
Exploring Radioiodinated Anastrozole and Epirubicin as AKT1-Targeted Radiopharmaceuticals in Breast Cancer: In Silico Analysis and Potential Therapeutic Effect with Functional Nuclear Imagining Implications

Abstract
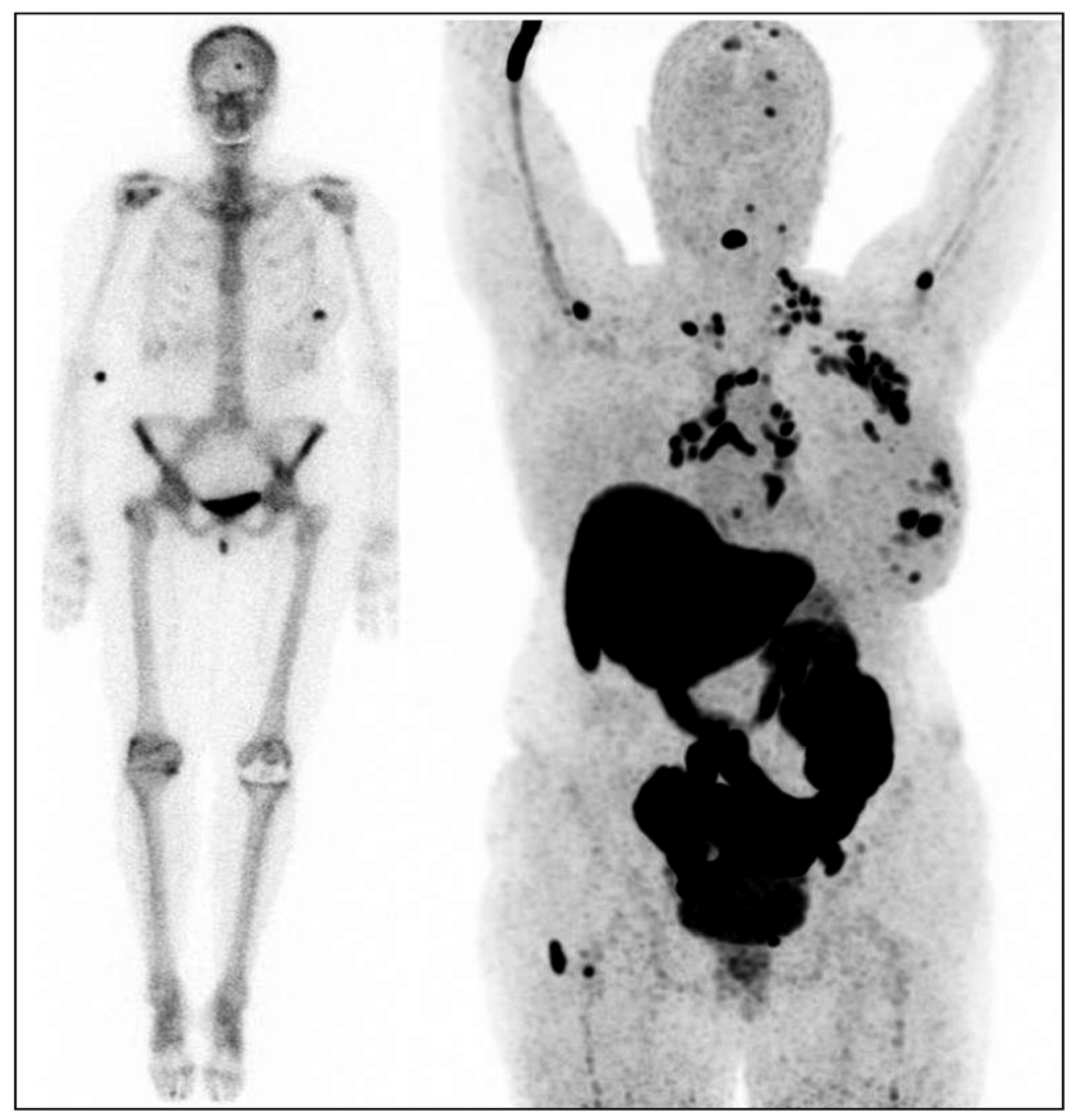
:1. Introduction
2. Results
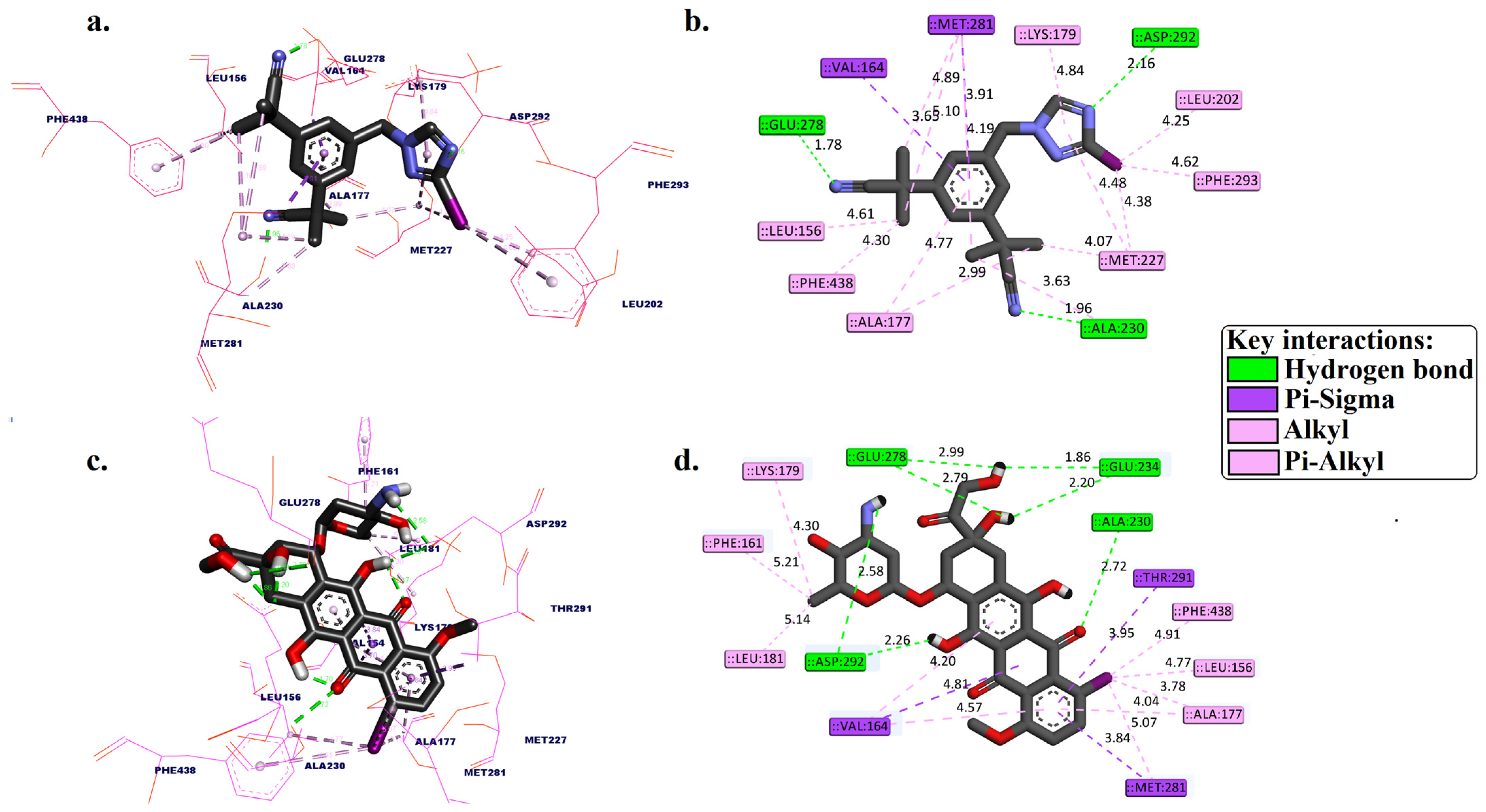
2.1. Molecular Docking
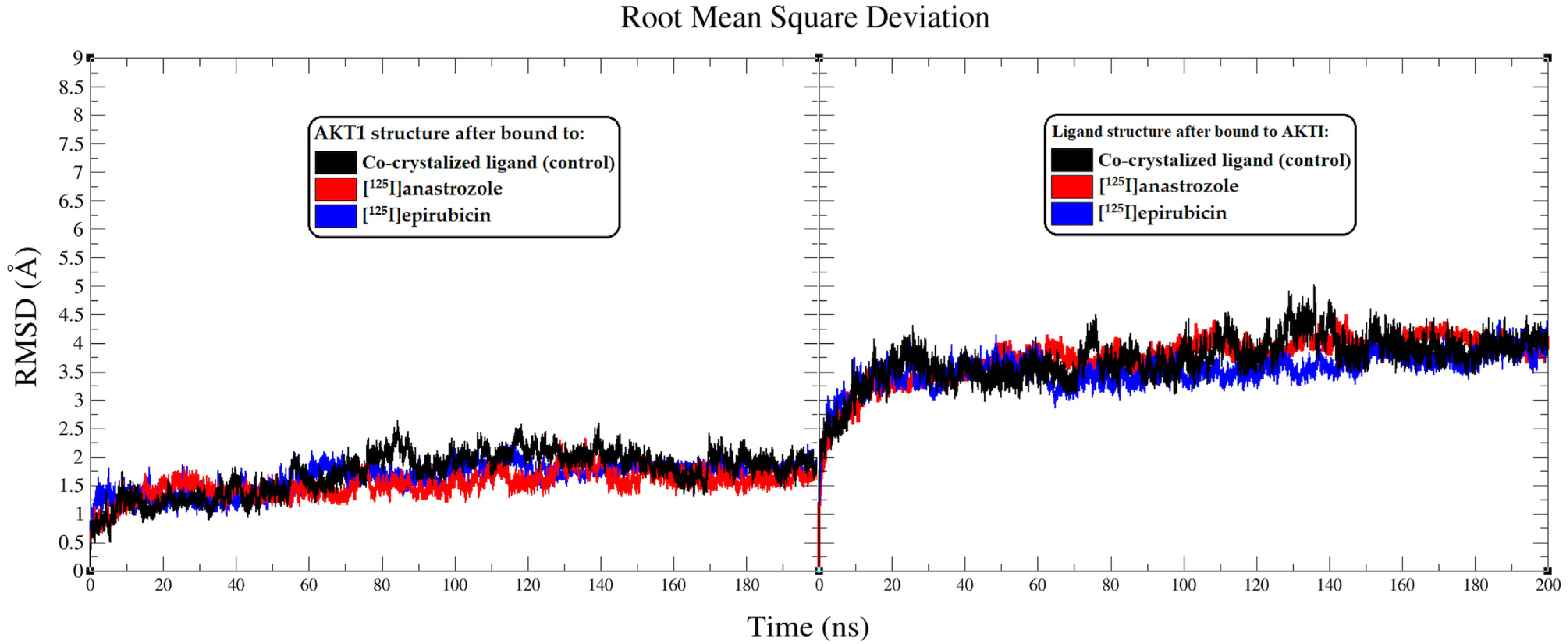
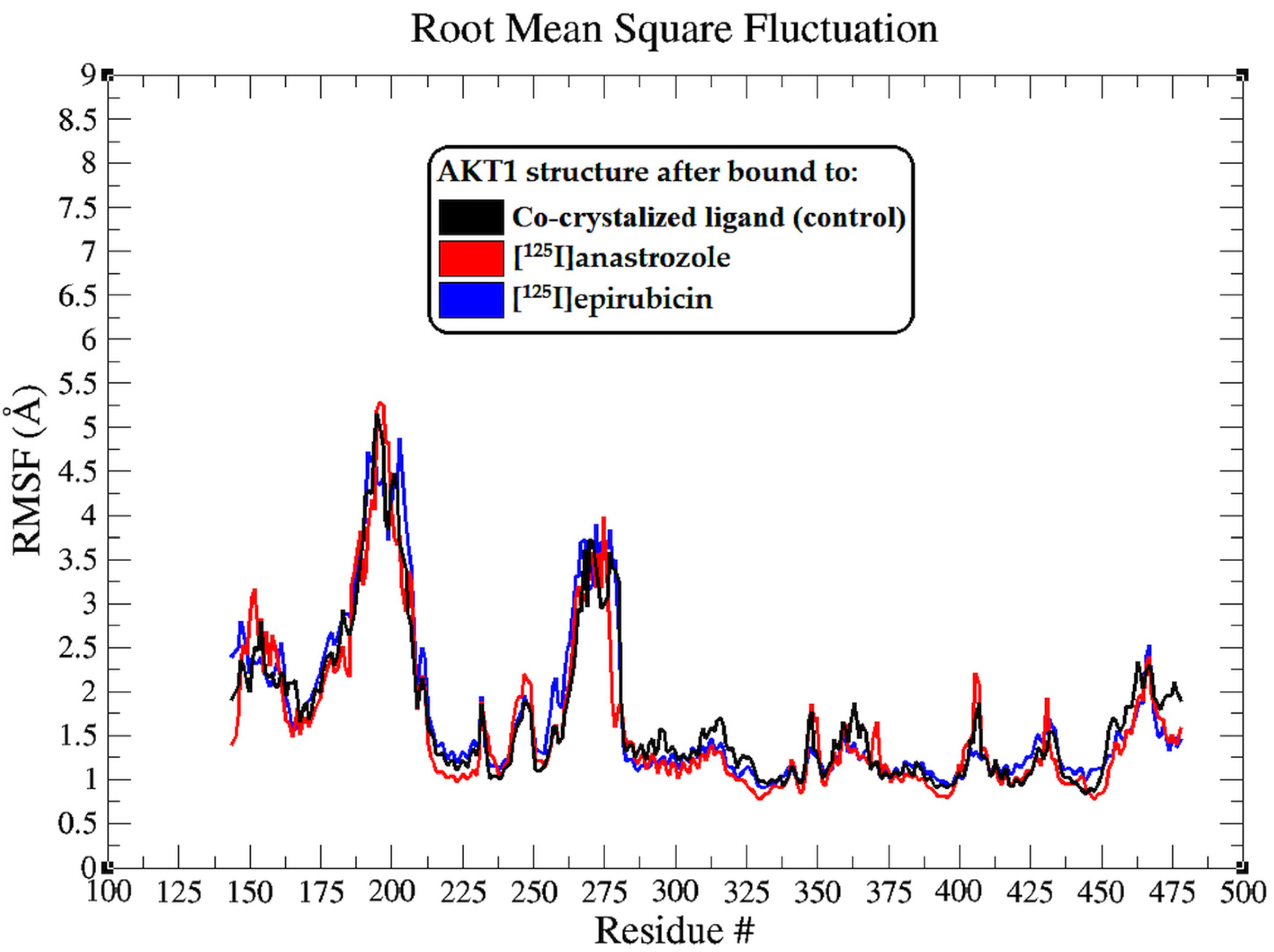
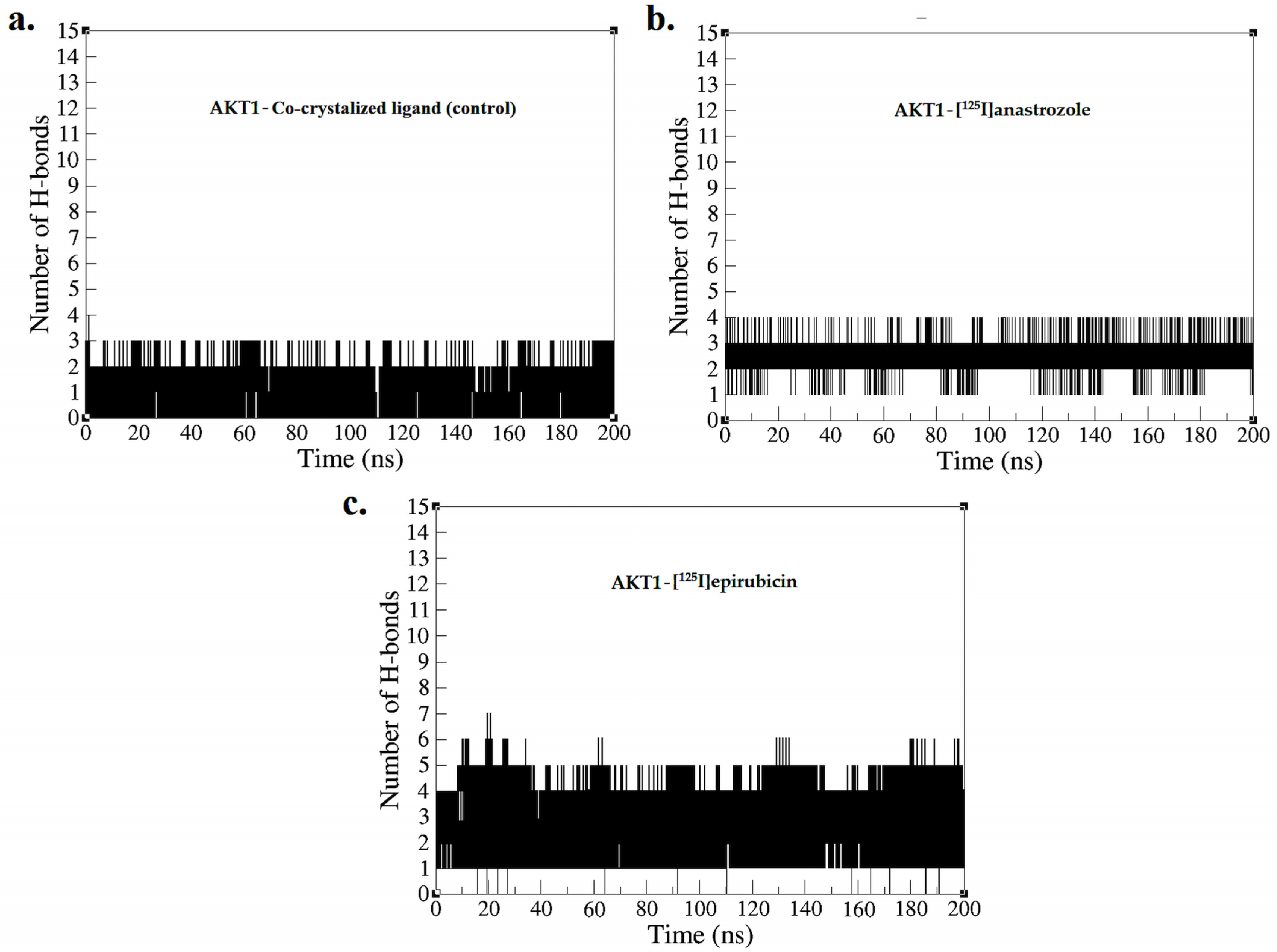
2.2. Molecular Dynamic Simulation
3. Discussion
3.1. Molecular Docking Simulation
3.2. Molecular Dynamic Simulation
4. Methodology
4.1. Molecular Docking Simulation
4.2. Molecular Dynamic Simulation
4.3. MM-PBSA Calculation
5. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Torre, L.A.; Islami, F.; Siegel, R.L.; Ward, E.M.; Jemal, A. Global cancer in women: Burden and trends. Cancer Epidemiol. Biomark. Prev. 2017, 26, 444–457. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Morgan, E.; Rumgay, H.; Mafra, A.; Singh, D.; Laversanne, M.; Vignat, J.; Gralow, J.R.; Cardoso, F.; Siesling, S. Current and future burden of breast cancer: Global statistics for 2020 and 2040. Breast 2022, 66, 15–23. [Google Scholar] [CrossRef]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Azubuike, S.O.; Muirhead, C.; Hayes, L.; McNally, R. Rising global burden of breast cancer: The case of sub-Saharan Africa (with emphasis on Nigeria) and implications for regional development: A review. World J. Surg. Oncol. 2018, 16, 1–13. [Google Scholar] [CrossRef]
- Bhushan, A.; Gonsalves, A.; Menon, J.U. Current state of breast cancer diagnosis, treatment, and theranostics. Pharmaceutics 2021, 13, 723. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.; GK, A.V.; Sakorikar, T.; Kamal, A.M.; Vaidya, J.S.; Pandya, H.J. Recent advances in biosensing approaches for point-of-care breast cancer diagnostics: Challenges and future prospects. Nanoscale Adv. 2021, 3, 5542–5564. [Google Scholar] [CrossRef]
- López-Portugués, C.; Montes-Bayón, M.; Díez, P. Biomarkers in Ovarian Cancer: Towards Personalized Medicine. Proteomes 2024, 12, 8. [Google Scholar] [CrossRef]
- Abeelh, E.A.; AbuAbeileh, Z. Comparative Effectiveness of Mammography, Ultrasound, and MRI in the Detection of Breast Carcinoma in Dense Breast Tissue: A Systematic Review. Cureus 2024, 16, e59054. [Google Scholar] [CrossRef]
- Rowe, S.P.; Pomper, M.G. Molecular imaging in oncology: Current impact and future directions. CA Cancer J. Clin. 2022, 72, 333–352. [Google Scholar] [CrossRef]
- Manafi-Farid, R.; Ranjbar, S.; Jamshidi Araghi, Z.; Pilz, J.; Schweighofer-Zwink, G.; Pirich, C.; Beheshti, M. Molecular imaging in primary staging of prostate cancer patients: Current aspects and future trends. Cancers 2021, 13, 5360. [Google Scholar] [CrossRef] [PubMed]
- Sgouros, G.; Bodei, L.; McDevitt, M.R.; Nedrow, J.R. Radiopharmaceutical therapy in cancer: Clinical advances and challenges. Nat. Rev. Drug Discov. 2020, 19, 589–608. [Google Scholar] [CrossRef] [PubMed]
- Barca, C.; Griessinger, C.M.; Faust, A.; Depke, D.; Essler, M.; Windhorst, A.D.; Devoogdt, N.; Brindle, K.M.; Schäfers, M.; Zinnhardt, B. Expanding theranostic radiopharmaceuticals for tumor diagnosis and therapy. Pharmaceuticals 2021, 15, 13. [Google Scholar] [CrossRef] [PubMed]
- Signore, A.; Lauri, C.; Auletta, S.; Varani, M.; Onofrio, L.; Glaudemans, A.W.; Panzuto, F.; Marchetti, P. Radiopharmaceuticals for Breast Cancer and Neuroendocrine Tumors: Two Examples of How Tissue Characterization May Influence the Choice of Therapy. Cancers 2020, 12, 781. [Google Scholar] [CrossRef] [PubMed]
- Crișan, G.; Moldovean-Cioroianu, N.S.; Timaru, D.-G.; Andrieș, G.; Căinap, C.; Chiș, V. Radiopharmaceuticals for PET and SPECT imaging: A literature review over the last decade. Int. J. Mol. Sci. 2022, 23, 5023. [Google Scholar] [CrossRef]
- Theodoropoulos, A.S.; Gkiozos, I.; Kontopyrgias, G.; Charpidou, A.; Kotteas, E.; Kyrgias, G.; Tolia, M. Modern radiopharmaceuticals for lung cancer imaging with positron emission tomography/computed tomography scan: A systematic review. SAGE Open Med. 2020, 8, 2050312120961594. [Google Scholar] [CrossRef]
- Alqahtani, F.F. SPECT/CT and PET/CT, related radiopharmaceuticals, and areas of application and comparison. Saudi Pharm. J. 2023, 31, 312–328. [Google Scholar] [CrossRef] [PubMed]
- Linden, H.M.; Stekhova, S.A.; Link, J.M.; Gralow, J.R.; Livingston, R.B.; Ellis, G.K.; Petra, P.H.; Peterson, L.M.; Schubert, E.K.; Dunnwald, L.K. Quantitative fluoroestradiol positron emission tomography imaging predicts response to endocrine treatment in breast cancer. J. Clin. Oncol. 2006, 24, 2793–2799. [Google Scholar] [CrossRef]
- Venema, C.M.; Apollonio, G.; Hospers, G.A.; Schröder, C.P.; Dierckx, R.A.; de Vries, E.F.; Glaudemans, A.W. Recommendations and technical aspects of 16α-[18F] fluoro-17β-estradiol PET to image the estrogen receptor in vivo: The Groningen experience. Clin. Nucl. Med. 2016, 41, 844–851. [Google Scholar] [CrossRef]
- Beauregard, J.-M.; Croteau, É.; Ahmed, N.; van Lier, J.E.; Bénard, F. Assessment of human biodistribution and dosimetry of 4-fluoro-11β-methoxy-16α-18F-fluoroestradiol using serial whole-body PET/CT. J. Nucl. Med. 2009, 50, 100–107. [Google Scholar] [CrossRef]
- McLarty, K.; Cornelissen, B.; Scollard, D.A.; Done, S.J.; Chun, K.; Reilly, R.M. Associations between the uptake of 111 In-DTPA-trastuzumab, HER2 density and response to trastuzumab (Herceptin) in athymic mice bearing subcutaneous human tumour xenografts. Eur. J. Nucl. Med. Mol. Imaging 2009, 36, 81–93. [Google Scholar] [CrossRef]
- Lin, M.; Paolillo, V.; Le, D.B.; Macapinlac, H.; Ravizzini, G.C. Monoclonal antibody based radiopharmaceuticals for imaging and therapy. Curr. Probl. Cancer 2021, 45, 100796. [Google Scholar] [CrossRef]
- Mankoff, D.A.; Peterson, L.M.; Tewson, T.J.; Link, J.M.; Gralow, J.R.; Graham, M.M.; Krohn, K.A. [18F] fluoroestradiol radiation dosimetry in human PET studies. J. Nucl. Med. 2001, 42, 679–684. [Google Scholar] [PubMed]
- Peterson, L.M.; Mankoff, D.A.; Lawton, T.; Yagle, K.; Schubert, E.K.; Stekhova, S.; Gown, A.; Link, J.M.; Tewson, T.; Krohn, K.A. Quantitative imaging of estrogen receptor expression in breast cancer with PET and 18F-fluoroestradiol. J. Nucl. Med. 2008, 49, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Hospers, G.; Helmond, F.; De Vries, E.; Dierckx, R.; De Vries, E. PET imaging of steroid receptor expression in breast and prostate cancer. Curr. Pharm. Des. 2008, 14, 3020–3032. [Google Scholar] [CrossRef]
- Dehdashti, F.; Mortimer, J.E.; Trinkaus, K.; Naughton, M.J.; Ellis, M.; Katzenellenbogen, J.A.; Welch, M.J.; Siegel, B.A. PET-based estradiol challenge as a predictive biomarker of response to endocrine therapy in women with estrogen-receptor-positive breast cancer. Breast Cancer Res. Treat. 2009, 113, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Dehdashti, F.; Flanagan, F.L.; Mortimer, J.E.; Katzenellenbogen, J.A.; Welch, M.J.; Siegel, B.A. Positron emission tomographic assessment of “metabolic flare” to predict response of metastatic breast cancer to antiestrogen therapy. Eur. J. Nucl. Med. 1999, 26, 51–56. [Google Scholar] [CrossRef]
- Kurland, B.F.; Peterson, L.M.; Lee, J.H.; Schubert, E.K.; Currin, E.R.; Link, J.M.; Krohn, K.A.; Mankoff, D.A.; Linden, H.M. Estrogen receptor binding (18F-FES PET) and glycolytic activity (18F-FDG PET) predict progression-free survival on endocrine therapy in patients with ER+ breast cancer. Clin. Cancer Res. 2017, 23, 407–415. [Google Scholar] [CrossRef]
- van Kruchten, M.; Glaudemans, A.W.; de Vries, E.F.; Beets-Tan, R.G.; Schröder, C.P.; Dierckx, R.A.; de Vries, E.G.; Hospers, G.A. PET imaging of estrogen receptors as a diagnostic tool for breast cancer patients presenting with a clinical dilemma. J. Nucl. Med. 2012, 53, 182–190. [Google Scholar] [CrossRef]
- Venema, C.; de Vries, E.; Glaudemans, A.; Poppema, B.; Hospers, G.; Schröder, C. 18F-FES PET has added value in staging and therapy decision making in patients with disseminated lobular breast cancer. Clin. Nucl. Med. 2017, 42, 612–614. [Google Scholar] [CrossRef]
- van Kruchten, M.; de Vries, E.G.; Glaudemans, A.W.; van Lanschot, M.C.; van Faassen, M.; Kema, I.P.; Brown, M.; Schröder, C.P.; de Vries, E.F.; Hospers, G.A. Measuring residual estrogen receptor availability during fulvestrant therapy in patients with metastatic breast cancer. Cancer Discov. 2015, 5, 72–81. [Google Scholar] [CrossRef]
- Yang, Z.; Sun, Y.; Xue, J.; Yao, Z.; Xu, J.; Cheng, J.; Shi, W.; Zhu, B.; Zhang, Y.; Zhang, Y. Can Positron Emission Tomography/Computed Tomography with the Dual Tracers Fluorine-18 Fluoroestradiol and Fluorodeoxyglucose Predict Neoadjuvant Chemotherapy Response of Breast Cancer?—A Pilot Study. PLoS ONE 2013, 8, e78192. [Google Scholar] [CrossRef] [PubMed]
- de Vries, E.; Venema, C.; Glaudemans, A.; Jager, A.; Garner, F.; O’Neill, A.; Patki, A.; van Menke-van der Houven, C. A phase 1 study of RAD1901, an oral selective estrogen receptor degrader, to determine changes in the F-18-FES uptake and tumor responses in ER-positive, HER2-negative, advanced breast cancer patients. Cancer Res. 2017, 77, P2-08-08. [Google Scholar] [CrossRef]
- van Kruchten, M.; de Vries, E.F.; Arts, H.J.; Jager, N.M.; Bongaerts, A.H.; Glaudemans, A.W.; Hollema, H.; de Vries, E.G.; Hospers, G.A.; Reyners, A.K. Assessment of estrogen receptor expression in epithelial ovarian cancer patients using 16α-18F-fluoro-17β-estradiol PET/CT. J. Nucl. Med. 2015, 56, 50–55. [Google Scholar] [CrossRef]
- Antunes, I.F.; Willemsen, A.T.; Sijbesma, J.W.; Boerema, A.S.; van Waarde, A.; Glaudemans, A.W.; Dierckx, R.A.; de Vries, E.G.; Hospers, G.A.; de Vries, E.F. In vivo quantification of Erβ expression by pharmacokinetic modeling: Studies with 18F-FHNP PET. J. Nucl. Med. 2017, 58, 1743–1748. [Google Scholar] [CrossRef] [PubMed]
- Güleç, B.A.; Yurt, F. Treatment with radiopharmaceuticals and radionuclides in breast cancer: Current options. Eur. J. Breast Health 2021, 17, 214. [Google Scholar] [CrossRef] [PubMed]
- Filippi, L.; Urso, L.; Ferrari, C.; Guglielmo, P.; Evangelista, L. The impact of PET imaging on triple negative breast cancer: An updated evidence-based perspective. Eur. J. Nucl. Med. Mol. Imaging 2024, 1–17. [Google Scholar] [CrossRef]
- Vito, A.; Rathmann, S.; Mercanti, N.; El-Sayes, N.; Mossman, K.; Valliant, J. Combined radionuclide therapy and immunotherapy for treatment of triple negative breast cancer. Int. J. Mol. Sci. 2021, 22, 4843. [Google Scholar] [CrossRef]
- Raguraman, R.; Srivastava, A.; Munshi, A.; Ramesh, R. Therapeutic approaches targeting molecular signaling pathways common to diabetes, lung diseases and cancer. Adv. Drug Deliv. Rev. 2021, 178, 113918. [Google Scholar] [CrossRef] [PubMed]
- Miricescu, D.; Totan, A.; Stanescu-Spinu, I.-I.; Badoiu, S.C.; Stefani, C.; Greabu, M. PI3K/AKT/mTOR signaling pathway in breast cancer: From molecular landscape to clinical aspects. Int. J. Mol. Sci. 2020, 22, 173. [Google Scholar] [CrossRef]
- Ortega, M.A.; Fraile-Martínez, O.; Asúnsolo, Á.; Buján, J.; García-Honduvilla, N.; Coca, S. Signal transduction pathways in breast cancer: The important role of PI3K/Akt/mTOR. J. Oncol. 2020, 2020, 9258396. [Google Scholar] [CrossRef]
- Mitsiades, C.S.; Mitsiades, N.; Koutsilieris, M. The Akt pathway: Molecular targets for anti-cancer drug development. Curr. Cancer Drug Targets 2004, 4, 235–256. [Google Scholar] [CrossRef]
- Kumar, A.; Rajendran, V.; Sethumadhavan, R.; Purohit, R. AKT kinase pathway: A leading target in cancer research. Sci. World J. 2013, 2013, 756134. [Google Scholar] [CrossRef]
- Nitulescu, G.M.; Margina, D.; Juzenas, P.; Peng, Q.; Olaru, O.T.; Saloustros, E.; Fenga, C.; Spandidos, D.A.; Libra, M.; Tsatsakis, A.M. Akt inhibitors in cancer treatment: The long journey from drug discovery to clinical use. Int. J. Oncol. 2016, 48, 869–885. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Sampath, D.; Nannini, M.A.; Lee, B.B.; Degtyarev, M.; Oeh, J.; Savage, H.; Guan, Z.; Hong, R.; Kassees, R. Targeting activated Akt with GDC-0068, a novel selective Akt inhibitor that is efficacious in multiple tumor models. Clin. Cancer Res. 2013, 19, 1760–1772. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Sun, M.M.; Zhang, G.G.; Yang, J.; Chen, K.S.; Xu, W.W.; Li, B. Targeting PI3K/Akt signal transduction for cancer therapy. Signal Transduct. Target. Ther. 2021, 6, 425. [Google Scholar] [CrossRef]
- Nandan, D.; Zhang, N.; Yu, Y.; Schwartz, B.; Chen, S.; Kima, P.E.; Reiner, N.E. Miransertib (ARQ 092), an orally-available, selective Akt inhibitor is effective against Leishmania. PLoS ONE 2018, 13, e0206920. [Google Scholar] [CrossRef]
- Ibrahim, A.; Sakr, T.; Khoweysa, O.; Motaleb, M.; Abd El-Bary, A.; El-Kolaly, M. Radioiodinated anastrozole and epirubicin as potential targeting radiopharmaceuticals for solid tumor imaging. J. Radioanal. Nucl. Chem. 2015, 303, 967–975. [Google Scholar] [CrossRef]
- Binmujlli, M.A. Radiological and Molecular Analysis of Radioiodinated Anastrozole and Epirubicin as Innovative Radiopharmaceuticals Targeting Methylenetetrahydrofolate Dehydrogenase 2 in Solid Tumors. Pharmaceutics 2024, 16, 616. [Google Scholar] [CrossRef] [PubMed]
- Tenenbaum, F.; Ricard, M. Peroperative detection probes. Evaluation and perspectives in endocrinology. In Annales d’Endocrinologie; Elsevier: Masson, The Netherlands, 1997; pp. 2213–3941. [Google Scholar]
- Mross, K.; Mayer, U.; Langenbuch, T.; Hamm, K.; Burk, K.; Hossfeld, D. Toxicity, pharmacokinetics and metabolism of iododoxorubicin in cancer patients. Eur. J. Cancer Clin. Oncol. 1990, 26, 1156–1162. [Google Scholar] [CrossRef]
- Twelves, C.; Dobbs, N.; Lawrence, M.; Ramirez, A.; Summerhayes, M.; Richards, M.; Towlson, K.; Rubens, R. Iododoxorubicin in advanced breast cancer: A phase II evaluation of clinical activity, pharmacology and quality of life. Br. J. Cancer 1994, 69, 726–731. [Google Scholar] [CrossRef]
- Formelli, F.; Carsana, R.; Pollini, C. Pharmacokinetics of 4′-deoxy-4′-iodo-doxorubicin in plasma and tissues of tumor-bearing mice compared with doxorubicin. Cancer Res. 1987, 47, 5401–5406. [Google Scholar] [PubMed]
- Amine, A.; Mohammadi, H.; Bourais, I.; Palleschi, G. Enzyme inhibition-based biosensors for food safety and environmental monitoring. Biosens. Bioelectron. 2006, 21, 1405–1423. [Google Scholar] [CrossRef]
- Copeland, R.A. Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists; John Wiley & Sons: New York, NY, USA, 2013. [Google Scholar]
- Alonso, H.; Bliznyuk, A.A.; Gready, J.E. Combining docking and molecular dynamic simulations in drug design. Med. Res. Rev. 2006, 26, 531–568. [Google Scholar] [CrossRef]
- Sanabria-Chanaga, E.E.; Betancourt-Conde, I.; Hernández-Campos, A.; Téllez-Valencia, A.; Castillo, R. In silico hit optimization toward AKT inhibition: Fragment-based approach, molecular docking and molecular dynamics study. J. Biomol. Struct. Dyn. 2019, 37, 4301–4311. [Google Scholar] [CrossRef]
- Kaur, T.; Madgulkar, A.; Bhalekar, M.; Asgaonkar, K. Molecular docking in formulation and development. Curr. Drug Discov. Technol. 2019, 16, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Alhawarri, M.B. Exploring the Anticancer Potential of Furanpydone A: A Computational Study on its Inhibition of MTHFD2 Across Diverse Cancer Cell Lines. Cell Biochem. Biophys. 2024, 1–18. [Google Scholar] [CrossRef]
- Ibrahim, M.M.; Azmi, M.N.; Alhawarri, M.B.; Kamal, N.N.S.N.M.; AbuMahmoud, H. Synthesis, characterization and bioactivity of new pyridine-2(H)-one, nicotinonitrile, and furo[2,3-b]pyridine derivatives. Mol. Divers. 2024, 1–19. [Google Scholar] [CrossRef]
- Alidmat, M.M.; Alhawarri, M.B.; Al-Refai, M.; Mansi, I.A.; Al-Balas, Q.; Ibrahim, M.M. Synthesis, Characterization and Glyoxalase inhibitory activity of 4,6-Diheteroarylpyrimidine-2-amine derivatives: In vitro and in silico studies. Egypt. J. Chem. 2024. [Google Scholar] [CrossRef]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular docking and structure-based drug design strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef]
- De Ruyck, J.; Brysbaert, G.; Blossey, R.; Lensink, M.F. Molecular docking as a popular tool in drug design, an in silico travel. Adv. Appl. Bioinform. Chem. 2016, 9, 1–11. [Google Scholar] [CrossRef]
- Mukherjee, S. Docking Platform and Validation Resources for Structure-based Drug Design. Ph.D. Thesis, State University of New York, Stony Brook, NY, USA, 2012. [Google Scholar]
- Alhawarri, M.B.; Al-Thiabat, M.G.; Dubey, A.; Tufail, A.; Fouad, D.; Alrimawi, B.H.; Dayoob, M. ADME profiling, molecular docking, DFT, and MEP analysis reveal cissamaline, cissamanine, and cissamdine from Cissampelos capensis Lf as potential anti-Alzheimer’s agents. RSC Adv. 2024, 14, 9878–9891. [Google Scholar] [CrossRef] [PubMed]
- Alhawarri, M.B.; Olimat, S. Potential Serotonin 5-HT2A Receptor Agonist of Psychoactive Components of Silene undulata Aiton: LC-MS/MS, ADMET, and Molecular Docking Studies. Curr. Pharm. Biotechnol. 2024. [Google Scholar] [CrossRef]
- Yunos, N.M.; Al-Thiabat, M.G.; Sallehudin, N.J. Quassinoids from Eurycoma longifolia as Potential Dihydrofolate Reductase Inhibitors: A Computational Study. Curr. Pharm. Biotechnol. 2024, 25, 2154–2165. [Google Scholar] [CrossRef] [PubMed]
- Yunos, N.M.; Wahab, H.A.; Al-Thiabat, M.G.; Sallehudin, N.J.; Jauri, M.H. In Vitro and In Silico Analysis of the Anticancer Effects of Eurycomanone and Eurycomalactone from Eurycoma longifolia. Plants 2023, 12, 2827. [Google Scholar] [CrossRef]
- Alhawarri, M.B.; Dianita, R.; Rawa, M.S.A.; Nogawa, T.; Wahab, H.A. Potential Anti-Cholinesterase Activity of Bioactive Compounds Extracted from Cassia grandis Lf and Cassia timoriensis DC. Plants 2023, 12, 344. [Google Scholar] [CrossRef] [PubMed]
- Amir Rawa, M.S.; Al-Thiabat, M.G.; Nogawa, T.; Futamura, Y.; Okano, A.; Wahab, H.A. Naturally Occurring 8ß, 13ß-kaur-15-en-17-al and Anti-Malarial Activity from Podocarpus polystachyus Leaves. Pharmaceuticals 2022, 15, 902. [Google Scholar] [CrossRef]
- Alidmat, M.M.; Khairuddean, M.; Kamal, N.N.S.N.M.; Muhammad, M.; Wahab, H.A.; Althiabat, M.G.; Alhawarri, M.B. Synthesis, Characterization, Molecular Docking and Cytotoxicity Evaluation of New Thienyl Chalcone Derivatives against Breast Cancer Cells. Syst. Rev. Pharm. 2022, 13, 1. [Google Scholar]
- Shalayel, M.H.F.; Al-Mazaideh, G.M.; Alanezi, A.A.; Almuqati, A.F.; Alotaibi, M. Diosgenin and Monohydroxy Spirostanol from Prunus amygdalus var amara Seeds as Potential Suppressors of EGFR and HER2 Tyrosine Kinases: A Computational Approach. Pharmaceuticals 2023, 16, 704. [Google Scholar] [CrossRef]
- Shalayel, M.H.F.; Al-Mazaideh, G.M.; Alanezi, A.A.; Almuqati, A.F.; Alotaibi, M. The Potential Anti-Cancerous Activity of Prunus amygdalus var. amara Extract. Processes 2023, 11, 1277. [Google Scholar] [CrossRef]
- Li, D.-D.; Wu, T.-T.; Yu, P.; Wang, Z.-Z.; Xiao, W.; Jiang, Y.; Zhao, L.-G. Molecular dynamics analysis of binding sites of epidermal growth factor receptor kinase inhibitors. ACS Omega 2020, 5, 16307–16314. [Google Scholar] [CrossRef]
- Janati-Fard, F.; Housaindokht, M.R.; Monhemi, H. Investigation of structural stability and enzymatic activity of glucose oxidase and its subunits. J. Mol. Catal. B Enzym. 2016, 134, 16–24. [Google Scholar] [CrossRef]
- da Fonseca, A.M.; Caluaco, B.J.; Madureira, J.M.C.; Cabongo, S.Q.; Gaieta, E.M.; Djata, F.; Colares, R.P.; Neto, M.M.; Fernandes, C.F.C.; Marinho, G.S. Screening of potential inhibitors targeting the main protease structure of SARS-CoV-2 via molecular docking, and approach with molecular dynamics, RMSD, RMSF, H-bond, SASA and MMGBSA. Mol. Biotechnol. 2023, 66, 1919–1933. [Google Scholar] [CrossRef]
- Ben-Shalom, I.Y.; Pfeiffer-Marek, S.; Baringhaus, K.-H.; Gohlke, H. Efficient approximation of ligand rotational and translational entropy changes upon binding for use in MM-PBSA calculations. J. Chem. Inf. Model. 2017, 57, 170–189. [Google Scholar] [CrossRef]
- Robertson, J.F.; Coleman, R.E.; Cheung, K.-L.; Evans, A.; Holcombe, C.; Skene, A.; Rea, D.; Ahmed, S.; Jahan, A.; Horgan, K. Proliferation and AKT activity biomarker analyses after capivasertib (AZD5363) treatment of patients with ER+ invasive breast cancer (STAKT). Clin. Cancer Res. 2020, 26, 1574–1585. [Google Scholar] [CrossRef]
- Luboff, A.J.; DeRemer, D.L. Capivasertib: A novel AKT inhibitor approved for hormone-receptor-positive, HER-2-negative metastatic breast cancer. Ann. Pharmacother. 2024, 10600280241241531. [Google Scholar] [CrossRef] [PubMed]
- Voorhees, P.M.; Spencer, A.; Sutherland, H.J.; O’Dwyer, M.E.; Huang, S.-Y.; Stewart, K.; Chari, A.; Rosenzwieg, M.; Nooka, A.K.; Rosenbaum, C.A. Novel AKT inhibitor afuresertib in combination with bortezomib and dexamethasone demonstrates favorable safety profile and significant clinical activity in patients with relapsed/refractory multiple myeloma. Blood 2013, 122, 283. [Google Scholar]
- Shariati, M.; Meric-Bernstam, F. Targeting AKT for cancer therapy. Expert Opin. Investig. Drugs 2019, 28, 977–988. [Google Scholar] [CrossRef]
- Arceci, R.J.; Allen, C.E.; Dunkel, I.; Jacobsen, E.D.; Whitlock, J.; Vassallo, R.; Borrello, I.M.; Oliff, A.; Morris, S.R.; Reedy, B.A.M. Evaluation of Afuresertib, an oral pan-AKT inhibitor, in patients with Langerhans cell histiocytosis. Blood 2013, 122, 2907. [Google Scholar] [CrossRef]
- Sharif Siam, M.K.; Sarker, A.; Sayeem, M.M.S. In silico drug design and molecular docking studies targeting Akt1 (RAC-alpha serine/threonine-protein kinase) and Akt2 (RAC-beta serine/threonine-protein kinase) proteins and investigation of CYP (cytochrome P450) inhibitors against MAOB (monoamine oxidase B) for OSCC (oral squamous cell carcinoma) treatment. J. Biomol. Struct. Dyn. 2021, 39, 6467–6479. [Google Scholar]
- Zhong, S.; Hou, Y.; Zhang, Z.; Guo, Z.; Yang, W.; Dou, G.; Lv, X.; Wang, X.; Ge, J.; Wu, B. Identification of novel natural inhibitors targeting AKT Serine/Threonine Kinase 1 (AKT1) by computational study. Bioengineered 2022, 13, 12003–12020. [Google Scholar] [CrossRef]
- Kong, W.; Zhu, L.; Li, T.; Chen, J.; Fan, B.; Ji, W.; Zhang, C.; Cai, X.; Hu, C.; Sun, X. Azeliragon inhibits PAK1 and enhances the therapeutic efficacy of AKT inhibitors in pancreatic cancer. Eur. J. Pharmacol. 2023, 948, 175703. [Google Scholar] [CrossRef] [PubMed]
- Halder, A.K.; Cordeiro, M.N.D. AKT inhibitors: The road ahead to computational modeling-guided discovery. Int. J. Mol. Sci. 2021, 22, 3944. [Google Scholar] [CrossRef] [PubMed]
- Westbrook, J.; Feng, Z.; Chen, L.; Yang, H.; Berman, H.M. The protein data bank and structural genomics. Nucleic Acids Res. 2003, 31, 489–491. [Google Scholar] [CrossRef]
- Blake, J.F.; Kallan, N.C.; Xiao, D.; Xu, R.; Bencsik, J.R.; Skelton, N.J.; Spencer, K.L.; Mitchell, I.S.; Woessner, R.D.; Gloor, S.L. Discovery of pyrrolopyrimidine inhibitors of Akt. Bioorganic Med. Chem. Lett. 2010, 20, 5607–5612. [Google Scholar] [CrossRef] [PubMed]
- Biovia, D.S. Dassault Systèmes; Discovery Studio Visualizer: San Diego, CA, USA, 2017; Volume 936, Available online: https://discover.3ds.com/discovery-studio-visualizer-download (accessed on 29 April 2024).
- Land, H.; Humble, M.S. YASARA: A tool to obtain structural guidance in biocatalytic investigations. In Protein Engineering; Springer: Berlin/Heidelberg, Germany, 2018; pp. 43–67. [Google Scholar]
- Abdelbagi, M.E.; Al-Mazaideh, G.M.; Ahmed, A.E.; Al-Rimawi, F.; Ayyal Salman, H.; Almutairi, A.; Abuilaiwi, F.A.; Wedian, F. Exploring Securigera securidaca Seeds as a Source of Potential CDK1 Inhibitors: Identification of Hippeastrine and Naringenin as Promising Hit Candidates. Processes 2023, 11, 1478. [Google Scholar] [CrossRef]
- Gordon, J.C.; Myers, J.B.; Folta, T.; Shoja, V.; Heath, L.S.; Onufriev, A. H++: A server for estimating p K as and adding missing hydrogens to macromolecules. Nucleic Acids Res. 2005, 33, W368–W371. [Google Scholar] [CrossRef] [PubMed]
- Norgan, A.P.; Coffman, P.K.; Kocher, J.-P.A.; Katzmann, D.J.; Sosa, C.P. Multilevel parallelization of AutoDock 4.2. J. Cheminform. 2011, 3, 1–9. [Google Scholar] [CrossRef]
- Hou, X.; Du, J.; Zhang, J.; Du, L.; Fang, H.; Li, M. How to improve docking accuracy of AutoDock 4.2: A case study using different electrostatic potentials. J. Chem. Inf. Model. 2013, 53, 188–200. [Google Scholar] [CrossRef]
- Jaramillo-Botero, A.; Naserifar, S.; Goddard, W.A., III. General multiobjective force field optimization framework, with application to reactive force fields for silicon carbide. J. Chem. Theory Comput. 2014, 10, 1426–1439. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 1–14. [Google Scholar] [CrossRef]
- Fuhrmann, J.; Rurainski, A.; Lenhof, H.P.; Neumann, D. A new Lamarckian genetic algorithm for flexible ligand-receptor docking. J. Comput. Chem. 2010, 31, 1911–1918. [Google Scholar] [CrossRef]
- Schmid, N.; Eichenberger, A.P.; Choutko, A.; Riniker, S.; Winger, M.; Mark, A.E.; Van Gunsteren, W.F. Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur. Biophys. J. 2011, 40, 843–856. [Google Scholar] [CrossRef] [PubMed]
- Rühle, V. Pressure coupling/barostats. J. Club. 2008, 1–5. Available online: https://www.mpip-mainz.mpg.de/en/home (accessed on 25 April 2024).
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Petersen, H.G. Accuracy and efficiency of the particle mesh Ewald method. J. Chem. Phys. 1995, 103, 3668–3679. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.; Fraaije, J.G. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Consortium, O.S.D.D.; Lynn, A. g_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Verma, S.; Grover, S.; Tyagi, C.; Goyal, S.; Jamal, S.; Singh, A.; Grover, A. Hydrophobic interactions are a key to MDM2 inhibition by polyphenols as revealed by molecular dynamics simulations and MM/PBSA free energy calculations. PLoS ONE 2016, 11, e0149014. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Free Binding Energy (kcal/mol) | Molecular Interactions Analysis within the AKT1 Active Binding Site | |||
|---|---|---|---|---|---|
| H-bond | Distance (Å) | Pi-Sigma | Hydrophobic Interaction | ||
| [125I]anastrozole | −10.68 | ALA230, GLU278, and ASP292 | 1.96, 1.78, and 2.16 | VAL164 and MET281 | LEU156, ALA177, LYS179, LEU202, MET227, MET281, PHE293, and PHE438 |
| [125I]epirubicin | −11.84 | ALA230, GLU234, GLU234, GLU278, GLU278, ASP292, and ASP292 | 2.72, 1.86, 2.20, 2.79, 2.99, 2.26, and 2.58 | VAL164, MET281, and THR291 | LEU156, PHE161, VAL164, ALA177, LYS179, LEU181, PHE281, and PHE348 |
| * Co-crystalized ligand (original pose) | −9.53 | ALA230 and GLU278 | 1.98 and 2.67 | - | VAL164, ALA177, LYS179, ALA230, and MET281 |
| System | ΔGbind (kcal/mol) | Electrostatic (kcal/mol) | Van der Waal (kcal/mol) | Polar Salvation (kcal/mol) | Non-Polar Salvation (kcal/mol) |
|---|---|---|---|---|---|
| AKT1-Co-crystallized ligand | −16.38 ± 0.14 | −11.28 ± 0.11 | −13.52 ± 0.12 | 19.11 ± 0.12 | −10.69 ± 0.13 |
| AKT1-[125I]anastrozole | −20.03 ± 0.15 | −12.86 ± 0.13 | −14.69 ± 0.11 | 19.84 ± 0.13 | −12.32 ± 0.12 |
| AKT1-[125I]epirubicin | −23.57 ± 0.14 | −14.73 ± 0.12 | −15.84 ± 0.14 | 19.86 ± 0.12 | −12.86 ± 0.13 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Binmujlli, M.A. Exploring Radioiodinated Anastrozole and Epirubicin as AKT1-Targeted Radiopharmaceuticals in Breast Cancer: In Silico Analysis and Potential Therapeutic Effect with Functional Nuclear Imagining Implications. Molecules 2024, 29, 4203. https://doi.org/10.3390/molecules29174203
Binmujlli MA. Exploring Radioiodinated Anastrozole and Epirubicin as AKT1-Targeted Radiopharmaceuticals in Breast Cancer: In Silico Analysis and Potential Therapeutic Effect with Functional Nuclear Imagining Implications. Molecules. 2024; 29(17):4203. https://doi.org/10.3390/molecules29174203
Chicago/Turabian StyleBinmujlli, Mazen Abdulrahman. 2024. "Exploring Radioiodinated Anastrozole and Epirubicin as AKT1-Targeted Radiopharmaceuticals in Breast Cancer: In Silico Analysis and Potential Therapeutic Effect with Functional Nuclear Imagining Implications" Molecules 29, no. 17: 4203. https://doi.org/10.3390/molecules29174203