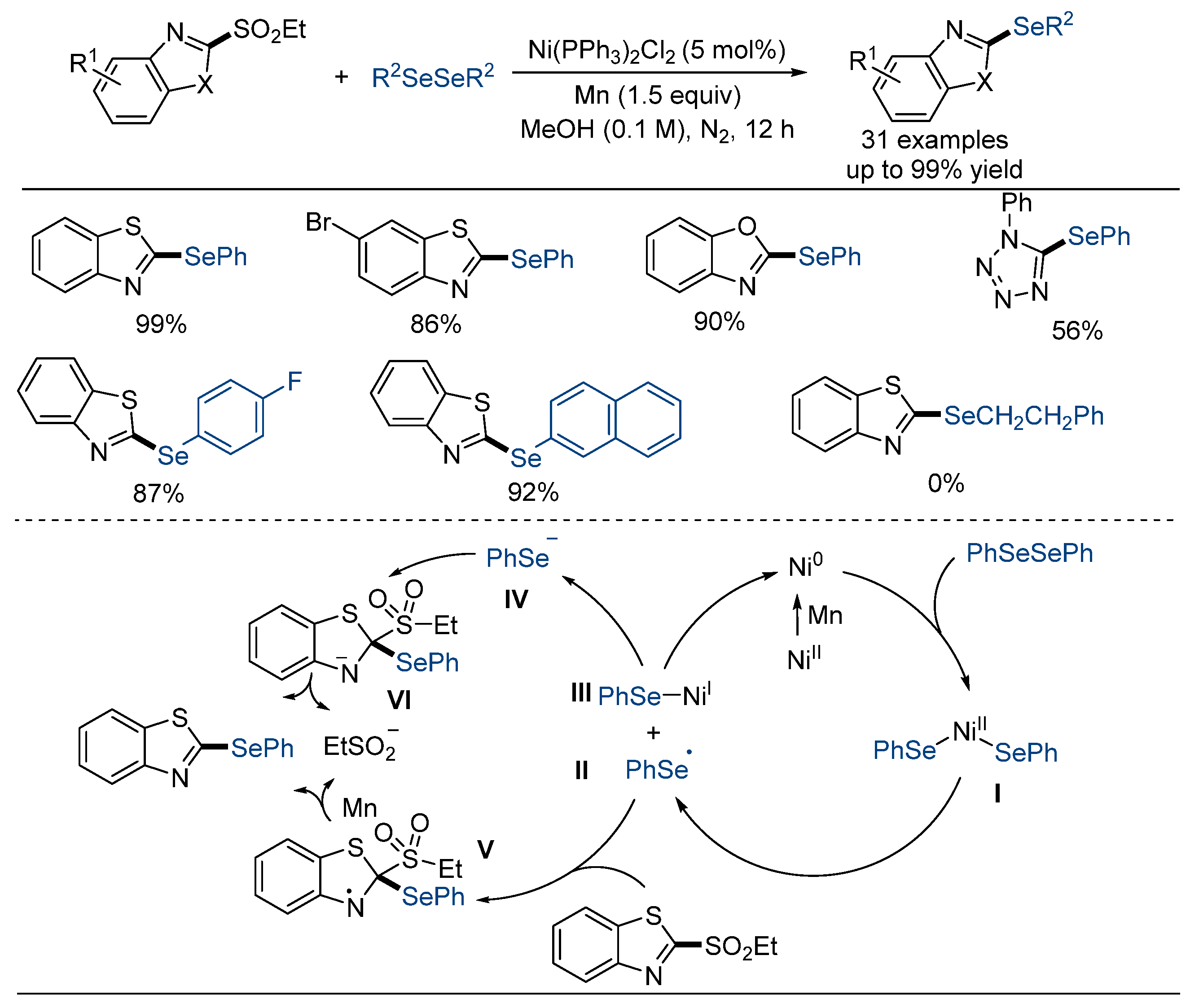
- Liu, X.-Y.; Dou, Y.-X.; Hasan, M.; Rao, W.; Shen, D.; Shen, S.; Wang, S.-Y. Ni-catalyzed cross-coupling of heteroaryl sulfones and diselenides via deheteroaromatization and heteroaromatization: Synthesis of heteroaryl selenides. Org. Chem. Front. 2024, 11, 1469–1472. [Google Scholar] [CrossRef]
- Romero, N.A.; Nicewicz, D.A. Organic Photoredox Catalysis. Chem. Rev. 2016, 116, 10075–10166. [Google Scholar] [CrossRef]
- Crisenza, G.E.M.; Melchiorre, P. Chemistry Glows Green with Photoredox Catalysis. Nat. Commun. 2020, 11, 803. [Google Scholar] [CrossRef] [PubMed]
- Kamijo, S.; Kamijo, K.; Murafuji, T. Synthesis of Alkylated Pyrimidines via Photoinduced Coupling Using Benzophenone as a Mediator. J. Org. Chem. 2017, 82, 2664–2671. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-J.; Zheng, S.; Matsui, J.K.; Lu, Z.; Molander, G.A. Desulfonative Photoredox Alkylation of N-Heteroaryl Sulfones—An Acid-Free Approach for Substituted Heteroarene Synthesis. Chem. Sci. 2019, 10, 4389–4393. [Google Scholar] [CrossRef]
- Kamijo, S.; Kamijo, K.; Murafuji, T. Aryl Ketone Mediated Photoinduced Radical Coupling for the Alkylation of Benzazoles Employing Saturated Heterocyclic Compounds. Synthesis 2019, 51, 3859–3864. [Google Scholar]
- Li, P.; Tu, J.-L.; Gao, H.; Shi, C.; Zhu, Y.; Guo, L.; Yang, C.; Xia, W. FeCl3-Catalyzed C(sp3)−H Heteroarylation Enabled by Photoinduced Ligand-to-Metal Charge Transfer. Adv. Synth. Catal. 2024, 366, 220–224. [Google Scholar] [CrossRef]
- Yonekura, K.; Murooka, M.; Aoki, K.; Shirakawa, E. Electrochemical Direct α-Arylation of Alkylamines with Sulfonylarenes. Org. Lett. 2023, 25, 6682–6687. [Google Scholar] [CrossRef]
- Meng, L.; Dong, J.; Tang, Y.; Yang, H.; Sun, L.; Chen, J.; Fan, B. Photoredox Catalytic Aminomethylation of Sulfonylthiazoles. Green Chem. 2024, 26, 6063–6067. [Google Scholar] [CrossRef]
- Yu, J.; Wu, Z.; Zhu, C. Efficient Docking–Migration Strategy for Selective Radical Difluoromethylation of Alkenes. Angew. Chem. Int. Ed. 2018, 57, 17156–17160. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wu, S.; Yu, J.; Lu, C.; Wu, Z.; Wu, X.; Xue, X.-S.; Zhu, C. Polarity Umpolung Strategy for the Radical Alkylation of Alkenes. Angew. Chem. Int. Ed. 2020, 59, 8195–8202. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Lee, K.; Kim, S.; Hong, S. Photoinduced Carboarylation of Alkenes by Using Bifunctional Reagents. Synlett 2023, 34, 1437–1441. [Google Scholar]

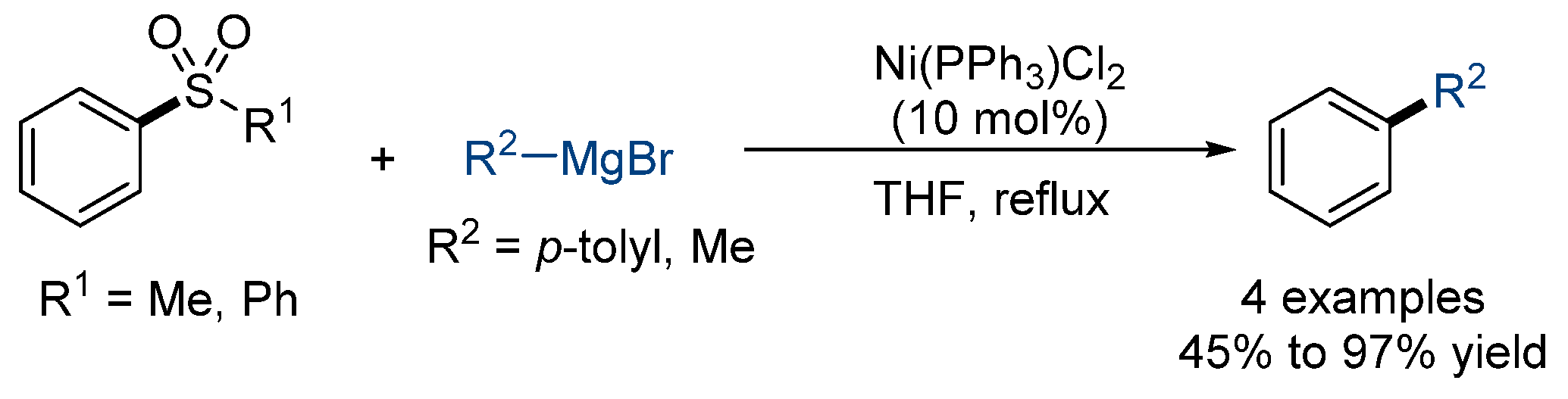












Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, R.; Gu, B.; Wang, M.; Zhao, Y.; Jiang, X. Desulfonylative Functionalization of Organosulfones via Inert (Hetero)Aryl C(sp2)–SO2 Bond Cleavage. Molecules 2024, 29, 4137. https://doi.org/10.3390/molecules29174137
Huang R, Gu B, Wang M, Zhao Y, Jiang X. Desulfonylative Functionalization of Organosulfones via Inert (Hetero)Aryl C(sp2)–SO2 Bond Cleavage. Molecules. 2024; 29(17):4137. https://doi.org/10.3390/molecules29174137
Chicago/Turabian StyleHuang, Rui, Boning Gu, Ming Wang, Yinsong Zhao, and Xuefeng Jiang. 2024. "Desulfonylative Functionalization of Organosulfones via Inert (Hetero)Aryl C(sp2)–SO2 Bond Cleavage" Molecules 29, no. 17: 4137. https://doi.org/10.3390/molecules29174137
APA StyleHuang, R., Gu, B., Wang, M., Zhao, Y., & Jiang, X. (2024). Desulfonylative Functionalization of Organosulfones via Inert (Hetero)Aryl C(sp2)–SO2 Bond Cleavage. Molecules, 29(17), 4137. https://doi.org/10.3390/molecules29174137