
Novel Autotaxin Inhibitor ATX-1d Significantly Enhances Potency of Paclitaxel—An In Silico and In Vitro Study

 , , , and
, , , and 
Abstract
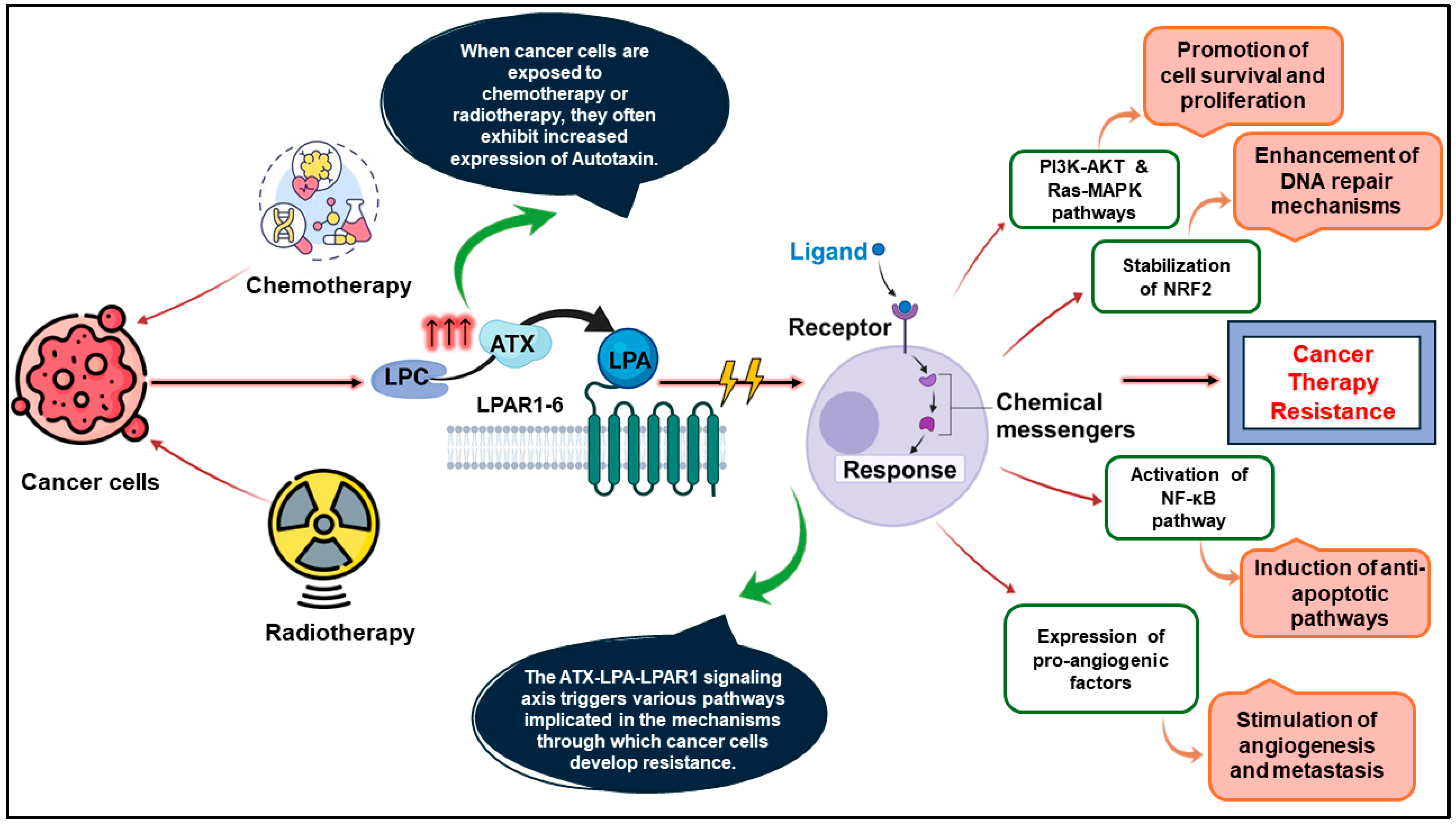
1. Introduction
2. Methods
3. Computational Details
3.1. Molecular Docking
3.2. Molecular Dynamics (MD) Simulations
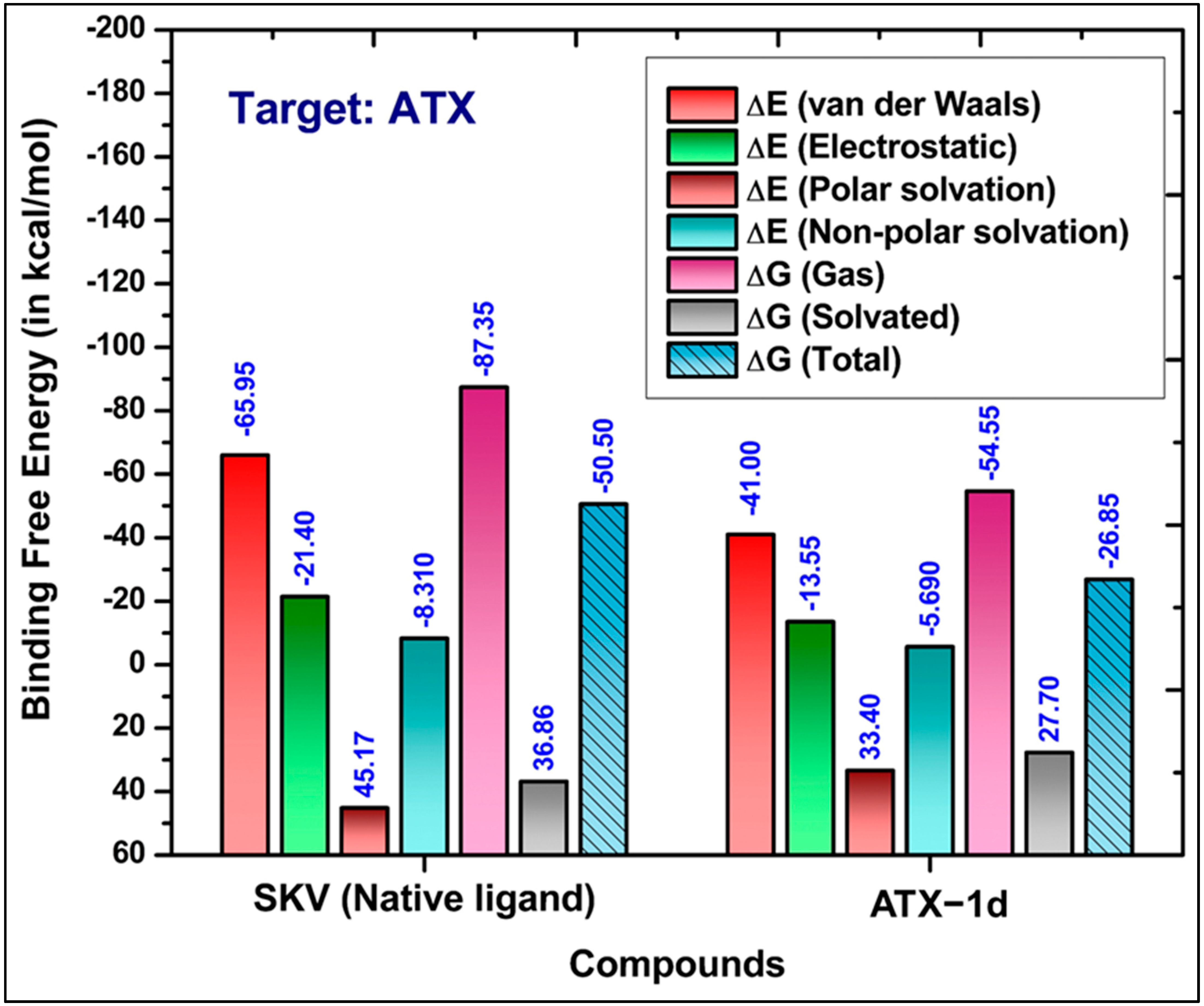
3.3. Binding Free Energy Calculations
3.4. QM/MM-GBSA Calculations
3.5. Symmetry-Adapted Perturbation Theory (SAPT) Calculations
3.6. Conceptual DFT Parameters
4. Experimental Details
4.1. Chemical Samples
4.2. In Vitro ATX Enzyme Inhibition Assay
4.3. Cell Viability Assay
5. Results and Discussion
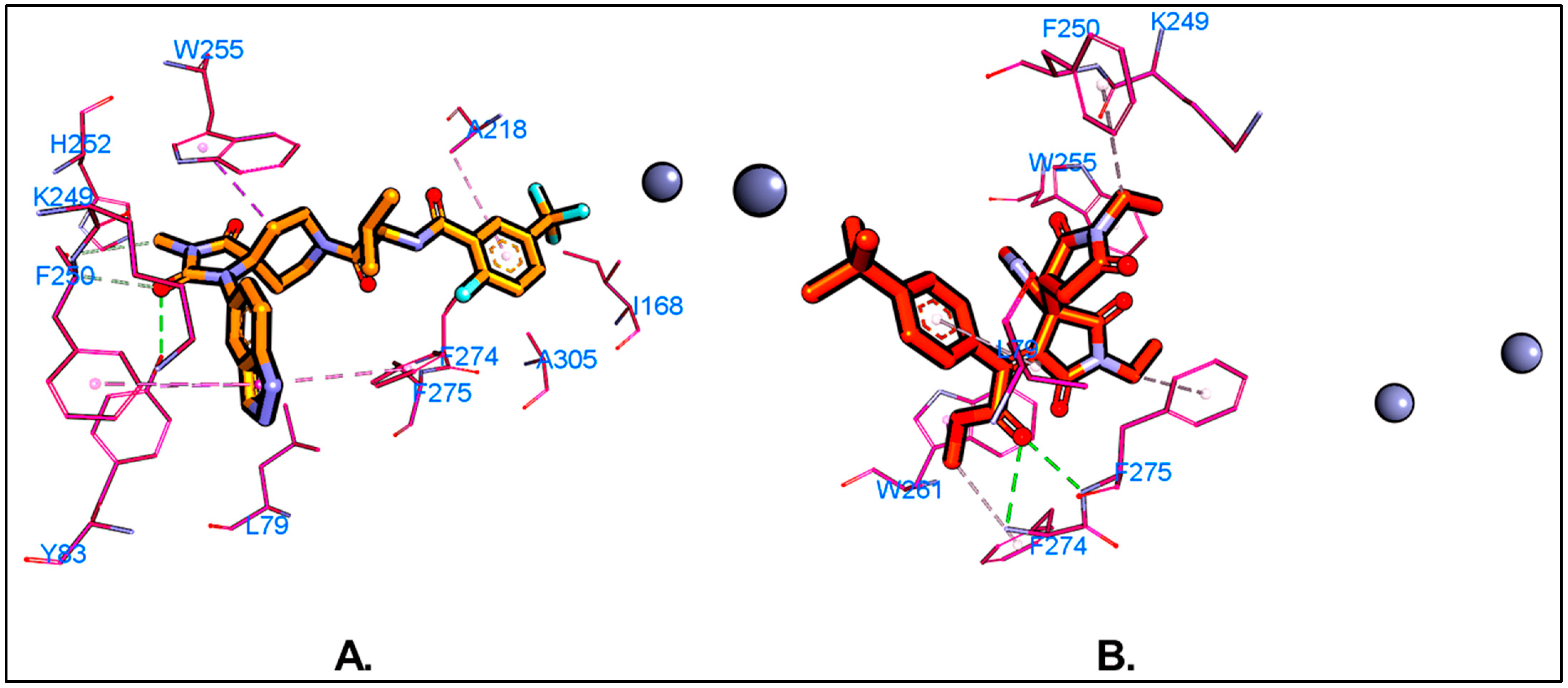
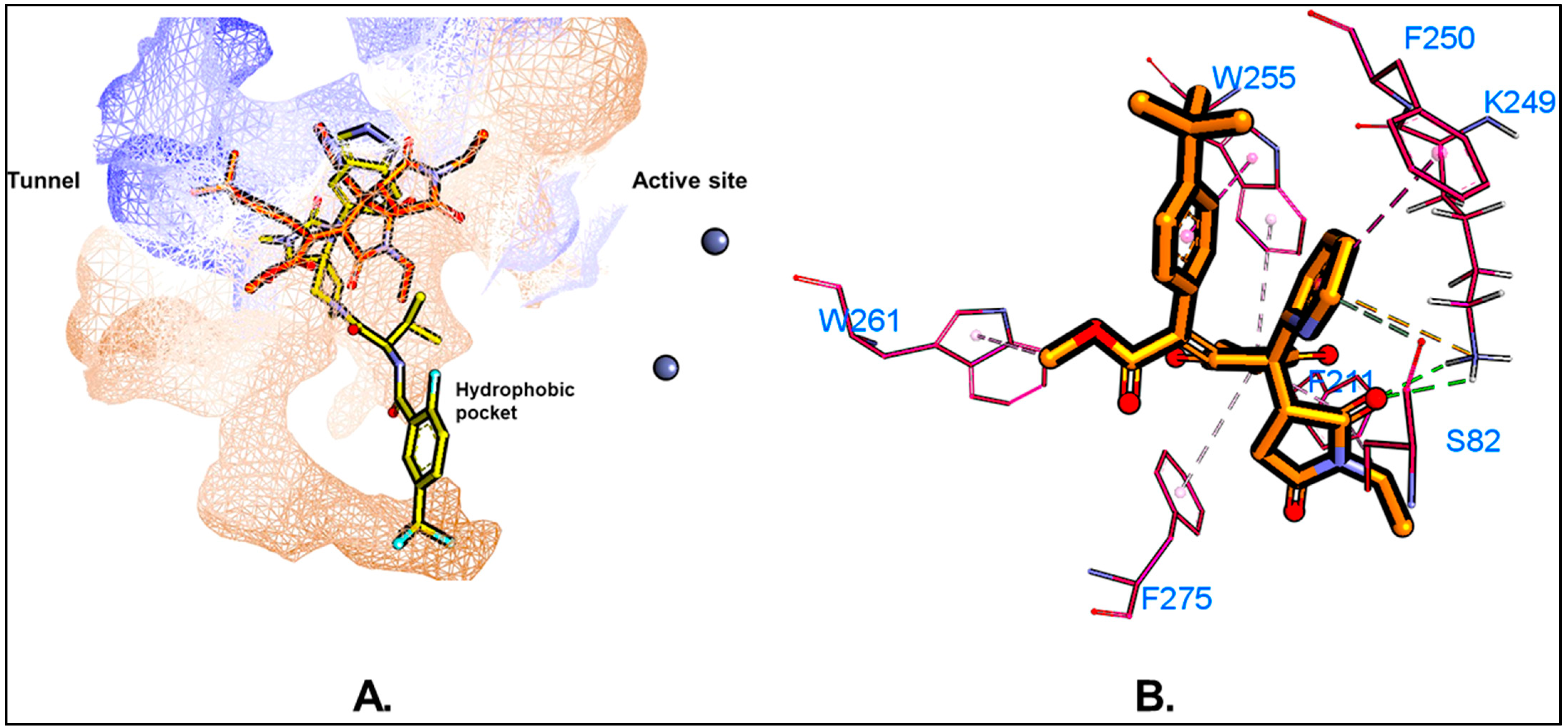
5.1. Molecular Docking
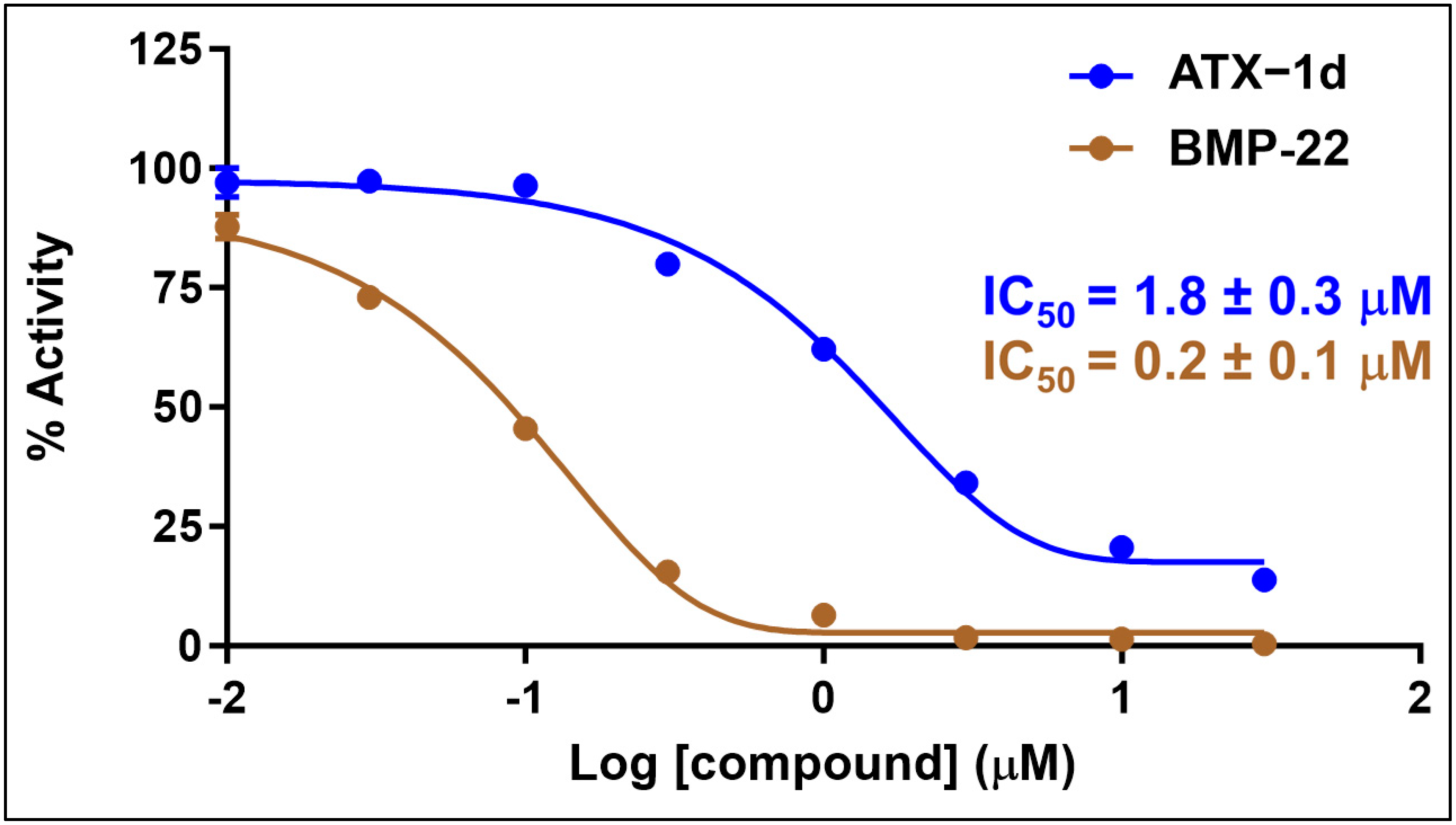
5.2. In Vitro Screening through ATX Enzyme Inhibition Assay
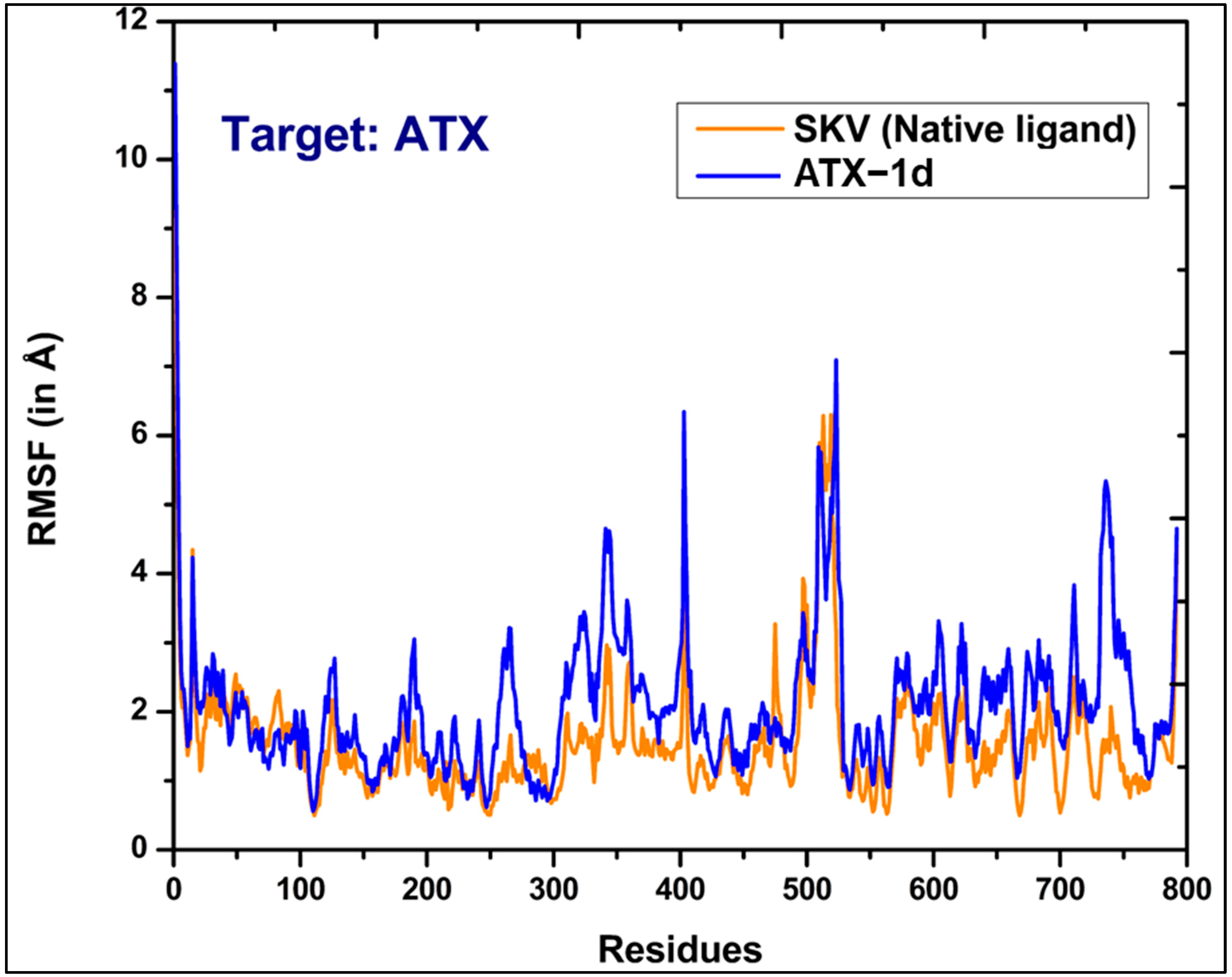
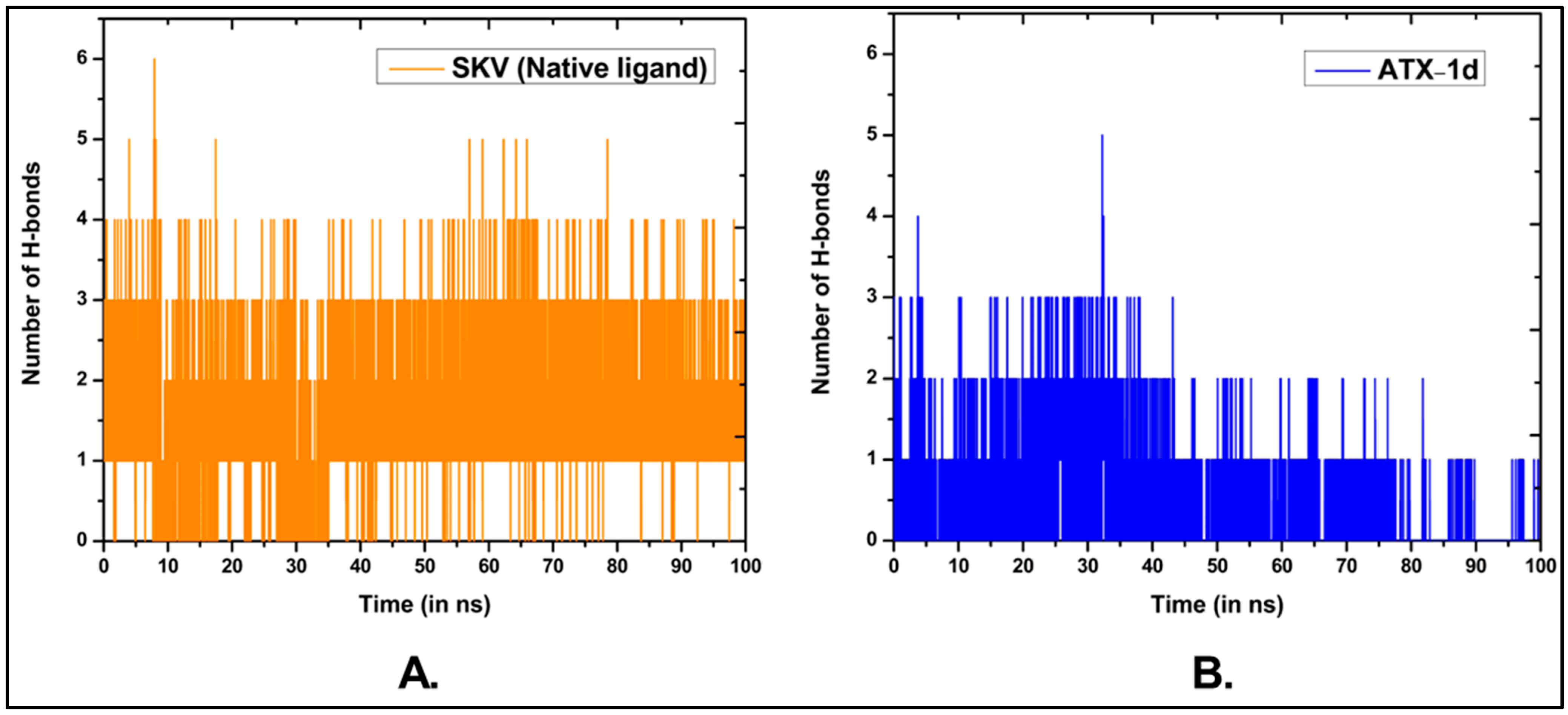
5.3. MD Simulations
5.4. SAPT Calculations
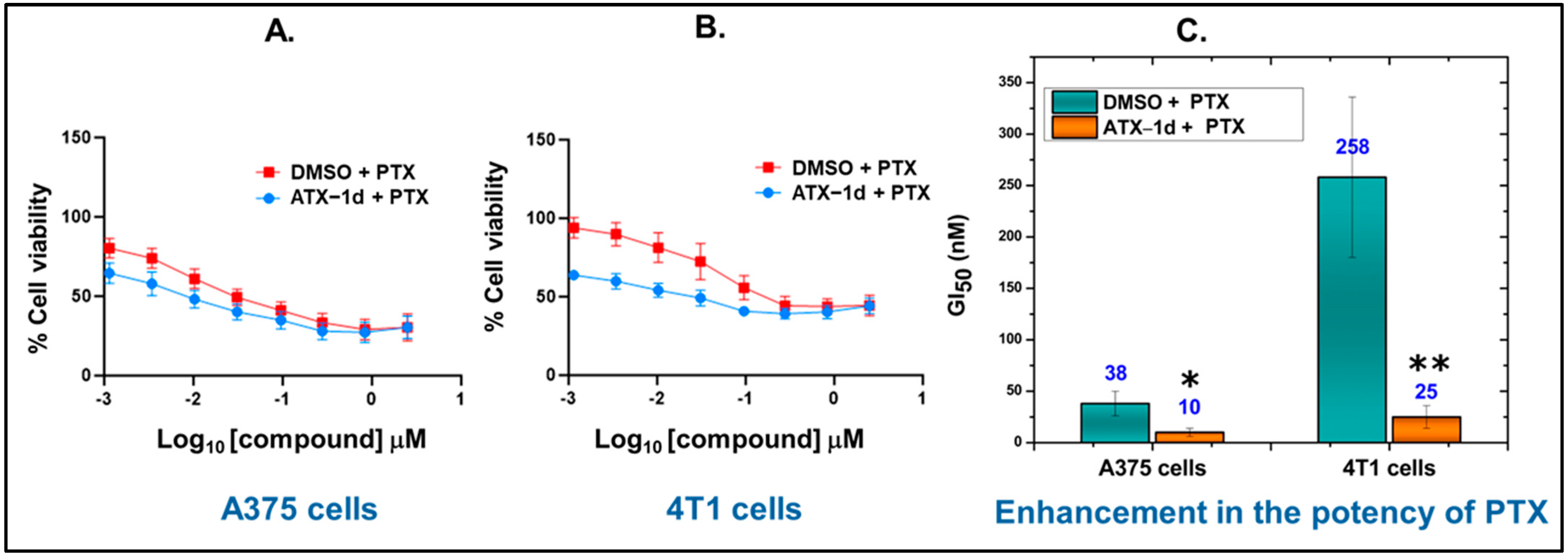
5.5. Cellular Activity of ATX-1d
5.6. ATX Inhibition by ATX-1d Improves Cellular Response to PTX
5.7. Conceptual DFT Parameters
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Perrakis, A.; Moolenaar, W.H. Autotaxin: Structure-Function and Signaling. J. Lipid Res. 2014, 55, 1010–1018. [Google Scholar] [CrossRef] [PubMed]
- van Meeteren, L.A.; Moolenaar, W.H. Regulation and Biological Activities of the Autotaxin–LPA Axis. Prog. Lipid Res. 2007, 46, 145–160. [Google Scholar] [CrossRef] [PubMed]
- Nakanaga, K.; Hama, K.; Aoki, J. Autotaxin—An LPA Producing Enzyme with Diverse Functions. J. Biochem. 2010, 148, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, M.; Yin, N.; Zhang, J. The Expression Regulation and Biological Function of Autotaxin. Cells 2021, 10, 939. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Murph, M.; Panupinthu, N.; Mills, G.B. ATX-LPA Receptor Axis in Inflammation and Cancer. Cell Cycle 2009, 8, 3695–3701. [Google Scholar] [CrossRef]
- Hemmings, D.G.; Brindley, D.N. Signalling by Lysophosphatidate and Its Health Implications. Essays Biochem. 2020, 64, 547–563. [Google Scholar] [CrossRef]
- Samadi, N.; Bekele, R.; Capatos, D.; Venkatraman, G.; Sariahmetoglu, M.; Brindley, D.N. Regulation of Lysophosphatidate Signaling by Autotaxin and Lipid Phosphate Phosphatases with Respect to Tumor Progression, Angiogenesis, Metastasis and Chemo-Resistance. Biochimie 2011, 93, 61–70. [Google Scholar] [CrossRef]
- Meng, L.; Zheng, Y.; Liu, H.; Fan, D. The Tumor Microenvironment: A Key Player in Multidrug Resistance in Cancer. Oncologie 2024, 26, 41–58. [Google Scholar] [CrossRef]
- Morin, P.J. The Microenvironment and Drug Resistance. In Cancer Drug Resistance; Humana Press: Totowa, NJ, USA, 2006; pp. 201–210. [Google Scholar]
- Dzobo, K.; Senthebane, D.A.; Dandara, C. The Tumor Microenvironment in Tumorigenesis and Therapy Resistance Revisited. Cancers 2023, 15, 376. [Google Scholar] [CrossRef]
- Lin, K.; Lee, S.C.; Dacheux, M.A.; Norman, D.D.; Balogh, A.; Bavaria, M.; Lee, H.; Tigyi, G. E2F7 Drives Autotaxin/Enpp2 Transcription via Chromosome Looping: Repression by P53 in Murine but Not in Human Carcinomas. FASEB J. 2023, 37, e23058. [Google Scholar] [CrossRef]
- Koirala, M.; DiPaola, M. Overcoming Cancer Resistance: Strategies and Modalities for Effective Treatment. Biomedicines 2024, 12, 1801. [Google Scholar] [CrossRef] [PubMed]
- Samadi, N.; Gaetano, C.; Goping, I.S.; Brindley, D.N. Autotaxin Protects MCF-7 Breast Cancer and MDA-MB-435 Melanoma Cells against Taxol-Induced Apoptosis. Oncogene 2009, 28, 1028–1039. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Wuest, M.; Benesch, M.G.K.; Dufour, J.; Zhao, Y.; Curtis, J.M.; Monjardet, A.; Heckmann, B.; Murray, D.; Wuest, F.; et al. Inhibition of Autotaxin with GLPG1690 Increases the Efficacy of Radiotherapy and Chemotherapy in a Mouse Model of Breast Cancer. Mol. Cancer Ther. 2020, 19, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Shuyu, E.; Tsukahara, R.; Valentine, W.J.; Durgam, G.; Gududuru, V.; Balazs, L.; Manickam, V.; Arsura, M.; Vanmiddlesworth, L.; et al. The Lysophosphatidic Acid Type 2 Receptor Is Required for Protection against Radiation-Induced Intestinal Injury. Gastroenterology 2007, 132, 1834–1851. [Google Scholar] [CrossRef]
- Weiner, J.A.; Chun, J. Schwann Cell Survival Mediated by the Signaling Phospholipid Lysophosphatidic Acid. Proc. Natl. Acad. Sci. USA 1999, 96, 5233–5238. [Google Scholar] [CrossRef]
- Liu, S.; Umezu-Goto, M.; Murph, M.; Lu, Y.; Liu, W.; Zhang, F.; Yu, S.; Stephens, L.C.; Cui, X.; Murrow, G.; et al. Expression of Autotaxin and Lysophosphatidic Acid Receptors Increases Mammary Tumorigenesis, Invasion, and Metastases. Cancer Cell 2009, 15, 539–550. [Google Scholar] [CrossRef]
- Yasuda, D.; Kobayashi, D.; Akahoshi, N.; Ohto-Nakanishi, T.; Yoshioka, K.; Takuwa, Y.; Mizuno, S.; Takahashi, S.; Ishii, S. Lysophosphatidic Acid–Induced YAP/TAZ Activation Promotes Developmental Angiogenesis by Repressing Notch Ligand Dll4. J. Clin. Investig. 2019, 129, 4332–4349. [Google Scholar] [CrossRef]
- Tigyi, G.J.; Yue, J.; Norman, D.D.; Szabo, E.; Balogh, A.; Balazs, L.; Zhao, G.; Lee, S.C. Regulation of Tumor Cell—Microenvironment Interaction by the Autotaxin-Lysophosphatidic Acid Receptor. Adv. Biol. Regul. 2019, 71, 183–193. [Google Scholar] [CrossRef]
- Venkatraman, G.; Benesch, M.G.K.; Tang, X.; Dewald, J.; McMullen, T.P.W.; Brindley, D.N. Lysophosphatidate Signaling Stabilizes Nrf2 and Increases the Expression of Genes Involved in Drug Resistance and Oxidative Stress Responses: Implications for Cancer Treatment. FASEB J. 2015, 29, 772–785. [Google Scholar] [CrossRef]
- Bekele, R.T.; Venkatraman, G.; Liu, R.-Z.; Tang, X.; Mi, S.; Benesch, M.G.K.; Mackey, J.R.; Godbout, R.; Curtis, J.M.; McMullen, T.P.W.; et al. Oxidative Stress Contributes to the Tamoxifen-Induced Killing of Breast Cancer Cells: Implications for Tamoxifen Therapy and Resistance. Sci. Rep. 2016, 6, 21164. [Google Scholar] [CrossRef]
- Su, S.-C.; Hu, X.; Kenney, P.A.; Merrill, M.M.; Babaian, K.N.; Zhang, X.-Y.; Maity, T.; Yang, S.-F.; Lin, X.; Wood, C.G. Autotaxin–Lysophosphatidic Acid Signaling Axis Mediates Tumorigenesis and Development of Acquired Resistance to Sunitinib in Renal Cell Carcinoma. Clin. Cancer Res. 2013, 19, 6461–6472. [Google Scholar] [CrossRef]
- Banerjee, S.; Lee, S.; Norman, D.D.; Tigyi, G.J. Designing Dual Inhibitors of Autotaxin-LPAR GPCR Axis. Molecules 2022, 27, 5487. [Google Scholar] [CrossRef]
- Li, S.; Wang, B.; Xu, Y.; Zhang, J. Autotaxin Is Induced by TSA through HDAC3 and HDAC7 Inhibition and Antagonizes the TSA-Induced Cell Apoptosis. Mol. Cancer 2011, 10, 18. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.; Wang, Y.; Qu, M.; Zhang, D.; Zhang, X.; Zhang, J. Combined Inhibition of EZH2 and the Autotaxin-LPA-LPA2 Axis Exerts Synergistic Antitumor Effects on Colon Cancer Cells. Cancer Lett. 2023, 566, 216226. [Google Scholar] [CrossRef]
- Centonze, M.; Di Conza, G.; Lahn, M.; Fabregat, I.; Dituri, F.; Gigante, I.; Serino, G.; Scialpi, R.; Carrieri, L.; Negro, R.; et al. Autotaxin Inhibitor IOA-289 Reduces Gastrointestinal Cancer Progression in Preclinical Models. J. Exp. Clin. Cancer Res. 2023, 42, 197. [Google Scholar] [CrossRef]
- Salgado-Polo, F.; Perrakis, A. The Structural Binding Mode of the Four Autotaxin Inhibitor Types That Differentially Affect Catalytic and Non-Catalytic Functions. Cancers 2019, 11, 1577. [Google Scholar] [CrossRef]
- Castagna, D.; Budd, D.C.; Macdonald, S.J.F.; Jamieson, C.; Watson, A.J.B. Development of Autotaxin Inhibitors: An Overview of the Patent and Primary Literature. J. Med. Chem. 2016, 59, 5604–5621. [Google Scholar] [CrossRef] [PubMed]
- Nishimasu, H.; Okudaira, S.; Hama, K.; Mihara, E.; Dohmae, N.; Inoue, A.; Ishitani, R.; Takagi, J.; Aoki, J.; Nureki, O. Crystal Structure of Autotaxin and Insight into GPCR Activation by Lipid Mediators. Nat. Struct. Mol. Biol. 2011, 18, 205–212. [Google Scholar] [CrossRef]
- Jones, S.B.; Pfeifer, L.A.; Bleisch, T.J.; Beauchamp, T.J.; Durbin, J.D.; Klimkowski, V.J.; Hughes, N.E.; Rito, C.J.; Dao, Y.; Gruber, J.M.; et al. Novel Autotaxin Inhibitors for the Treatment of Osteoarthritis Pain: Lead Optimization via Structure-Based Drug Design. ACS Med. Chem. Lett. 2016, 7, 857–861. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, Y.; Zhou, Q.; Fan, H.; Liu, X.; Hu, J. Recent Research Advances in ATX Inhibitors: An Overview of Primary Literature. Bioorg. Med. Chem. 2023, 90, 117374. [Google Scholar] [CrossRef]
- Halgren, T.A.; Nachbar, R.B. Merck Molecular Force Field. IV. Conformational Energies and Geometries for MMFF94. J. Comput. Chem. 1996, 17, 587–615. [Google Scholar] [CrossRef]
- Wolber, G.; Langer, T. LigandScout: 3-D Pharmacophores Derived from Protein-Bound Ligands and Their Use as Virtual Screening Filters. J. Chem. Inf. Model 2005, 45, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Cuozzo, J.W. A New Autotaxin Inhibitor for the Treatment of Idiopathic Pulmonary Fibrosis: A Clinical Candidate Discovered Using DNA-Encoded Chemistry. Available online: https://www.wwpdb.org/pdb?id=pdb_00006w35 (accessed on 12 June 2024).
- Chrencik, J.E.; Roth, C.B.; Terakado, M.; Kurata, H.; Omi, R.; Kihara, Y.; Warshaviak, D.; Nakade, S.; Asmar-Rovira, G.; Mileni, M.; et al. Crystal Structure of Human Lysophosphatidic Acid Receptor 1 in Complex with ONO-9910539. Cell 2015, 161, 1633–1643. [Google Scholar] [CrossRef] [PubMed]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-Pdb Viewer: An Environment for Comparative Protein Modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef] [PubMed]
- Korb, O.; Stützle, T.; Exner, T.E. Empirical Scoring Functions for Advanced Protein–Ligand Docking with PLANTS. J. Chem. Inf. Model. 2009, 49, 84–96. [Google Scholar] [CrossRef]
- Biovia, D.S.; Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Richmond, T.J. Discovery Studio Visualizer; Dassault Systèmes BIOVIA: San Diego, CA, USA, 2020. [Google Scholar]
- Bell, E.W.; Zhang, Y. DockRMSD: An Open-Source Tool for Atom Mapping and RMSD Calculation of Symmetric Molecules through Graph Isomorphism. J. Cheminform. 2019, 11, 40. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Huang, J.; MacKerell, A.D. CHARMM36 All-Atom Additive Protein Force Field: Validation Based on Comparison to NMR Data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM General Force Field: A Force Field for Drug-like Molecules Compatible with the CHARMM All-atom Additive Biological Force Fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Valiente, P.A.; Moreno, E. Gmx_MMPBSA: A New Tool to Perform End-State Free Energy Calculations with GROMACS. J. Chem. Theory Comput. 2021, 17, 6281–6291. [Google Scholar] [CrossRef]
- Onufriev, A.; Bashford, D.; Case, D.A. Exploring Protein Native States and Large-scale Conformational Changes with a Modified Generalized Born Model. Proteins Struct. Funct. Bioinform. 2004, 55, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Weiser, J.; Shenkin, P.S.; Still, W.C. Approximate Atomic Surfaces from Linear Combinations of Pairwise Overlaps (LCPO). J. Comput. Chem. 1999, 20, 217–230. [Google Scholar] [CrossRef]
- Fogolari, F.; Brigo, A.; Molinari, H. Protocol for MM/PBSA Molecular Dynamics Simulations of Proteins. Biophys. J. 2003, 85, 159–166. [Google Scholar] [CrossRef]
- Korth, M. Third-Generation Hydrogen-Bonding Corrections for Semiempirical QM Methods and Force Fields. J. Chem. Theory Comput. 2010, 6, 3808–3816. [Google Scholar] [CrossRef]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of Multiple Amber Force Fields and Development of Improved Protein Backbone Parameters. Proteins Struct. Funct. Bioinform. 2006, 65, 712–725. [Google Scholar] [CrossRef]
- Parrish, R.M.; Sherrill, C.D. Spatial Assignment of Symmetry Adapted Perturbation Theory Interaction Energy Components: The Atomic SAPT Partition. J. Chem. Phys. 2014, 141, 044115. [Google Scholar] [CrossRef] [PubMed]
- Parrish, R.M.; Parker, T.M.; Sherrill, C.D. Chemical Assignment of Symmetry-Adapted Perturbation Theory Interaction Energy Components: The Functional-Group SAPT Partition. J. Chem. Theory Comput. 2014, 10, 4417–4431. [Google Scholar] [CrossRef]
- Smith, D.G.A.; Burns, L.A.; Simmonett, A.C.; Parrish, R.M.; Schieber, M.C.; Galvelis, R.; Kraus, P.; Kruse, H.; Di Remigio, R.; Alenaizan, A.; et al. PSI4 1.4: Open-source software for high-throughput quantum chemistry. J. Chem. Phys. 2020, 152, 184108. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian Basis Sets for Use in Correlated Molecular Calculations. I. The Atoms Boron through Neon and Hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Gonthier, J.F.; Sherrill, C.D. Density-Fitted Open-Shell Symmetry-Adapted Perturbation Theory and Application to π -Stacking in Benzene Dimer Cation and Ionized DNA Base Pair Steps. J. Chem. Phys. 2016, 145, 134106. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Chattaraj, P.K.; Sarkar, U.; Roy, D.R. Electrophilicity Index. Chem. Rev. 2006, 106, 2065–2091. [Google Scholar] [CrossRef] [PubMed]
- Geerlings, P.; De Proft, F.; Langenaeker, W. Conceptual Density Functional Theory. Chem. Rev. 2003, 103, 1793–1874. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Maiti, D.; Das, R.; Prabakar, T.; Sen, S. Blue LED Induced Solvent-Free Multicomponent Reactions among Aryl Diazoacetates, Pyridine Derivatives and Maleimides: Direct Eco-Friendly Synthesis of Densely Functionalized Itaconimides. Green Chem. 2022, 24, 3001–3008. [Google Scholar] [CrossRef]
- Maiti, D.; Saha, A.; Guin, S.; Maiti, D.; Sen, S. Unveiling Catalyst-Free Electro-Photochemical Reactivity of Aryl Diazoesters and Facile Synthesis of Oxazoles, Imide-Fused Pyrroles and Tetrahydro-Epoxy-Pyridines via Carbene Radical Anions. Chem. Sci. 2023, 14, 6216–6225. [Google Scholar] [CrossRef]
- Khan, H.; Guha, S.; Baruah, M.; Yadav, S.; Maheshwari, S.; Sainani, S.; Maiti, D.; Sen, S. Blue LED Induced Three Component Reactions for the Generation of 4,6-Dioxo-hexahydro-1H-furo [3, 4-c] Pyrrole: Their Evaluation as Anticancer Agents through PARP-1 Inhibition. Chem. Asian J. 2023, 18, e202300420. [Google Scholar] [CrossRef]
- Singh, S.; Gremaud, L.; Sen, S.; Maiti, D. A Combination of Computational and Experimental Studies to Correlate Electronic Structure and Reactivity of Donor–Acceptor Singlet Carbenes. Synlett 2024, 35, 319–324. [Google Scholar] [CrossRef]
- Maurya, R.A.; Reddy, C.N.; Mani, G.S.; Kapure, J.S.; Adiyala, P.R.; Nanubolu, J.B.; Singarapu, K.K.; Kamal, A. Highly Diastereoselective Synthesis of Spiro[Cyclopropane-1,3′-Indolin]-2′-Ones via Catalyst-Free Cyclopropanation Using Ethyl Diazoacetate. Tetrahedron 2014, 70, 4709–4717. [Google Scholar] [CrossRef]
- Rath, S.; Maiti, D.; Modi, M.; Pal, P.; Munan, S.; Mohanty, B.; Bhatia, A.; Bhowal, R.; Priyadarshini, R.; Samanta, A.; et al. Metal-Free Synthesis and Study of Glycine Betaine Derivatives in Water for Antimicrobial and Anticancer Applications. iScience 2023, 26, 107285. [Google Scholar] [CrossRef]
- North, E.J.; Howard, A.L.; Wanjala, I.W.; Pham, T.C.T.; Baker, D.L.; Parrill, A.L. Pharmacophore Development and Application Toward the Identification of Novel, Small-Molecule Autotaxin Inhibitors. J. Med. Chem. 2010, 53, 3095–3105. [Google Scholar] [CrossRef]
- Chen, Y.-C. Beware of Docking! Trends Pharmacol. Sci. 2015, 36, 78–95. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.K.; Koča, J. Assessing the Performance of MM/PBSA, MM/GBSA, and QM–MM/GBSA Approaches on Protein/Carbohydrate Complexes: Effect of Implicit Solvent Models, QM Methods, and Entropic Contributions. J. Phys. Chem. B 2018, 122, 8113–8121. [Google Scholar] [CrossRef] [PubMed]
- Wichapong, K.; Rohe, A.; Platzer, C.; Slynko, I.; Erdmann, F.; Schmidt, M.; Sippl, W. Application of Docking and QM/MM-GBSA Rescoring to Screen for Novel Myt1 Kinase Inhibitors. J. Chem. Inf. Model 2014, 54, 881–893. [Google Scholar] [CrossRef] [PubMed]
- Pu, C.; Yan, G.; Shi, J.; Li, R. Assessing the Performance of Docking Scoring Function, FEP, MM-GBSA, and QM/MM-GBSA Approaches on a Series of PLK1 Inhibitors. Medchemcomm 2017, 8, 1452–1458. [Google Scholar] [CrossRef]
- Bamdad, F.; Farrokhpour, H.; Ashrafizaadeh, M.; Najafi, B. Decomposition of the Interaction Energy of Several Flavonoids with Escherichia Coli DNA Gyr Using the SAPT (DFT) Method: The Relation between the Interaction Energy Components, Ligand Structure, and Biological Activity. Biochim. Biophys. Acta Gen. Subj. 2022, 1866, 130111. [Google Scholar] [CrossRef]
- Stasyuk, O.A.; Jakubec, D.; Vondrášek, J.; Hobza, P. Noncovalent Interactions in Specific Recognition Motifs of Protein–DNA Complexes. J. Chem. Theory Comput. 2017, 13, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Ding, T.; Karlov, D.S.; Pino-Angeles, A.; Tikhonova, I.G. Intermolecular Interactions in G Protein-Coupled Receptor Allosteric Sites at the Membrane Interface from Molecular Dynamics Simulations and Quantum Chemical Calculations. J. Chem. Inf. Model 2022, 62, 4736–4747. [Google Scholar] [CrossRef]
- Matczak, P. Description of Weak Halogen Bonding Using Various Levels of Symmetry-Adapted Perturbation Theory Combined with Effective Core Potentials. J. Chem. 2017, 2017, 9031494. [Google Scholar] [CrossRef]
- Pitoňák, M.; Riley, K.E.; Neogrády, P.; Hobza, P. Highly Accurate CCSD(T) and DFT–SAPT Stabilization Energies of H-Bonded and Stacked Structures of the Uracil Dimer. Chem. Phys. Chem. 2008, 9, 1636–1644. [Google Scholar] [CrossRef]
- Jansen, G. Symmetry-adapted Perturbation Theory Based on Density Functional Theory for Noncovalent Interactions. WIREs Comput. Mol. Sci. 2014, 4, 127–144. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. PkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Gfeller, D.; Grosdidier, A.; Wirth, M.; Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: A Web Server for Target Prediction of Bioactive Small Molecules. Nucleic Acids Res. 2014, 42, W32–W38. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Norman, D.D.; Lee, S.C.; Parrill, A.L.; Pham, T.C.T.; Baker, D.L.; Tigyi, G.J.; Miller, D.D. Highly Potent Non-Carboxylic Acid Autotaxin Inhibitors Reduce Melanoma Metastasis and Chemotherapeutic Resistance of Breast Cancer Stem Cells. J. Med. Chem. 2017, 60, 1309–1324. [Google Scholar] [CrossRef] [PubMed]
- Iwaki, Y.; Ohhata, A.; Nakatani, S.; Hisaichi, K.; Okabe, Y.; Hiramatsu, A.; Watanabe, T.; Yamamoto, S.; Nishiyama, T.; Kobayashi, J.; et al. ONO-8430506: A Novel Autotaxin Inhibitor That Enhances the Antitumor Effect of Paclitaxel in a Breast Cancer Model. ACS Med. Chem. Lett. 2020, 11, 1335–1341. [Google Scholar] [CrossRef]
- Brindley, D.N.; Lin, F.-T.; Tigyi, G.J. Role of the Autotaxin–Lysophosphatidate Axis in Cancer Resistance to Chemotherapy and Radiotherapy. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2013, 1831, 74–85. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Residues | van der Waals | Electrostatic | Polar Solv. | Non-Polar Solv. | TOTAL | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Avg. | Std. Err. of Mean | Avg. | Std. Err. of Mean | Avg. | Std. Err. of Mean | Avg. | Std. Err. of Mean | Avg. | Std. Err. of Mean | |
| GLN:67 | −0.09 | 0.00 | −0.46 | 0.00 | 0.45 | 0.00 | −0.04 | 0.00 | −0.14 | 0.00 |
| LEU:79 | −1.29 | 0.01 | 0.06 | 0.00 | −0.21 | 0.00 | −1.22 | 0.00 | −2.66 | 0.01 |
| SER:82 | −0.47 | 0.00 | −0.06 | 0.00 | −0.08 | 0.00 | −0.43 | 0.00 | −1.03 | 0.01 |
| TYR:83 | −0.78 | 0.01 | −0.55 | 0.01 | 0.13 | 0.00 | −0.65 | 0.01 | −1.85 | 0.02 |
| PHE:211 | −1.52 | 0.01 | −0.39 | 0.00 | 0.11 | 0.00 | −1.33 | 0.01 | −3.13 | 0.01 |
| TYR:215 | −0.93 | 0.01 | −0.20 | 0.00 | 0.02 | 0.00 | −0.72 | 0.00 | −1.83 | 0.01 |
| LEU:244 | −0.74 | 0.00 | 0.21 | 0.00 | −0.26 | 0.00 | −0.70 | 0.00 | −1.48 | 0.01 |
| ARG:245 | −0.09 | 0.00 | 0.20 | 0.00 | −0.20 | 0.00 | 0.00 | 0.00 | −0.10 | 0.01 |
| LYS:249 | −1.83 | 0.00 | −5.32 | 0.03 | 4.15 | 0.02 | −1.54 | 0.00 | −4.53 | 0.03 |
| PHE:250 | −1.78 | 0.00 | −0.27 | 0.00 | 0.17 | 0.00 | −1.45 | 0.00 | −3.34 | 0.01 |
| TRP:255 | −2.63 | 0.01 | −0.60 | 0.00 | −0.13 | 0.00 | −1.92 | 0.01 | −5.28 | 0.01 |
| TRP:261 | −1.43 | 0.01 | −0.10 | 0.00 | −0.09 | 0.00 | −1.25 | 0.00 | −2.86 | 0.01 |
| ILE:262 | −0.26 | 0.00 | 0.00 | 0.00 | 0.01 | 0.00 | −0.22 | 0.00 | −0.46 | 0.00 |
| THR:273 | −0.18 | 0.00 | −0.02 | 0.00 | 0.10 | 0.00 | −0.15 | 0.00 | −0.25 | 0.00 |
| PHE:274 | −0.41 | 0.00 | −0.08 | 0.00 | 0.06 | 0.00 | −0.18 | 0.00 | −0.60 | 0.00 |
| PHE:275 | −2.44 | 0.01 | −0.66 | 0.00 | 0.06 | 0.00 | −2.02 | 0.00 | −5.06 | 0.01 |
| Compounds | van der Waals | Electrostatics (SCF) | Polar Solv. | Non-Polar Solv. | TOTAL | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Avg. | Std. Err. of Mean | Avg. | Std. Err. of Mean | Avg. | Std. Err. of Mean | Avg. | Std. Err. of Mean | Avg. | Std. Err. of Mean | |
| SKV (native ligand, PDB ID: 6W35) vs. ATX | −5.85 | 0.01 | −68.09 | 0.11 | 58.78 | 0.11 | −7.79 | 0.01 | −22.95 | 0.06 |
| ATX−1d vs. ATX | −7.06 | 0.06 | −65.58 | 0.43 | 69.86 | 0.46 | −5.6 | 0.02 | −8.37 | 0.11 |
| Compounds | Total SAPT0 Protein–Ligand Interaction Energy (in kcal/mol) |
|---|---|
| SKV (native ligand, PDB ID: 6W35) vs. ATX | −65.80 |
| ATX-1d vs. ATX | −49.78 |
| 4T1 Breast Cancer Cells | A375 Melanoma Cells | ||
|---|---|---|---|
| Compounds | GI50 (in nM) | Compounds | GI50 (in nM) |
| Paclitaxel | 62 ± 18 | Paclitaxel | 4 ± 1 |
| ATX-1d | >20,000 | ATX-1d | >20,000 |
| Compound | ATX−1d |
|---|---|
| ELUMO | −0.041 |
| EHOMO | −0.299 |
| Energy Gap (Eg) {Eg = ELUMO − EHOMO} | 0.258 |
| Ionization Potential (IP) {IP = −EHOMO} | 0.299 |
| Electron Affinity (EA) {EA = −ELUMO} | 0.041 |
| Electronegativity (χ) {χ = | 0.170 |
| Chemical Potential (μ) {μ = | −0.170 |
| Global Hardness (η) {η = } | 0.129 |
| Global Softness (S) {S = } | 3.875 |
| Electrophilicity Index (ω) {ω = } | 0.112 |
| Nucleophilicity Index (ε) {ε = } | 8.964 |
| Maximum Charge Transfer Index (ΔNmax) {ΔNmax = } | 1.315 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rai, P.; Clark, C.J.; Womack, C.B.; Dearing, C.; Thammathong, J.; Norman, D.D.; Tigyi, G.J.; Sen, S.; Bicker, K.; Weissmiller, A.M.; et al. Novel Autotaxin Inhibitor ATX-1d Significantly Enhances Potency of Paclitaxel—An In Silico and In Vitro Study. Molecules 2024, 29, 4285. https://doi.org/10.3390/molecules29184285
Rai P, Clark CJ, Womack CB, Dearing C, Thammathong J, Norman DD, Tigyi GJ, Sen S, Bicker K, Weissmiller AM, et al. Novel Autotaxin Inhibitor ATX-1d Significantly Enhances Potency of Paclitaxel—An In Silico and In Vitro Study. Molecules. 2024; 29(18):4285. https://doi.org/10.3390/molecules29184285
Chicago/Turabian StyleRai, Prateek, Christopher J. Clark, Carl B. Womack, Curtis Dearing, Joshua Thammathong, Derek D. Norman, Gábor J. Tigyi, Subhabrata Sen, Kevin Bicker, April M. Weissmiller, and et al. 2024. "Novel Autotaxin Inhibitor ATX-1d Significantly Enhances Potency of Paclitaxel—An In Silico and In Vitro Study" Molecules 29, no. 18: 4285. https://doi.org/10.3390/molecules29184285
APA StyleRai, P., Clark, C. J., Womack, C. B., Dearing, C., Thammathong, J., Norman, D. D., Tigyi, G. J., Sen, S., Bicker, K., Weissmiller, A. M., & Banerjee, S. (2024). Novel Autotaxin Inhibitor ATX-1d Significantly Enhances Potency of Paclitaxel—An In Silico and In Vitro Study. Molecules, 29(18), 4285. https://doi.org/10.3390/molecules29184285