Theoretical Study of Metal–Ligand Interactions in Lead Complexes with Radiopharmaceutical Interest †


Abstract
:
1. Introduction
2. Results and Discussion
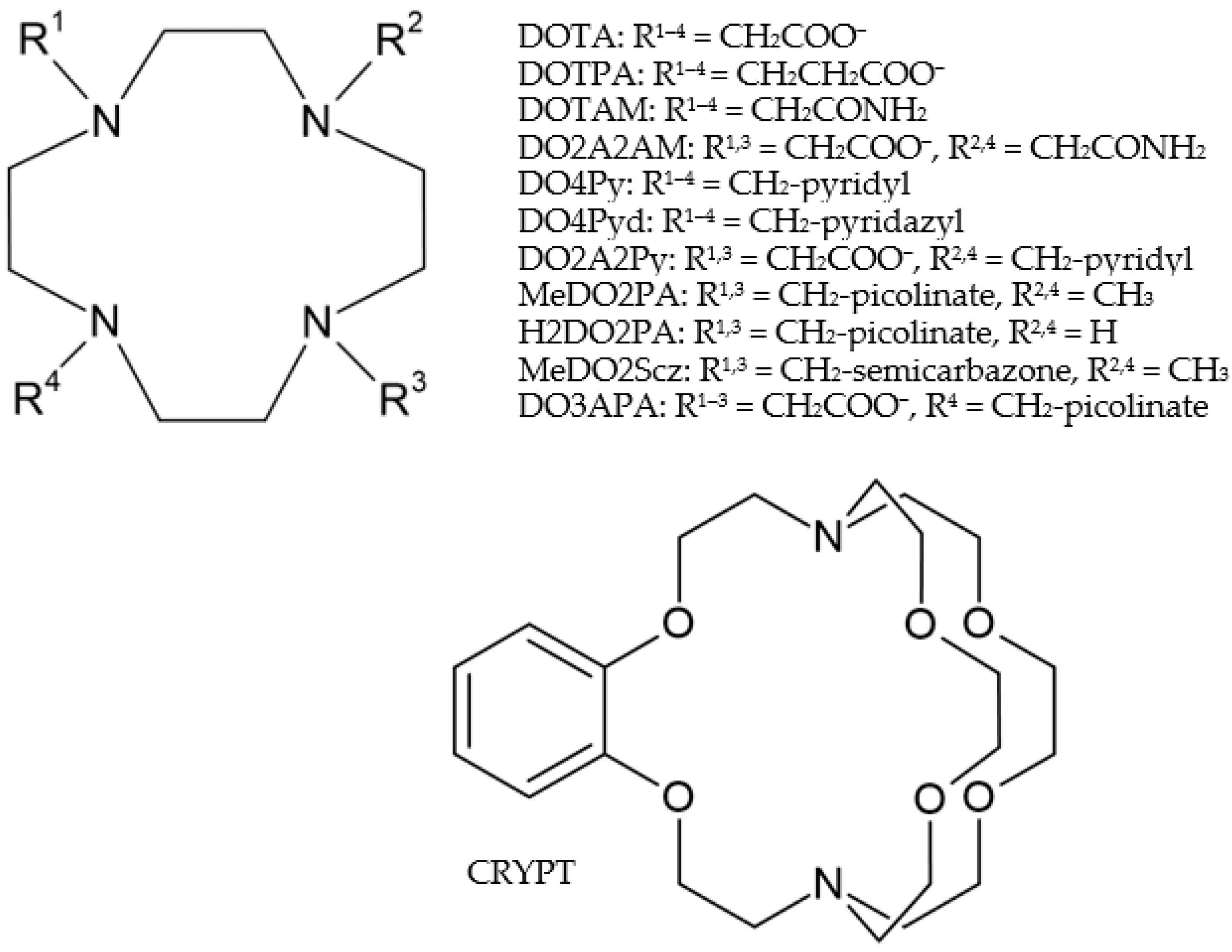
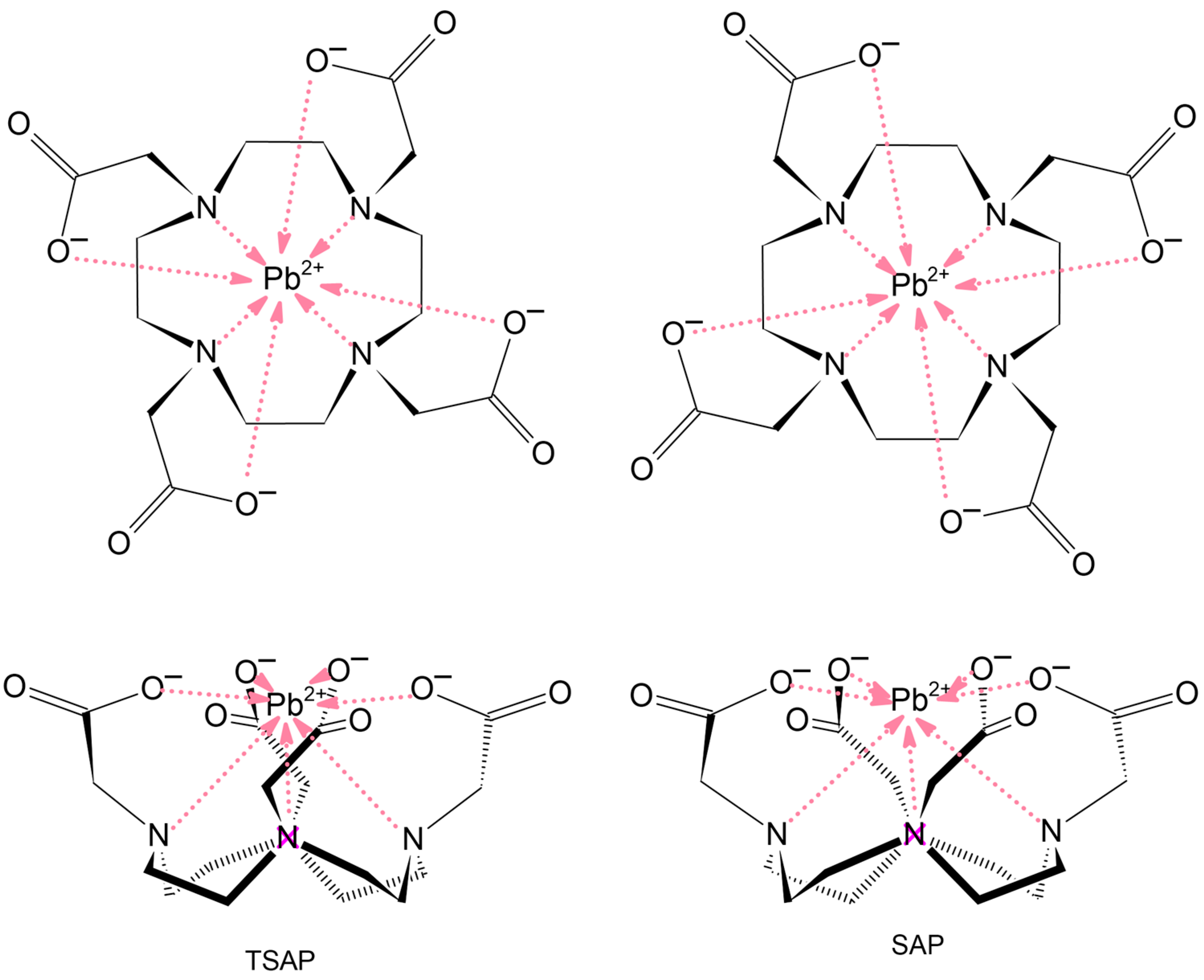
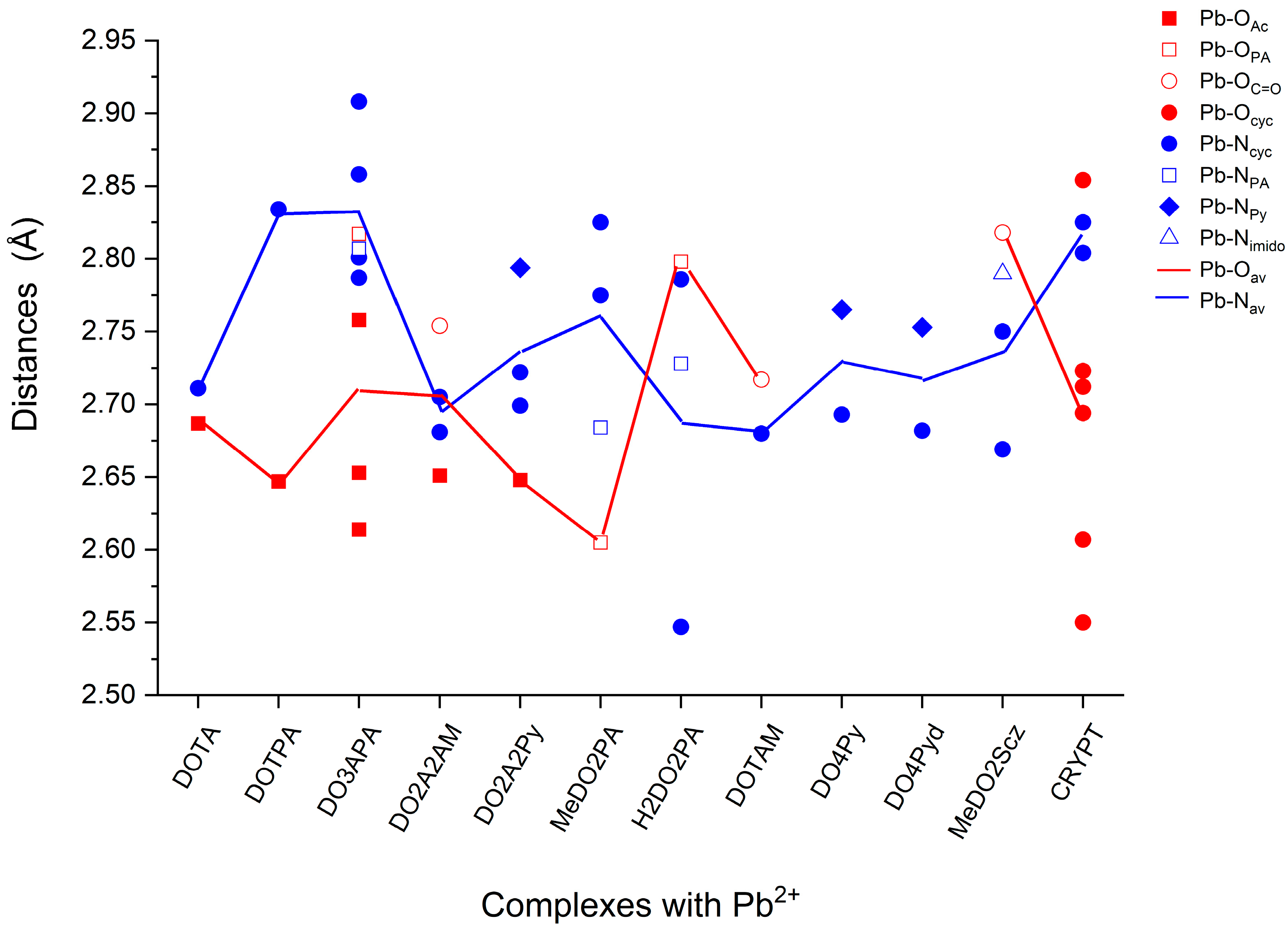
2.1. Structures
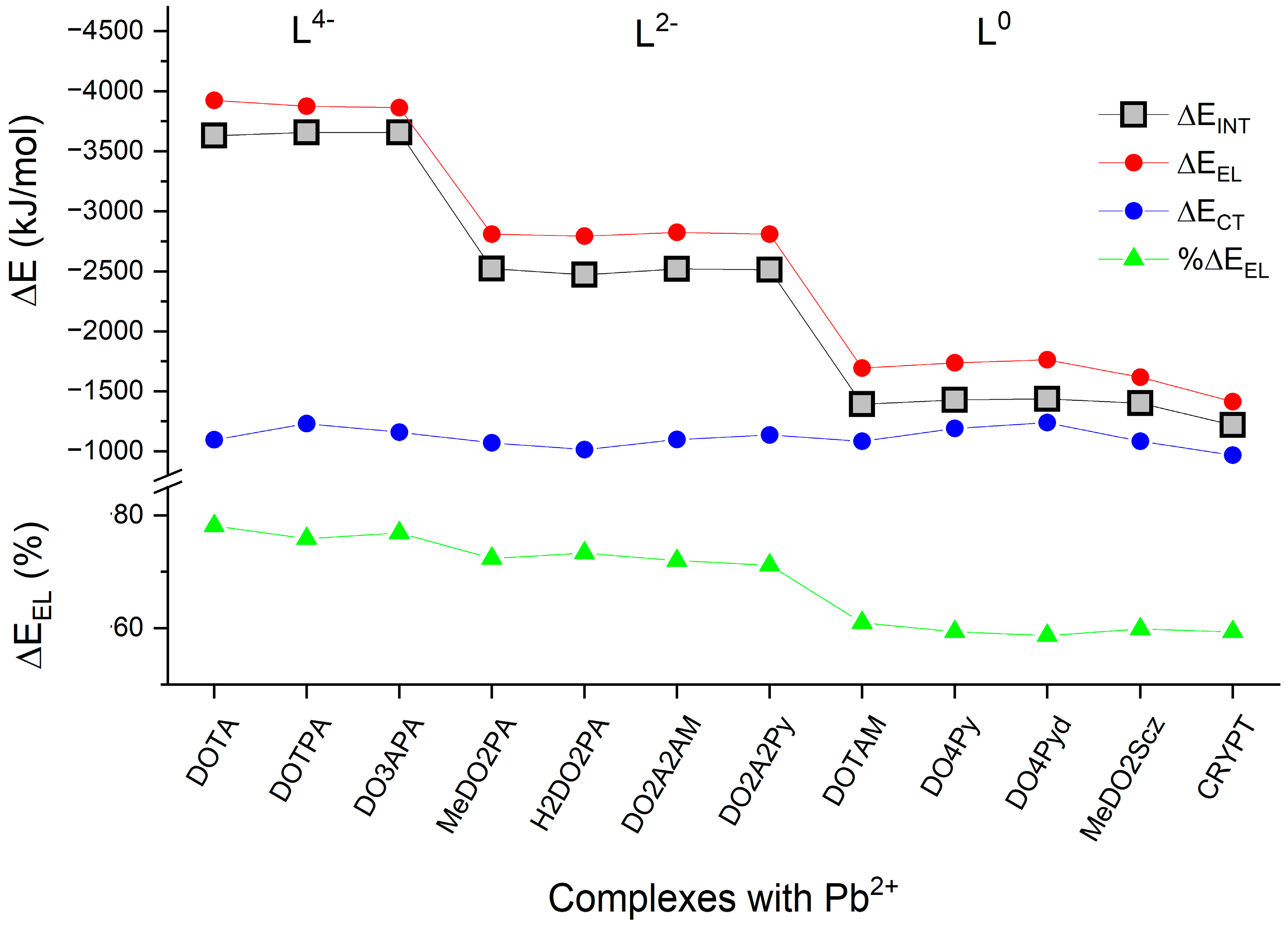
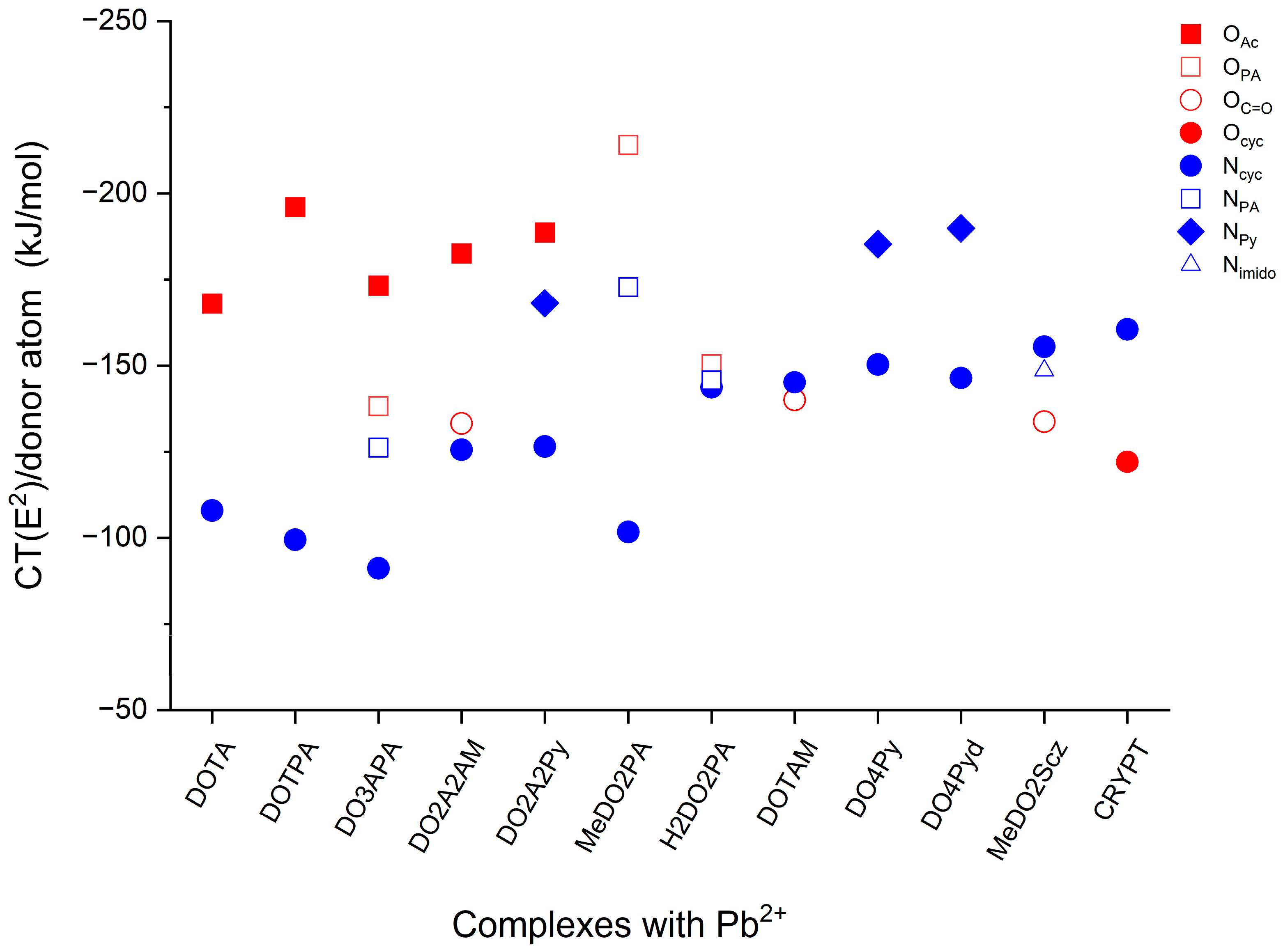
2.2. Bonding Analysis
3. Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kvassheim, M.; Revheim, M.-E.R.; Stokke, C. Quantitative SPECT/CT imaging of lead-212: A phantom study. EJNMMI Phys. 2022, 9, 52. [Google Scholar] [CrossRef] [PubMed]
- ICRP. Nuclear Decay Data for Dosimetric Calculations; ICRP Publication 107, Annals of the ICRP; International Commission on Radiological Protection: Ottawa, ON, Canada, 2008; Volume 38. [Google Scholar]
- Price, E.W.; Orvig, C. Matching chelators to radiometals for radiopharmaceuticals. Chem. Soc. Rev. 2014, 43, 260–290. [Google Scholar] [CrossRef]
- Boros, E.; Packard, A.B. Radioactive Transition Metals for Imaging and Therapy. Chem. Rev. 2019, 119, 870–901. [Google Scholar] [CrossRef]
- Morgan, K.A.; Rudd, S.E.; Noor, A.; Donnelly, P.S. Theranostic Nuclear Medicine with Gallium-68, Lutetium-177, Copper-64/67, Actinium-225, and Lead-212/203 Radionuclides. Chem. Rev. 2023, 123, 12004–12035. [Google Scholar] [CrossRef] [PubMed]
- Ramogida, C.F.; Orvig, C. Tumour targeting with radiometals for diagnosis and therapy. Chem. Commun. 2013, 49, 4720–4739. [Google Scholar] [CrossRef] [PubMed]
- Pearson, R.G. Hard and Soft Acids and Bases. J. Am. Chem. Soc. 1963, 85, 3533–3539. [Google Scholar] [CrossRef]
- Pearson, R.G. Hard and soft acids and bases, HSAB, part 1: Fundamental principles. J. Chem. Educ. 1968, 45, 581–587. [Google Scholar] [CrossRef]
- Pearson, R.G. Hard and soft acids and bases, HSAB, part II: Underlying theories. J. Chem. Educ. 1968, 45, 643–648. [Google Scholar] [CrossRef]
- Parr, J. Some recent coordination chemistry of lead(II). Polyhedron 1997, 16, 551–566. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised Effective Ionic Radii and Systematic Studies of Interatomic Distances in Halides and Chalcogenides. Acta Cryst. 1976, A32, 751–767. [Google Scholar] [CrossRef]
- Nugent, J.W.; Lee, H.-S.; Reibenspies, J.H.; Hancock, R.D. Spectroscopic, structural, and thermodynamic aspects of the stereochemically active lone pair on lead(II): Structure of the lead(II) dota complex. Polyhedron 2015, 91, 120–127. [Google Scholar] [CrossRef]
- Moncomble, A.; Cornard, J.-P.; Meyer, M. A quantum chemistry evaluation of the stereochemical activity of the lone pair in PbII complexes with sequestering ligands. J. Mol. Model. 2017, 23, 24. [Google Scholar] [CrossRef]
- Mirzadeh, S.; Kumar, K.; Gansow, O.A. The Chemical Fate of 212Bi-DOTA Formed by β− Decay of 212Pb(DOTA)2−. Radiochim. Acta 1993, 60, 1–10. [Google Scholar] [CrossRef]
- Pippin, C.G.; McMurry, T.J.; Brechbiel, M.W.; McDonald, M.; Lambrecht, R.; Milenic, D.E.; Roselli, M.; Colcher, D.; Gansow, O.A. Lead(II) complexes of 1,4,7,10-tetraazacyclododecane-N,N′,N″,N‴-tetraacetate: Solution chemistry and application to tumor localization with 203Pb labeled monoclonal antibodies. Inorg. Chim. Acta 1995, 239, 43–51. [Google Scholar] [CrossRef]
- Maumela, H.; Hancock, R.D.; Carlton, L.; Reibenspies, J.H.; Wainwright, K.P. The Amide Oxygen as a Donor Group. Metal Ion Complexing Properties of Tetra-N-acetamide Substituted Cyclen: A Crystallographic, NMR, Molecular Mechanics, and Thermodynamic Study. J. Am. Chem. Soc. 1995, 117, 6698–6707. [Google Scholar] [CrossRef]
- Chappell, L.L.; Dadachova, E.; Milenic, D.E.; Garmestani, K.; Wu, C.; Brechbiel, M.W. Synthesis, characterization, and evaluation of a novel bifunctional chelating agent for the lead isotopes 203Pb and 212Pb. Nucl. Med. Biol. 2000, 27, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.D.; Siddons, C.J.; Oscarson, K.A.; Reibenspies, J.M. The structure of the 11-coordinate barium complex of the pendant-donor macrocycle 1,4,7,10-tetrakis(carbamoylmethyl)-1,4,7,10-tetraazacyclododecane: An analysis of the coordination numbers of barium(II) in its complexes. Inorg. Chim. Acta 2004, 357, 723–727. [Google Scholar] [CrossRef]
- Cuenot, F.; Meyer, M.; Espinosa, E.; Bucaille, A.; Burgat, R.; Guilard, R.; Marichal-Westrich, C. New Insights into the Complexation of Lead(II) by 1,4,7,10-Tetrakis(carbamoylmethyl)-1,4,7,10-tetraazacyclododecane (DOTAM): Structural, Thermodynamic, and Kinetic Studies. Eur. J. Inorg. Chem. 2008, 2008, 267–283. [Google Scholar] [CrossRef]
- Lima, L.M.P.; Beyler, M.; Delgado, R.; Platas-Iglesias, C.; Tripier, R. Investigating the Complexation of the Pb2+/Bi3+ Pair with Dipicolinate Cyclen Ligands. Inorg. Chem. 2015, 54, 7045–7057. [Google Scholar] [CrossRef]
- Lange, J.L.; Davey, P.R.W.J.; Ma, M.T.; White, J.M.; Morgenstern, A.; Bruchertseifer, F.; Blower, P.J.; Paterson, B.M. An octadentate bis(semicarbazone) macrocycle: A potential chelator for lead and bismuth radiopharmaceuticals. Dalton Trans. 2020, 49, 14962–14974. [Google Scholar] [CrossRef]
- Ingham, A.; Kostelnik, T.I.; McNeil, B.L.; Patrick, B.O.; Choudhary, N.; de Guadalupe Jaraquemada-Peláez, M.; Orvig, C. Getting a lead on Pb2+-amide chelators for 203/212Pb radiopharmaceuticals. Dalton Trans. 2021, 50, 11579–11595. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, A.W.; McNeil, B.L.; Patrick, B.O.; Ramogida, C.F. Synthesis and Evaluation of Bifunctional [2.2.2]-Cryptands for Nuclear Medicine Applications. Inorg. Chem. 2021, 60, 10030–10037. [Google Scholar] [CrossRef] [PubMed]
- McNeil, B.L.; Robertson, A.K.H.; Fu, W.; Yang, H.; Hoehr, C.; Ramogida, C.F.; Schaffer, P. Production, purification, and radiolabeling of the 203Pb/212Pb theranostic pair. EJNMMI Radiopharm. Chem. 2021, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Tosato, M.; Lazzari, L.; Marco, V.D. Revisiting Lead(II)-1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic Acid Coordination Chemistry in Aqueous Solutions: Evidence of an Underestimated Thermodynamic Stability. ACS Omega 2022, 7, 15596–15602. [Google Scholar] [CrossRef]
- McNeil, B.L.; Kadassery, K.J.; McDonagh, A.W.; Zhou, W.; Schaffer, P.; Wilson, J.J.; Ramogida, C.F. Evaluation of the Effect of Macrocyclic Ring Size on [203Pb]Pb(II) Complex Stability in Pyridyl-Containing Chelators. Inorg. Chem. 2022, 61, 9638–9649. [Google Scholar] [CrossRef]
- Grieve, M.L.; Paterson, B.M. The Evolving Coordination Chemistry of Radiometals for Targeted Alpha Therapy. Aust. J. Chem. 2022, 75, 65–88. [Google Scholar] [CrossRef]
- Li, M.; Baumhover, N.J.; Liu, D.; Cagle, B.S.; Boschetti, F.; Paulin, G.; Lee, D.; Dai, Z.; Obot, E.R.; Marks, B.M.; et al. Preclinical Evaluation of a Lead Specific Chelator (PSC) Conjugated to Radiopeptides for 203Pb and 212Pb-Based Theranostics. Pharmaceutics 2023, 15, 414. [Google Scholar] [CrossRef]
- Blei, M.K.; Waurick, L.; Reissig, F.; Kopka, K.; Stumpf, T.; Drobot, B.; Kretzschmar, J.; Mamat, C. Equilibrium Thermodynamics of Macropa Complexes with Selected Metal Isotopes of Radiopharmaceutical Interest. Inorg. Chem. 2023, 62, 20699–20709. [Google Scholar] [CrossRef]
- Kovács, A.; Varga, Z. Metal–ligand interactions in complexes of cyclen-based ligands with Bi and Ac. Struct. Chem. 2021, 32, 1719–1731. [Google Scholar] [CrossRef]
- Glendening, E.D.; Streitwieser, A. Natural energy decomposition analysis: An energy partitioning procedure for molecular interactions with application to weak hydrogen bonding, strong ionic, and moderate donor–acceptor interactions. J. Chem. Phys. 1994, 100, 2900–2909. [Google Scholar] [CrossRef]
- Glendening, E.D. Natural Energy Decomposition Analysis: Extension to Density Functional Methods and Analysis of Cooperative Effects in Water Clusters. J. Phys. Chem. A 2005, 109, 11936–11940. [Google Scholar] [CrossRef] [PubMed]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Peters, J.A.; Djanashvili, K.; Geraldes, C.F.G.C.; Platas-Iglesias, C. The chemical consequences of the gradual decrease of the ionic radius along the Ln-series. Coord. Chem. Rev. 2020, 406, 213146. [Google Scholar] [CrossRef]
- Viola-Villegas, N.; Doyle, R.P. The coordination chemistry of 1,4,7,10-tetraazacyclododecane-N,N′,N″,N‴-tetraacetic acid (H4DOTA): Structural overview and analyses on structure-stability relationships. Coord. Chem. Rev. 2009, 253, 1906–1925. [Google Scholar] [CrossRef]
- Spirlet, M.R.; Rebizant, J.; Desreux, J.F.; Loncin, M.F. Crystal and molecular structure of sodium aqua(1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetato)europate(III) tetrahydrate Na+(EuDOTA.H2O)−.4H2O, and its relevance to NMR studies of the conformational behavior of the lanthanide complexes formed by the macrocyclic ligand DOTA. Inorg. Chem. 1984, 23, 359–363. [Google Scholar] [CrossRef]
- Benetollo, F.; Bombieri, G.; Calabi, L.; Aime, S.; Botta, M. Structural Variations Across the Lanthanide Series of Macrocyclic DOTA Complexes: Insights into the Design of Contrast Agents for Magnetic Resonance Imaging. Inorg. Chem. 2003, 42, 148–157. [Google Scholar] [CrossRef]
- Chang, C.A.; Francesconi, L.C.; Malley, M.F.; Kumar, K.; Gougoutas, J.Z.; Tweedle, M.F.; Lee, D.W.; Wilson, L.J. Synthesis, characterization, and crystal structures of M(DO3A) (M = iron, gadolinium) and Na[M(DOTA)] (M = Fe, yttrium, Gd). Inorg. Chem. 1993, 32, 3501–3508. [Google Scholar] [CrossRef]
- Aime, S.; Barge, A.; Botta, M.; Fasano, M.; Ayala, J.D.; Bombieri, G. Crystal structure and solution dynamics of the lutetium(III) chelate of DOTA. Inorg. Chim. Acta 1996, 246, 423–429. [Google Scholar] [CrossRef]
- Parker, D.; Pulukkody, K.; Smith, F.C.; Batsanov, A.; Howard, J.A.K. Structures of the yttrium complexes of 1,4,7,10-tetraazacyclododecane-N,N′,N″,N‴-tetraacetic acid (H4dota) and N,N″-bis(benzylcarbamoylmethyl)diethylenetriamine-N,N′,N″-triacetic acid and the solution structure of a zirconium complex of H4dota. J. Chem. Soc. Dalton Trans. 1994, 689–693. [Google Scholar] [CrossRef]
- Benetollo, F.; Bombieri, G.; Aime, S.; Botta, M. A holmium complex of a macrocyclic ligand (DOTA) and its isostructural europium analogue. Acta Crystallogr. C Cryst. Struct. Commun. 1999, 55, 353–356. [Google Scholar] [CrossRef]
- Burai, L.; Tóth, E.; Moreau, G.; Sour, A.; Scopelliti, R.; Merbach, A.E. Novel macrocyclic EuII complexes: Fast water exchange related to an extreme M-Owater distance. Chem. Eur. J. 2003, 9, 1394–1404. [Google Scholar] [CrossRef] [PubMed]
- Aime, S.; Barge, A.; Benetollo, F.; Bombieri, G.; Botta, M.; Uggeri, F. A Novel Compound in the Lanthanide(III) DOTA Series. X-ray Crystal and Molecular Structure of the Complex Na[La(DOTA)La(HDOTA)]·10H2O. Inorg. Chem. 1997, 36, 4287–4289. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D. 01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865, Erratum in Phys. Rev. Lett. 1997, 78, 1396. [Google Scholar] [CrossRef] [PubMed]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Mardirossian, N.; Head-Gordon, M. Thirty years of density functional theory in computational chemistry: An overview and extensive assessment of 200 density functionals. Mol. Phys. 2017, 115, 2315–2372. [Google Scholar] [CrossRef]
- Bursch, M.; Mewes, J.-M.; Hansen, A.; Grimme, S. Best-Practice DFT Protocols for Basic Molecular Computational Chemistry. Angew. Chem. Int. Ed. 2022, 61, e202205735. [Google Scholar] [CrossRef]
- Bühl, M.; Reimann, C.; Pantazis, D.A.; Bredow, T.; Neese, F. Geometries of Third-Row Transition-Metal Complexes from Density-Functional Theory. J. Chem. Theory Comput. 2008, 4, 1449–1459. [Google Scholar] [CrossRef]
- Morgenstern, A.; Lilley, L.M.; Stein, B.W.; Kozimor, S.A.; Batista, E.R.; Yang, P. Computer-Assisted Design of Macrocyclic Chelators for Actinium-225 Radiotherapeutics. Inorg. Chem. 2021, 60, 623–632. [Google Scholar] [CrossRef]
- Metz, B.; Stoll, H.; Dolg, M. Small-core multiconfiguration-Dirac—Hartree—Fock-adjusted pseudopotentials for post-d main group elements: Application to PbH and PbO. J. Chem. Phys. 2000, 113, 2563–2569. [Google Scholar] [CrossRef]
- Peterson, K.A. Systematically convergent basis sets with relativistic pseudopotentials. I. Correlation consistent basis sets for the post-d group 13-15 elements. J. Chem. Phys. 2003, 119, 11099–11112. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parameterization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef]
- Scalmani, G.; Frisch, M.J. Continuous surface charge polarizable continuum models of solvation. I. General formalism. J. Chem. Phys. 2010, 132, 114110. [Google Scholar] [CrossRef] [PubMed]
- Dennington, R.; Keith, T.; Millam, J. GaussView, Version 5; Semichem Inc.: Shawnee Mission, KS, USA, 2009. [Google Scholar]
- Glendening, E.D.; Landis, C.R.; Weinhold, F. NBO 7.0: New vistas in localized and delocalized chemical bonding theory. J. Comput. Chem. 2019, 40, 2234–2241. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C. 01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Fiszbein, D.J.; Brown, V.; Thiele, N.A.; Woods, J.J.; Wharton, L.; MacMillan, S.N.; Radchenko, V.; Ramogida, C.F.; Wilson, J.J. Tuning the Kinetic Inertness of Bi3+ Complexes: The Impact of Donor Atoms on Diaza-18-Crown-6 Ligands as Chelators for 213Bi Targeted Alpha Therapy. Inorg. Chem. 2021, 60, 9199–9211. [Google Scholar] [CrossRef] [PubMed]
- Cieslik, P.; Kubeil, M.; Zarschler, K.; Ullrich, M.; Brandt, F.; Anger, K.; Wadepohl, H.; Kopka, K.; Bachmann, M.; Pietzsch, J.; et al. Toward Personalized Medicine: One Chelator for Imaging and Therapy with Lutetium-177 and Actinium-225. J. Am. Chem. Soc. 2022, 144, 21555–21567. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | ΔETSAP-SAP | Pb-Oav | Pb-Nav | Symmetry |
|---|---|---|---|---|
| Pb(DOTA)2− | −22.8 | 2.687 | 2.711 | C4 |
| Pb(DOTPA)2− | 43.7 | 2.647 | 2.834 | C4 |
| Pb(DOTAM)2+ | −12.8 | 2.717 | 2.680 | C4 |
| Pb(DO2A2AM) | −18.9 | 2.702 | 2.693 | C2 |
| Pb(DO4Py)2+ | −19.0 | - | 2.729 | C4 |
| Pb(DO4Pyd)2+ | −25.1 | - | 2.718 | C4 |
| Pb(DO2A2Py) | −17.7 | 2.648 | 2.738 | C2 |
| Pb(MeDO2PA) | −8.5 | 2.605 | 2.761 | C2 |
| Pb(H2DO2PA) | −7.4 | 2.798 | 2.687 | C2 |
| Pb(MeDO2Scz)2+ | −14.7 | 2.818 | 2.736 | C2 |
| Pb(DO3APA)2− | −18.6 | 2.711 | 2.832 | C1 |
| Pb(CRYPT)2+ | - | 2.690 | 2.815 | C1 |
| Complex | NEDA | CT(E2) | CT(e) | ||||
|---|---|---|---|---|---|---|---|
| ΔEINT | ΔEEL | ΔECORE | ΔECT | O → Pb2+ | N → Pb2+ | ||
| Pb(DOTA)2− | −3630 | −3924 | 1391 | −1097 | −672.1 | −431.4 | 0.49 |
| Pb(DOTPA)2− | −3657 | −3877 | 1451 | −1232 | −784.1 | −397.9 | 0.51 |
| Pb(DO3APA)2− | −3657 | −3863 | 1367 | −1161 | −658.0 | −490.6 | 0.52 |
| Pb(MeDO2PA) | −2523 | −2810 | 1358 | −1072 | −428.0 | −754.2 | 0.50 |
| Pb(H2DO2PA) | −2471 | −2794 | 1338 | −1015 | −300.7 | −866.2 | 0.51 |
| Pb(DO2A2AM) | −2520 | −2825 | 1404 | −1100 | −631.4 | −502.2 | 0.50 |
| Pb(DO2A2Py) | −2513 | −2810 | 1435 | −1138 | −377.1 | −842.2 | 0.53 |
| Pb(DOTAM)2+ | −1394 | −1694 | 1384 | −1084 | −559.8 | −580.4 | 0.49 |
| Pb(DO4Py)2+ | −1429 | −1739 | 1501 | −1191 | - | −1342.2 | 0.56 |
| Pb(DO4Pyd)2+ | −1438 | −1763 | 1565 | −1240 | - | −1344.9 | 0.56 |
| Pb(MeDO2Scz)2+ | −1403 | −1619 | 1302 | −1086 | −267.3 | −919.0 | 0.54 |
| Pb(CRYPT)2+ | −1218 | −1414 | 1164 | −968 | −731.9 | −321.0 | 0.46 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kovács, A.; Varga, Z. Theoretical Study of Metal–Ligand Interactions in Lead Complexes with Radiopharmaceutical Interest. Molecules 2024, 29, 4198. https://doi.org/10.3390/molecules29174198
Kovács A, Varga Z. Theoretical Study of Metal–Ligand Interactions in Lead Complexes with Radiopharmaceutical Interest. Molecules. 2024; 29(17):4198. https://doi.org/10.3390/molecules29174198
Chicago/Turabian StyleKovács, Attila, and Zoltán Varga. 2024. "Theoretical Study of Metal–Ligand Interactions in Lead Complexes with Radiopharmaceutical Interest" Molecules 29, no. 17: 4198. https://doi.org/10.3390/molecules29174198
APA StyleKovács, A., & Varga, Z. (2024). Theoretical Study of Metal–Ligand Interactions in Lead Complexes with Radiopharmaceutical Interest. Molecules, 29(17), 4198. https://doi.org/10.3390/molecules29174198