
Chemical Modification of Pactamycin Leads to New Compounds with Retained Antimicrobial Activity and Reduced Toxicity

 ,
, 
Abstract
:1. Introduction
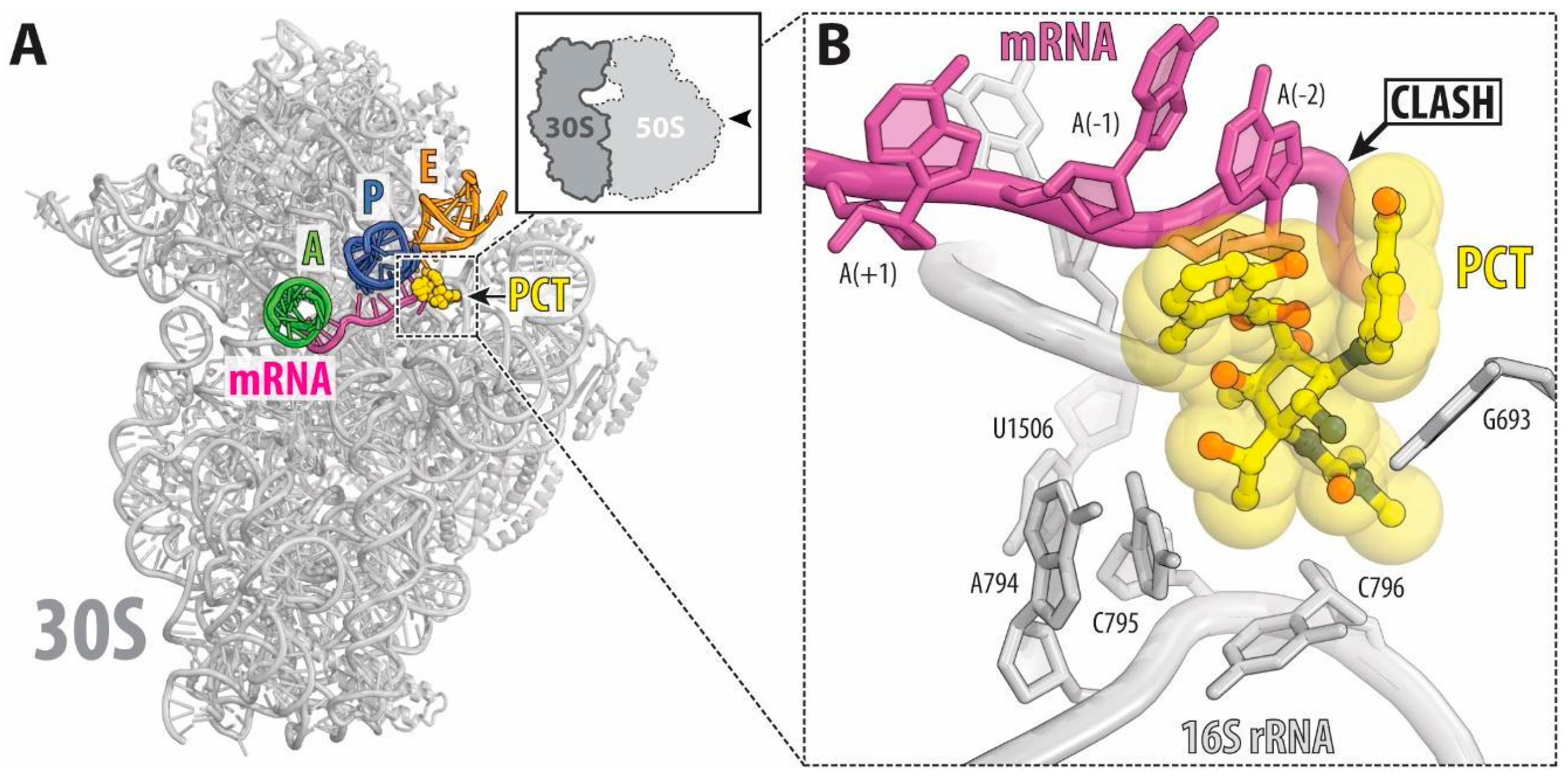
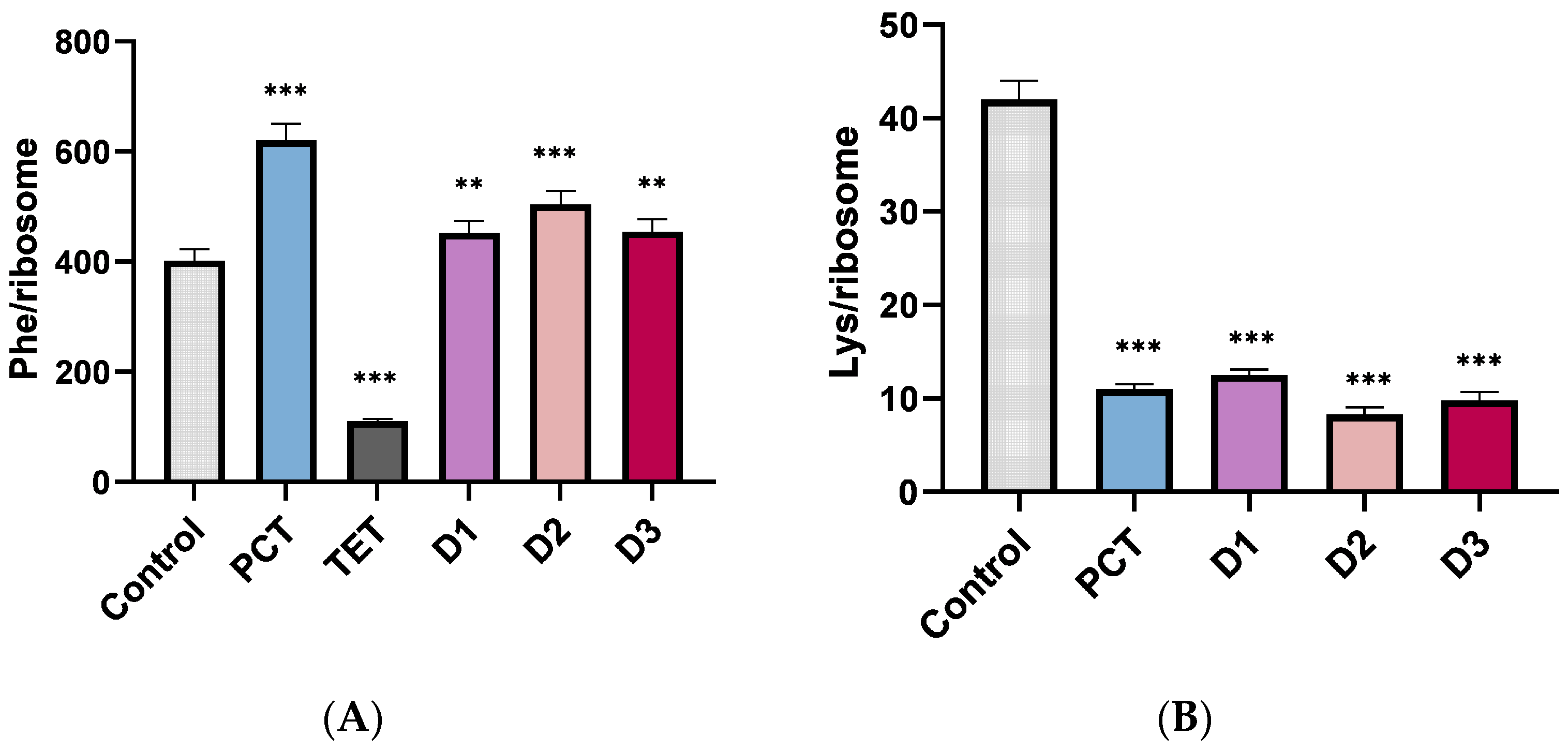
2. Results
2.1. Chemical Synthesis
2.2. Antibacterial Activity
3. Discussion
4. Materials and Methods
4.1. General Methods
4.2. Experimental Procedures
4.2.1. General Procedure for the Synthesis of Protected Pactamycin Derivatives
- Pactamycin (1) was added (1.0 eq to an ice-cold solution of N-protected amino acids 2–4 (1.1 eq) in DCM (0.18 M), DIPEA (1.1 eq) and HBTU (1.1 eq). The reaction mixture was stirred at room temperature for 24 h, and the progress of the reaction was monitored by TLC. After completion of the reaction, the mixture was evaporated to dryness under vacuum, and the residue thus obtained was diluted with AcOEt and washed with 5% aqueous citric acid, H2O, and brine. The organic layer was dried over Na2SO4, filtered, and evaporated to dryness under vacuum. The residue was subjected to FCC to obtain the pure product as a pale-yellow oil (75–82%).
- ((1S,2R,3R,4S,5S)-5-((3-acetylphenyl)amino)-4-((S)-2,6-bis((tert-butoxycarbonyl)amino)hexanamido)-3-(3,3-dimethylureido)-1,2-dihydroxy-3-((S)-1-hydroxyethyl)-2-methylcyclopentyl)methyl 2-hydroxy-6-methylbenzoate (5): 79%; Rf (PhMe/AcOEt 4:6) 0.10; MS (ESI, 30 eV): m/z 888.5 [M + H]; 1H NMR (CDCl3) δ 7.39–7.36 (m, 1H), 7.23–7.18 (m, 3H), 6.83 (d, J = 7.6 Hz, 1H), 6.77 (d, J = 7.6 Hz, 1H), 6.59 (d, J = 6.7 Hz, 1H), 4.95 (d, J = 12.4 Hz, 1H), 4.77 (d, J = 12.4 Hz, 1H), 4.69 (s, 1H), 4.18 (s, 1H), 3.76–3.71 (m, 2H), 3.54–3.46 (m, 2H), 2.94 (s, 6H), 2.87 (s, 3H), 2.79 (s, 3H), 2.49 (s, 2H), 2.28 (s, 2H), 2.25–2.14 (m, 2H), 1.55 (s, 3H), 1.42 (s, 3H), 1.35 (d, J = 24 Hz, 18H); 13C NMR (CDCl3) δ 172.8, 158.8, 157.2, 137.9, 134.3, 129.3, 128.2, 126.4, 126.1, 122.9, 119.3, 118.5, 117.2, 115.5, 111.1, 71.3, 66.1, 64.4, 63.7, 54.2, 49.4, 49.3, 42.6, 40.3, 36.8, 35.8, 33.7, 30.5, 29.7, 28.4, 26.6, 25.5, 24.8, 20.9, 18.5, 13.7.
- ((1S,2R,3R,4S,5S)-5-((3-acetylphenyl)amino)-4-((S)-2,5-bis((tert-butoxycarbonyl)amino)pentanamido)-3-(3,3-dimethylureido)-1,2-dihydroxy-3-((S)-1-hydroxyethyl)-2-methylcyclopentyl)methyl 2-hydroxy-6-methylbenzoate (6): 75%; Rf (PhMe/AcOEt 3:7) 0.11; MS (ESI, 30 eV): m/z 896.5 [M + Na]; 1H NMR (CDCl3) δ 7.74 (d, J = 12.0 Hz, 1H) 7.65 (d, J = 6.0 Hz, 1H), 7.38–7.34 (m, 2H), 7.21–7.15 (m, 3H), 7.07 (br s, 1H), 6.81–6.74 (m, 2H), 6.56 (br s, 1H), 4.92 (d, J = 12.4 Hz, 1H), 4.77 (d, J = 12.4 Hz, 1H), 4.22–4.19 (m, 1H), 4.06 (t, J = 6.7 Hz, 1H), 3.78 (t, J = 5.8 Hz, 2H), 3.72 (s, 3H), 3.64 (t, J = 5.8 Hz, 2H), 3.06 (s, 1H), 2.94 (s, 6H), 2.48 (s, 2H), 1.54 (s, 3H), 1.41 (d, J = 13.7 Hz, 18H), 1.35 (d, J = 4.0 Hz, 6H); 13C NMR (CDCl3) δ 172.8, 158.8, 157.2, 137.9, 133.6, 129.3, 128.2, 126.4, 126.0, 122.9, 118.5, 117.2, 115.5, 111.1, 71.3, 63.7, 49.3, 42.6, 36.8, 33.7, 29.7, 28.4, 26.6, 25.5, 24.8, 23.3, 20.9, 18.5, 13.7.
- ((1S,2R,3R,4S,5S)-5-((3-acetylphenyl)amino)-3-(3,3-dimethylureido)-1,2-dihydroxy-3-((S)-1-hydroxyethyl)-2-methyl-4-((S)-3-(1-trityl-1H-imidazol-4-yl)-2-(tritylamino)propanamido)cyclopentyl)methyl 2-hydroxy-6-methylbenzoate (7): 82%; Rf (PhMe/AcOEt 7:3) 0.09; MS (ESI, 30 eV): m/z 1181.6 [M + H]; 1H NMR (CD3OD) δ 7.28–7.19 (m, 16H), 7.08–7.04 (m, 5H), 6.98–6.95 (m, 6H), 6.88–6.86 (m, 4H), 6.78–6.65 (m, 4H), 6.54–6.43 (m, 1H), 4.70–4.62 (m, 3H), 3.37–3.33 (m, 2H), 3.25 (s, 6H), 3.21 (s, 3H), 2.71 (s, 3H), 2.19 (s, 1H), 2.10 (d, J = 6.1 Hz, 2H), 1.45 (s, 6H); 13C NMR (CD3OD) δ 172.8, 158.8, 157.2, 150.2, 137.9, 134.2, 129.3, 128.2, 126.4, 126.0, 122.9, 118.5, 117.2, 115.5, 111.1, 71.3, 66.1, 64.4, 63.7, 49.3, 42.6, 36.8, 33.7, 29.7, 28.4, 26.6, 25.5, 24.8, 23.3, 20.9, 18.5, 13.7.
4.2.2. General Procedure for the Deprotection of Pactamycin Derivatives
- ((1S,2R,3R,4S,5S)-5-((3-acetylphenyl)amino)-4-((S)-2,6-diaminohexanamido)-3-(3,3-dimethylureido)-1,2-dihydroxy-3-((S)-1-hydroxyethyl)-2-methylcyclopentyl)methyl 2-hydroxy-6-methylbenzoate (D1): 92%; Rf (DCM/MeOH/NH3: 9:1:0.1) 0.12; HR-ESI: m/z 687,3745; [M + H+] for the compound C34H51N6O9 requires 687.3712; 1H NMR (CD3OD) δ 7.20–7.15 (m, 3H), 7.07–7.04 (m, 2H), 6.67 (d, J = 6 Hz, 1H), 6.61 (d, J = 6 Hz, 1H), 5.13–5.09 (m, 3H), 4.83–4.79 (m, 3H), 4.46 (d, J = 12 Hz, 1H), 3.94–3.89 (m, 3H), 3.35–3.33 (m, 4H), 3.21–3.15 (m, 3H), 2.43 (s, 3H), 2.24 (s, 6H), 1.52 (s, 6H); 13C NMR (CD3OD) δ 202.3, 171.7, 150.1, 141.8, 139.5, 134.3, 131.0, 130.3, 129.6, 124.1, 120.6, 118.5, 116.1, 112.6, 84.2, 79.5, 74.6, 69.6, 68.2, 66.6, 60.4, 55.4, 42.0, 33.6, 27.6, 24.5, 23.7, 22.9, 18.8, 17.5, 15.3.
- ((1S,2R,3R,4S,5S)-5-((3-acetylphenyl)amino)-4-((S)-2,5-diaminopentanamido)-3-(3,3-dimethylureido)-1,2-dihydroxy-3-((S)-1-hydroxyethyl)-2-methylcyclopentyl)methyl 2-hydroxy-6-methylbenzoate (D2): 94%; Rf (DCM/MeOH/NH3: 9:1:0.1) 0.1; HR-ESI: m/z 673,3586; [M + H+] for the compound C33H48N6O9 requires 673,3556; 1H NMR (CD3OD) δ 7.12–7.09 (m, 2H), 7.01–6.98 (m, 2H), 6.63–6.56 (m, 3H), 4.73–4.70 (m, 2H), 3.29 (s, 6H), 3.13 (d, J = 6 Hz, 2H), 3.01–2.97 (m, 2H), 2.90–2.87 (m, 2H), 2.79 (s, 3H), 2.39 (s, 3H), 2.18 (d, J = 6 Hz, 4H), 1.79–1.76 (m, 4H), 1.39 (s, 6H); 13C NMR (CD3OD) δ 202.1, 174.3, 171.5, 141.6, 134.3, 131.0, 129.2, 124.1, 120.3, 119.4, 116.0, 89.5, 84.9, 83.7, 77.9, 75.2, 75.0, 66.6, 44.7, 39.7, 38.0, 33.5, 29.6, 27.6, 25.9, 24.5, 22.6, 18.3, 16.6, 15.2.
- ((1S,2R,3R,4S,5S)-5-((3-acetylphenyl)amino)-4-((S)-2-amino-3-(1H-imidazol-4-yl)propanamido)-3-(3,3-dimethylureido)-1,2-dihydroxy-3-((S)-1-hydroxyethyl)-2-methylcyclopentyl)methyl 2-hydroxy-6-methylbenzoate (D3): 89%; Rf (DCM/MeOH/NH3: 9:1:0.1) 0.09; HR-ESI: m/z 696,3376; [M + H+] for the compound C34H45N7O9 requires 696,3352; 1H NMR (CD3OD) δ 7.18–7.13 (m, 4H), 7.06–7.03 (m, 3H), 6.66 (d, J = 12 Hz, 1H), 6.59 (d, J = 12 Hz, 1H), 4.81–4.77 (m, 3H), 3.90–3.86 (m, 3H), 3.61 (s, 1H), 3.36 (s, 3H), 3.32 (s, 2H), 2.41 (s, 6H), 2.23 (s, 3H), 1.5 (s, 6H); 13C NMR (CD3OD) δ 202.0, 167.1, 130.6, 130.1, 129.4, 128.8, 119.4, 114.8, 113.7, 110.8, 106.3, 96.0, 80.2, 70.3, 68.9, 66.1, 56.7, 44.6, 35.6, 31.5, 30.0, 27.5, 26.8, 20.9, 19.6, 18.8, 18.2.
4.3. Materials
4.4. Bacterial Strains
4.5. Biochemical Preparations
4.6. MIC
4.7. Inhibition of Translation Using In Vitro Cell-Free Expression System
4.8. Poly Phenylalanine Synthesis
4.9. Poly Lysine
4.10. Misincorporation
4.11. Puromycin Reaction
4.12. Resazurin Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Argoudelis, A.D. Pactamycin, a New Antitumor Antibiotic. II. Isolation and Characterization. Antimicrob. Agents Chemother. 1962, 191–197. [Google Scholar]
- Wiley, P.F.; Jahnke, H.K.; MacKellar, F.; Kelly, R.B.; Argoudelis, A.D. The Structure of Pactamycin. J. Org. Chem. 1970, 35, 1420–1425. [Google Scholar] [CrossRef]
- BHUYAN, B.K. Pactamycin Production by Streptomyces Pactum. Appl. Microbiol. 1962, 10, 302–304. [Google Scholar] [CrossRef] [PubMed]
- Iwatsuki, M.; Nishihara-Tsukashima, A.; Ishiyama, A.; Namatame, M.; Watanabe, Y.; Handasah, S.; Pranamuda, H.; Marwoto, B.; Matsumoto, A.; Takahashi, Y.; et al. Jogyamycin, a New Antiprotozoal Aminocyclopentitol Antibiotic, Produced by Streptomyces sp. a-WM-JG-16.2. J. Antibiot. 2012, 65, 169–171. [Google Scholar] [CrossRef] [PubMed]
- Taber, R.; Rekosh, D.; Baltimore, D. Effect of Pactamycin on Synthesis of Poliovirus Proteins: A Method for Genetic Mapping. J. Virol. 1971, 8, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Brodersen, D.E.; Clemons, W.M.J.; Carter, A.P.; Morgan-Warren, R.J.; Wimberly, B.T.; Ramakrishnan, V. The Structural Basis for the Action of the Antibiotics Tetracycline, Pactamycin, and Hygromycin B on the 30S Ribosomal Subunit. Cell 2000, 103, 1143–1154. [Google Scholar] [CrossRef] [PubMed]
- Dinos, G.; Wilson, D.N.; Teraoka, Y.; Szaflarski, W.; Fucini, P.; Kalpaxis, D.; Nierhaus, K.H. Dissecting the Ribosomal Inhibition Mechanisms of Edeine and Pactamycin: The Universally Conserved Residues G693 and C795 Regulate P-Site RNA Binding. Mol. Cell 2004, 13, 113–124. [Google Scholar] [CrossRef]
- Polikanov, Y.S.; Osterman, I.A.; Szal, T.; Tashlitsky, V.N.; Serebryakova, M.V.; Kusochek, P.; Bulkley, D.; Malanicheva, I.A.; Efimenko, T.A.; Efremenkova, O.V.; et al. Amicoumacin a Inhibits Translation by Stabilizing MRNA Interaction with the Ribosome. Mol. Cell 2014, 56, 531–540. [Google Scholar] [CrossRef]
- Kappen, L.S.; Suzuki, H.; Goldberg, I.H. Inhibition of Reticulocyte Peptide-Chain Initiation by Pactamycin: Accumulation of Inactive Ribosomal Initiation Complexes. Proc. Natl. Acad. Sci. USA 1973, 70, 22–26. [Google Scholar] [CrossRef]
- Garreau de Loubresse, N.; Prokhorova, I.; Holtkamp, W.; Rodnina, M.V.; Yusupova, G.; Yusupov, M. Structural Basis for the Inhibition of the Eukaryotic Ribosome. Nature 2014, 513, 517–522. [Google Scholar] [CrossRef]
- Orelle, C.; Carlson, S.; Kaushal, B.; Almutairi, M.M.; Liu, H.; Ochabowicz, A.; Quan, S.; Pham, V.C.; Squires, C.L.; Murphy, B.T.; et al. Tools for Characterizing Bacterial Protein Synthesis Inhibitors. Antimicrob. Agents Chemother. 2013, 57, 5994–6004. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Laslop, N.; Mankin, A.S. Context-Specific Action of Ribosomal Antibiotics. Annu. Rev. Microbiol. 2018, 72, 185–207. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Laslop, N.; Mankin, A.S. How Macrolide Antibiotics Work. Trends Biochem. Sci. 2018, 43, 668–684. [Google Scholar] [CrossRef]
- Tourigny, D.S.; Fernández, I.S.; Kelley, A.C.; Vakiti, R.R.; Chattopadhyay, A.K.; Dorich, S.; Hanessian, S.; Ramakrishnan, V. Crystal Structure of a Bioactive Pactamycin Analog Bound to the 30S Ribosomal Subunit. J. Mol. Biol. 2013, 425, 3907–3910. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Alanzi, A.R.; Abugrain, M.E.; Ito, T.; Mahmud, T. Global and Pathway-Specific Transcriptional Regulations of Pactamycin Biosynthesis in Streptomyces Pactum. Appl. Microbiol. Biotechnol. 2018, 102, 10589–10601. [Google Scholar] [CrossRef]
- Hanessian, S.; Vakiti, R.R.; Dorich, S.; Banerjee, S.; Lecomte, F.; DelValle, J.R.; Zhang, J.; Deschênes-Simard, B. Total Synthesis of Pactamycin. Angew. Chem. Int. Ed. Engl. 2011, 50, 3497–3500. [Google Scholar] [CrossRef]
- Malinowski, J.T.; Sharpe, R.J.; Johnson, J.S. Enantioselective Synthesis of Pactamycin, a Complex Antitumor Antibiotic. Science 2013, 340, 180–182. [Google Scholar] [CrossRef]
- Hanessian, S.; Vakiti, R.R.; Chattopadhyay, A.K.; Dorich, S.; Lavallée, C. Probing Functional Diversity in Pactamycin toward Antibiotic, Antitumor, and Antiprotozoal Activity. Bioorg. Med. Chem. 2013, 21, 1775–1786. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Roongsawang, N.; Mahmud, T. Biosynthetic Studies and Genetic Engineering of Pactamycin Analogs with Improved Selectivity toward Malarial Parasites. Chem. Biol. 2011, 18, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Roongsawang, N.; Shirasaka, N.; Lu, W.; Flatt, P.M.; Kasanah, N.; Miranda, C.; Mahmud, T. Deciphering Pactamycin Biosynthesis and Engineered Production of New Pactamycin Analogues. Chembiochem 2009, 10, 2253–2265. [Google Scholar] [CrossRef]
- Tereshchenkov, A.G.; Dobosz-Bartoszek, M.; Osterman, I.A.; Marks, J.; Sergeeva, V.A.; Kasatsky, P.; Komarova, E.S.; Stavrianidi, A.N.; Rodin, I.A.; Konevega, A.L.; et al. Binding and Action of Amino Acid Analogs of Chloramphenicol upon the Bacterial Ribosome. J. Mol. Biol. 2018, 430, 842–852. [Google Scholar] [CrossRef]
- Tsirogianni, A.; Kournoutou, G.G.; Bougas, A.; Poulou-Sidiropoulou, E.; Dinos, G.; Athanassopoulos, C.M. New Chloramphenicol Derivatives with a Modified Dichloroacetyl Tail as Potential Antimicrobial Agents. Antibiotics 2021, 10, 394. [Google Scholar] [CrossRef] [PubMed]
- Demirci, H.; Murphy, F., 4th; Murphy, E.; Gregory, S.T.; Dahlberg, A.E.; Jogl, G. A Structural Basis for Streptomycin-Induced Misreading of the Genetic Code. Nat. Commun. 2013, 4, 1355. [Google Scholar] [CrossRef] [PubMed]
- Ogle, J.M.; Murphy, F.V.; Tarry, M.J.; Ramakrishnan, V. Selection of TRNA by the Ribosome Requires a Transition from an Open to a Closed Form. Cell 2002, 111, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Dinos, G.; Kalpaxis, D.L.; Wilson, D.N.; Nierhaus, K.H. Deacylated TRNA Is Released from the E Site upon A Site Occupation but before GTP Is Hydrolyzed by EF-Tu. Nucleic Acids Res. 2005, 33, 5291–5296. [Google Scholar] [CrossRef]
- Svetlov, M.S.; Plessa, E.; Chen, C.W.; Bougas, A.; Krokidis, M.G.; Dinos, G.P.; Polikanov, Y.S. High-Resolution Crystal Structures of Ribosome-Bound Chloramphenicol and Erythromycin Provide the Ultimate Basis for Their Competition. RNA 2019, 25, 600–606. [Google Scholar] [CrossRef]
- Hansen, J.L.; Moore, P.B.; Steitz, T.A. Structures of Five Antibiotics Bound at the Peptidyl Transferase Center of the Large Ribosomal Subunit. J. Mol. Biol. 2003, 330, 1061–1075. [Google Scholar] [CrossRef]
- Tsirogianni, A.; Kournoutou, G.G.; Mpogiatzoglou, M.; Dinos, G.; Athanassopoulos, C.M. Chloramphenicol Derivatization in Its Primary Hydroxyl Group with Basic Amino Acids Leads to New Pharmacophores with High Antimicrobial Activity. Antibiotics 2023, 12, 832. [Google Scholar] [CrossRef]
- Magoulas, G.E.; Kostopoulou, O.N.; Garnelis, T.; Athanassopoulos, C.M.; Kournoutou, G.G.; Leotsinidis, M.; Dinos, G.P.; Papaioannou, D.; Kalpaxis, D.L. Synthesis and Antimicrobial Activity of Chloramphenicol-Polyamine Conjugates. Bioorganic Med. Chem. 2015, 23, 3163–3174. [Google Scholar] [CrossRef]
- Aminov, R.I. A Brief History of the Antibiotic Era: Lessons Learned and Challenges for the Future. Front. Microbiol. 2010, 1, 134. [Google Scholar] [CrossRef]
- Davies, J.; Davies, D. Origins and Evolution of Antibiotic Resistance. Microbiol. Mol. Biol. Rev. 2010, 74, 417–433. [Google Scholar] [CrossRef] [PubMed]
- Santos-Lopez, A.; Marshall, C.W.; Haas, A.L.; Turner, C.; Rasero, J.; Cooper, V.S. The Roles of History, Chance, and Natural Selection in the Evolution of Antibiotic Resistance. eLife 2021, 10, e70676. [Google Scholar] [CrossRef] [PubMed]
- Kokkori, S.; Apostolidi, M.; Tsakris, A.; Pournaras, S.; Stathopoulos, C.; Dinos, G. Linezolid-Dependent Function and Structure Adaptation of Ribosomes in a Staphylococcus Epidermidis Strain Exhibiting Linezolid Dependence. Antimicrob. Agents Chemother. 2014, 5, 4651–4656. [Google Scholar] [CrossRef] [PubMed]
- Blaha, G.; Stelzl, U.; Spahn, C.M.T.; Agrawal, R.K.; Frank, J.; Nierhaus, K.H. Preparation of Functional Ribosomal Complexes and Effect of Buffer Conditions on TRNA Positions Observed by Cryoelectron Microscopy. Methods Enzymol. 2000, 317, 292–306. [Google Scholar] [CrossRef]
- Rheinberger, H.J.; Geigenmüller, U.; Wedde, M.; Nierhaus, K.H. Parameters for the Preparation of Escherichia Coli Ribosomes and Ribosomal Subunits Active in TRNA Binding. Methods Enzymol. 1988, 164, 658–670. [Google Scholar] [CrossRef] [PubMed]
- Bommer, U.; Burkhardt, N.; Junemann, R.; Spahn, C.M.T.; Triana-Alonso, F.J.; Nierhaus, K.H. Ribosomes and Polysomes. In Subcellular Fractionation. A Practical Approach; Graham, J., Rickwoods, D., Eds.; IRL Press at Oxford University Press: Oxford, UK, 1996; pp. 271–301. [Google Scholar]
- Lavogina, D.; Lust, H.; Tahk, M.-J.; Laasfeld, T.; Vellama, H.; Nasirova, N.; Vardja, M.; Eskla, K.-L.; Salumets, A.; Rinken, A.; et al. Revisiting the Resazurin-Based Sensing of Cellular Viability: Widening the Application Horizon. Biosensors 2022, 12, 196. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | MIC (μM) | MIC (μgr/mL) | ||
|---|---|---|---|---|
| E. coli | S. epidermidis | E. coli | S. epidermidis | |
| PCT | 8 | 4 | 4.47 | 2.23 |
| D1, PCT-Lysine | 16 | 8 | 18.29 | 9.14 |
| D2, PCT-Ornithine | 16 | 8 | 18.08 | 9.04 |
| D3, PCT-Histidine | 32 | 16 | 36.86 | 18.43 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsirogianni, A.; Ntinou, N.; Karampatsou, K.; Dinos, G.; Kournoutou, G.G.; Athanassopoulos, C.M. Chemical Modification of Pactamycin Leads to New Compounds with Retained Antimicrobial Activity and Reduced Toxicity. Molecules 2024, 29, 4169. https://doi.org/10.3390/molecules29174169
Tsirogianni A, Ntinou N, Karampatsou K, Dinos G, Kournoutou GG, Athanassopoulos CM. Chemical Modification of Pactamycin Leads to New Compounds with Retained Antimicrobial Activity and Reduced Toxicity. Molecules. 2024; 29(17):4169. https://doi.org/10.3390/molecules29174169
Chicago/Turabian StyleTsirogianni, Artemis, Nikolina Ntinou, Konstantina Karampatsou, George Dinos, Georgia G. Kournoutou, and Constantinos M. Athanassopoulos. 2024. "Chemical Modification of Pactamycin Leads to New Compounds with Retained Antimicrobial Activity and Reduced Toxicity" Molecules 29, no. 17: 4169. https://doi.org/10.3390/molecules29174169