
An Unprecedented 4,8-Cycloeudesmane, Further New Sesquiterpenoids, a Triterpene, Steroids, and a Lignan from the Resin of Commiphora myrrha and Their Anti-Inflammatory Activity In Vitro


Abstract
:1. Introduction
2. Results
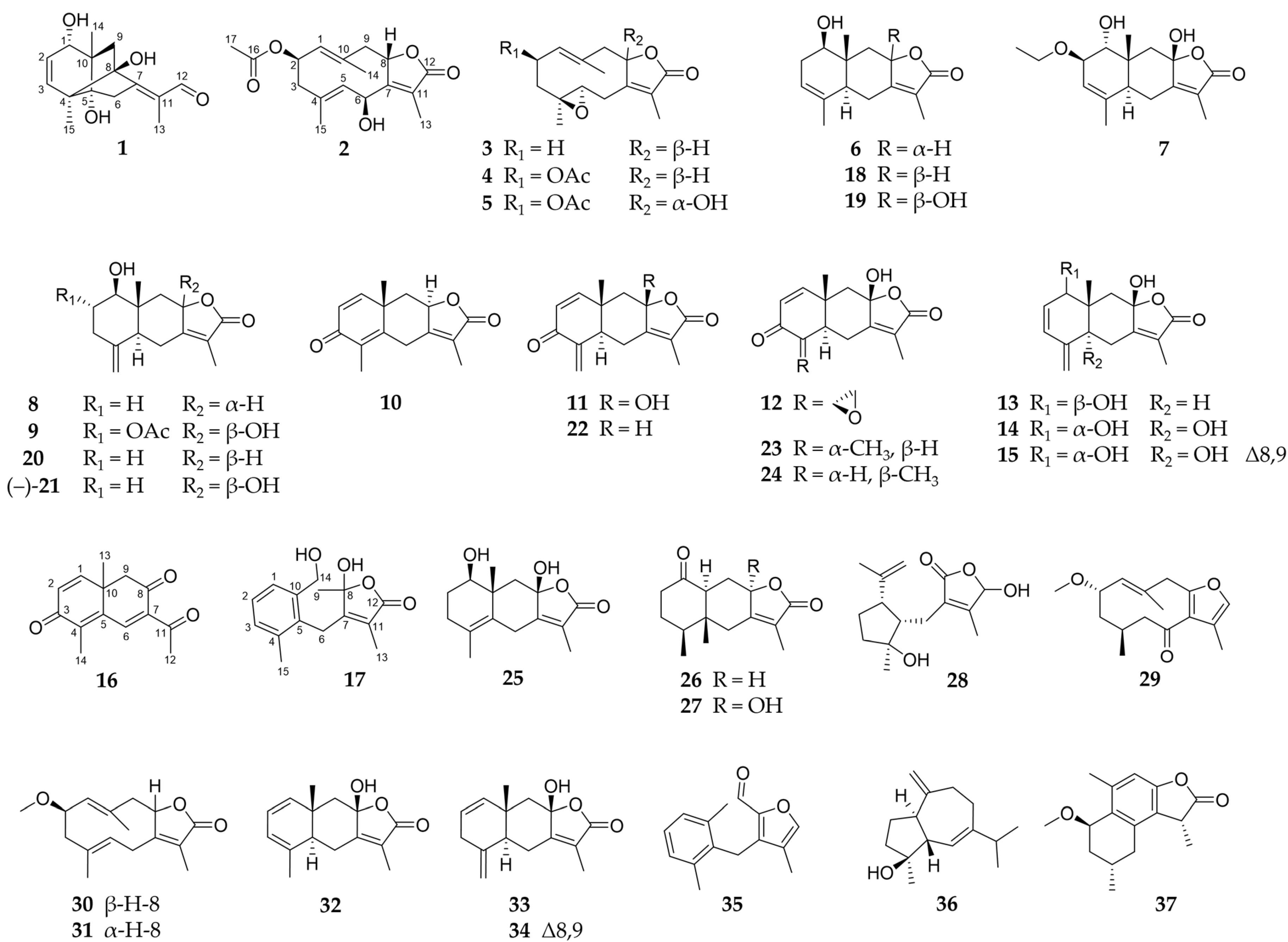
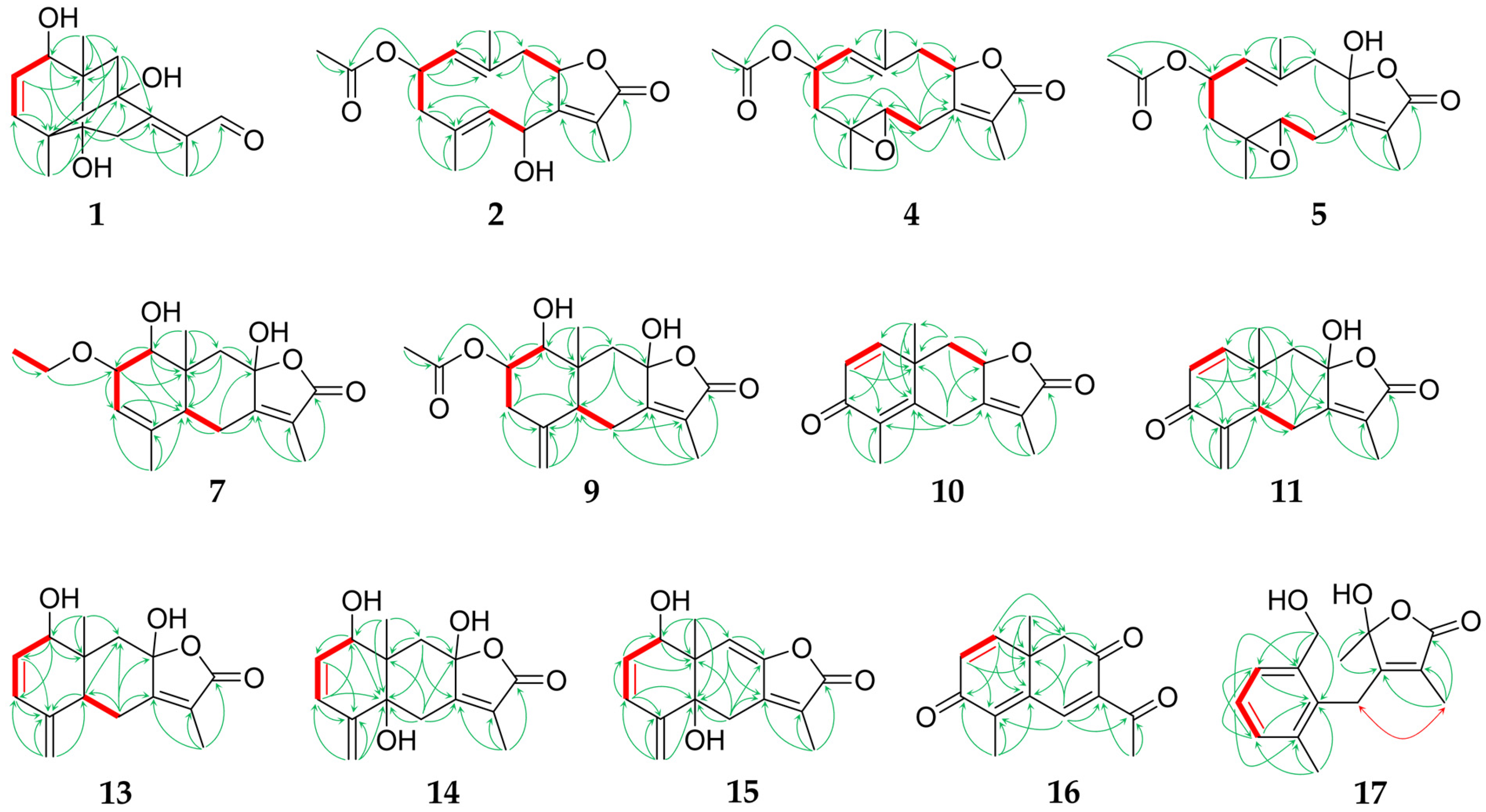
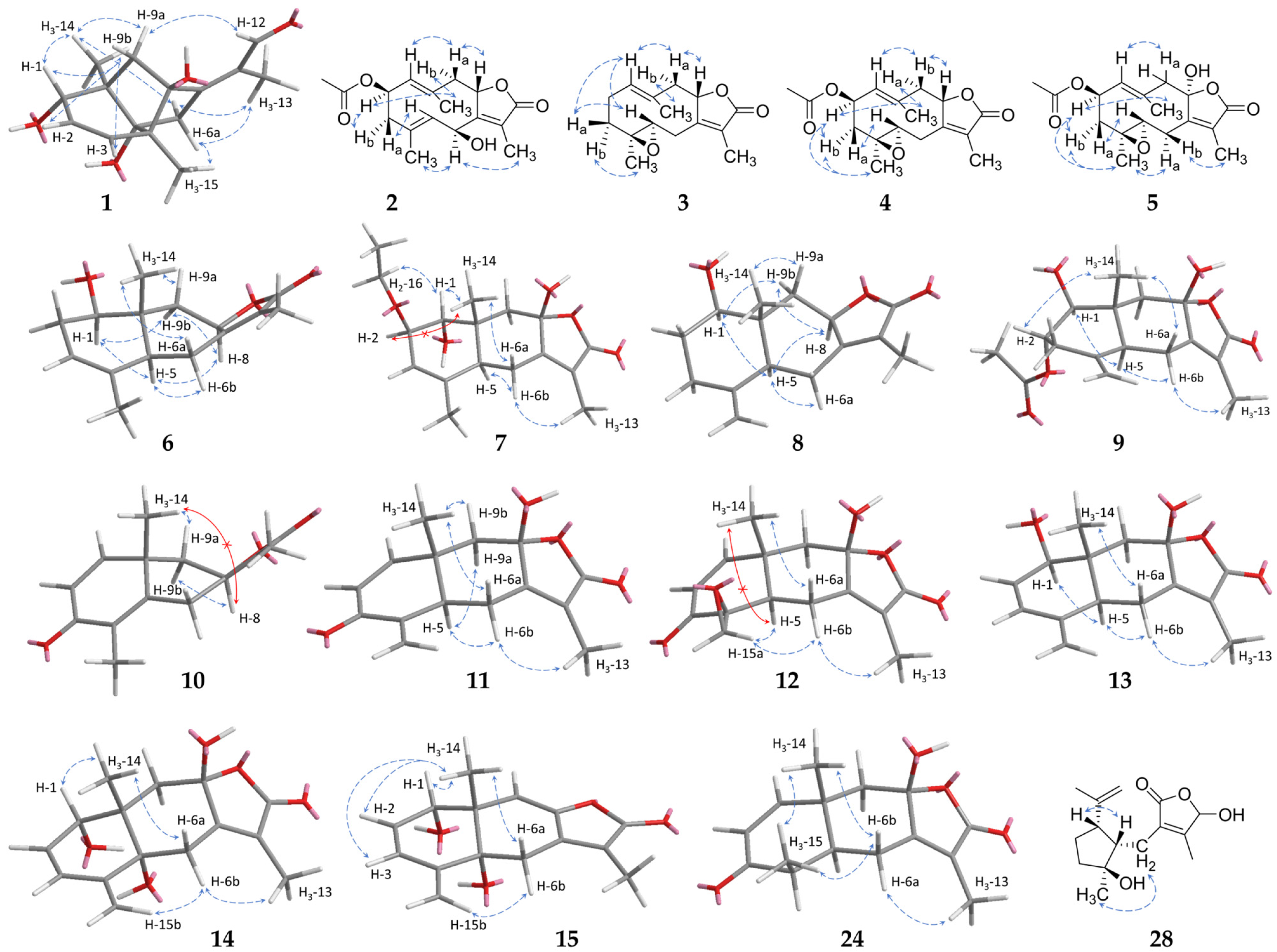
2.1. Sesquiterpenoids
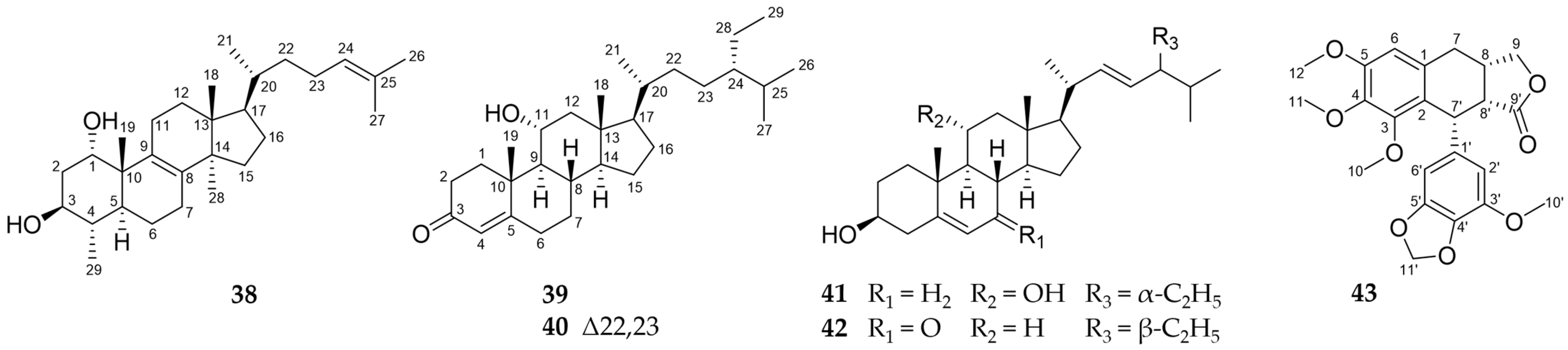
2.2. Further Structural Types
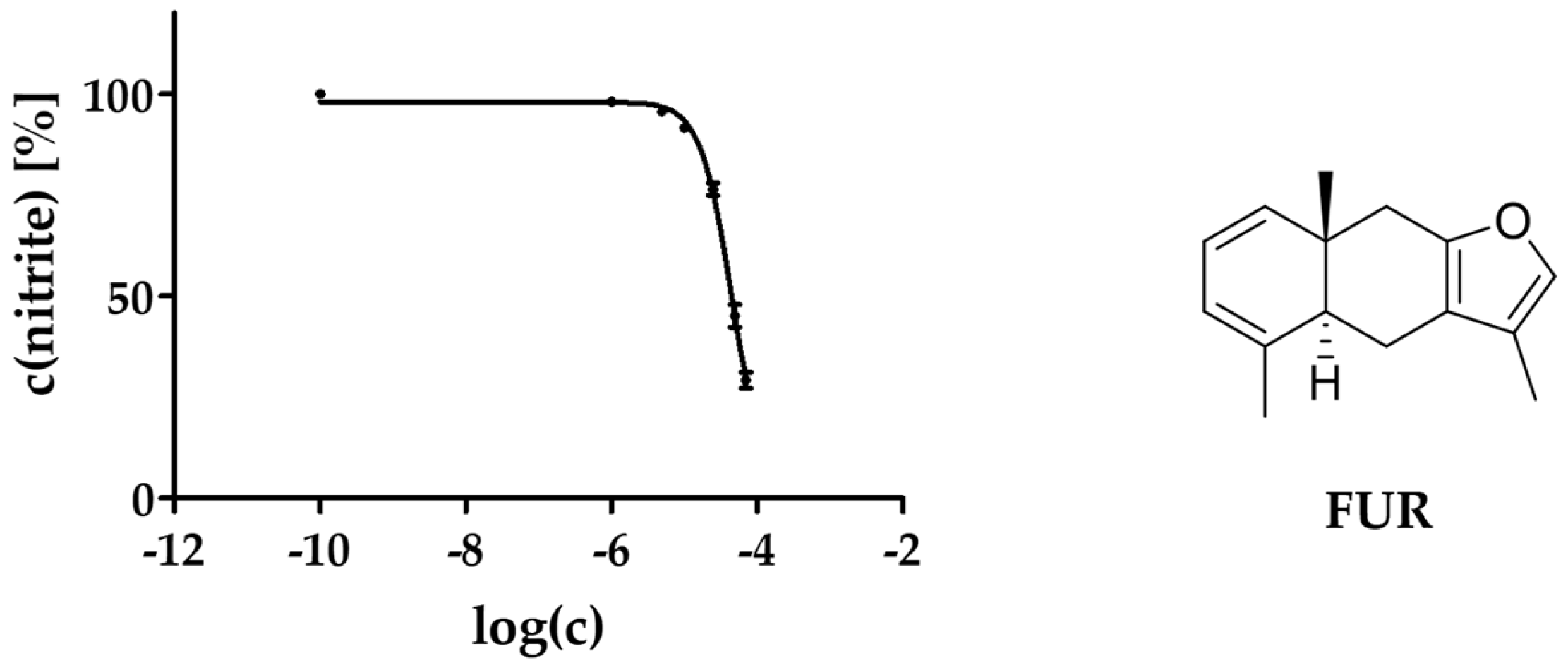
2.3. Activity against NO Production
3. Discussion and Conclusions
3.1. Structural Diversity
3.2. Anti-Inflammatory Activity
4. Materials and Methods
4.1. Chemicals
4.2. Plant Material and Extraction
4.3. Isolation
4.3.1. Liquid–Liquid Partition
4.3.2. Flash Chromatography and CPC of the HEP Fraction
4.3.3. Solid Phase Extraction and Flash Chromatography of the MeOH Fraction
4.3.4. CPC of Fraction M1.2
4.3.5. Flash Chromatography of M1.4 and the Resulting Fraction M1.4R1F7
4.3.6. Thin-Layer Chromatography (TLC)
4.3.7. Preparative HPLC
4.4. Compound Characterisation
4.5. Isolated Compounds
4.6. RAW 264.7 Experiments
4.6.1. MTT Assay
4.6.2. Griess Assay
4.6.3. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mahr, D. Commiphora: An introduction to the genus. Cactus Succul. J. 2012, 84, 140–154. [Google Scholar] [CrossRef]
- Tucker, A.O. Frankincense and myrrh. Econ. Bot. 1986, 40, 425–433. [Google Scholar] [CrossRef]
- Hanuš, L.O.; Řezanka, T.; Dembitsky, V.M.; Moussaieff, A. Myrrh-Commiphora chemistry. Biomed. Pap. 2005, 149, 3–28. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-W.; Zhang, M.-Y.; Yan, Y.-M.; Wei, X.-Y.; Dong, L.; Zhu, Y.-X.; Cheng, Y.-X. Characterization of sesquiterpene dimers from Resina Commiphora that promote adipose-derived stem cell proliferation and differentiation. J. Org. Chem. 2018, 83, 2725–2733. [Google Scholar] [CrossRef] [PubMed]
- Kuck, K.; Unterholzner, A.; Lipowicz, B.; Schwindl, S.; Jürgenliemk, G.; Schmidt, T.J.; Heilmann, J. Terpenoids from myrrh and their cytotoxic activity against HeLa cells. Molecules 2023, 28, 1637. [Google Scholar] [CrossRef]
- Wang, C.-C.; Liang, N.-Y.; Xia, H.; Wang, R.-Y.; Zhang, Y.-F.; Huo, H.-X.; Zhao, Y.-F.; Song, Y.-L.; Zheng, J.; Tu, P.-F. Cytotoxic sesquiterpenoid dimers from the resin of Commiphora myrrha Engl. Phytochemistry 2022, 204, 113443. [Google Scholar] [CrossRef]
- Ge, C.-Y.; Zhang, J.-L. Bioactive sesquiterpenoids and steroids from the resinous exudates of Commiphora myrrha. Nat. Prod. Res. 2019, 33, 309–315. [Google Scholar] [CrossRef]
- El-Razek, A.; Mohamed, T.A.; Abdel-Halim, S.; Bata, S.M.; Kubacy, T.M. Comprehensive NMR reassignments of lignans derived from Commiphora myrrha. Egypt. J. Chem. 2023, 66, 45–57. [Google Scholar] [CrossRef]
- Shen, T.; Li, G.-H.; Wang, X.-N.; Lou, H.-X. The genus Commiphora: A review of its traditional uses, phytochemistry and pharmacology. J. Ethnopharmacol. 2012, 142, 319–330. [Google Scholar] [CrossRef]
- Tariq, M.; Ageel, A.M.; Al-Yahya, M.A.; Mossa, J.S.; Al-Said, M.S.; Parmar, N.S. Anti-inflammatory activity of Commiphora molmol. Agents Actions 1985, 17, 381–382. [Google Scholar] [CrossRef]
- Su, S.; Duan, J.; Chen, T.; Huang, X.; Shang, E.; Yu, L.; Wei, K.; Zhu, Y.; Guo, J.; Guo, S.; et al. Frankincense and myrrh suppress inflammation via regulation of the metabolic profiling and the MAPK signaling pathway. Sci. Rep. 2015, 5, 13668. [Google Scholar] [CrossRef] [PubMed]
- Hamad, G.M.; Taha, T.H.; Alshehri, A.; El-Deeb, N.M. Myrrh as a functional food with therapeutic properties against colon cancer in traditional meals. J. Food Process. Preserv. 2017, 41, e12963. [Google Scholar] [CrossRef]
- Rahman, M.M.; Garvey, M.; Piddock, L.J.V.; Gibbons, S. Antibacterial terpenes from the oleo-resin of Commiphora molmol (Engl.). Phytother. Res. 2008, 22, 1356–1360. [Google Scholar] [CrossRef] [PubMed]
- Alhussaini, M.S.; Saadabi, A.M.; Mohammed, I. Alghonaim; Khalid Elfakki Ibrahim. An evaluation of the antimicrobial activity of Commiphora myrrha Nees (Engl.) oleo-gum resins from Saudi Arabia. J. Med. Sci. 2015, 15, 198–203. [Google Scholar] [CrossRef]
- Dolara, P.; Luceri, C.; Ghelardini, C.; Monserrat, C.; Aiolli, S.; Luceri, F.; Lodovici, M.; Menichetti, S.; Romanelli, M.N. Analgesic effects of myrrh. Nature 1996, 379, 29. [Google Scholar] [CrossRef]
- Germano, A.; Occhipinti, A.; Barbero, F.; Maffei, M.E. A pilot study on bioactive constituents and analgesic effects of MyrLiq®, a Commiphora myrrha extract with a high furanodiene content. BioMed Res. Int. 2017, 2017, 3804356. [Google Scholar] [CrossRef]
- Fatani, A.J.; Alrojayee, F.S.; Parmar, M.Y.; Abuohashish, H.M.; Ahmed, M.M.; Al-Rejaie, S.S. Myrrh attenuates oxidative and inflammatory processes in acetic acid-induced ulcerative colitis. Exp. Ther. Med. 2016, 12, 730–738. [Google Scholar] [CrossRef]
- Langhorst, J.; Varnhagen, I.; Schneider, S.B.; Albrecht, U.; Rueffer, A.; Stange, R.; Michalsen, A.; Dobos, G.J. Randomised clinical trial: A herbal preparation of myrrh, chamomile and coffee charcoal compared with mesalazine in maintaining remission in ulcerative colitis—A double-blind, double-dummy study. Aliment. Pharmacol. Ther. 2013, 38, 490–500. [Google Scholar] [CrossRef]
- Fraternale, D.; Sosa, S.; Ricci, D.; Genovese, S.; Messina, F.; Tomasini, S.; Montanari, F.; Marcotullio, M.C. Anti-inflammatory, antioxidant and antifungal furanosesquiterpenoids isolated from Commiphora erythraea (Ehrenb.) Engl. resin. Fitoterapia 2011, 82, 654–661. [Google Scholar] [CrossRef]
- Messina, F.; Gigliarelli, G.; Palmier, A.; C Marcotullio, M. Furanodienone: An emerging bioactive furanosesquiterpenoid. Curr. Org. Chem. 2017, 21, 305–310. [Google Scholar] [CrossRef]
- Kuck, K.; Jürgenliemk, G.; Lipowicz, B.; Heilmann, J. Sesquiterpenes from myrrh and their ICAM-1 inhibitory activity in vitro. Molecules 2020, 26, 42. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Wei, X.-C.; Xu, X.-R.; Zhang, H.-Z.; Luo, C.-H.; Feng, B.; Xu, R.-C.; Zhao, S.-Y.; Du, X.-J.; Han, L.; et al. Seeing the unseen of the combination of two natural resins, frankincense and myrrh: Changes in chemical constituents and pharmacological activities. Molecules 2019, 24, 3076. [Google Scholar] [CrossRef] [PubMed]
- Bogdan, C. Nitric oxide and the immune response. Nat. Immunol. 2001, 2, 907–916. [Google Scholar] [CrossRef] [PubMed]
- Boughton-Smith, N.K.; Evans, S.M.; Whittle, B.J.; Moncada, S.; Hawkey, C.J.; Cole, A.T.; Balsitis, M. Nitric oxide synthase activity in ulcerative colitis and Crohn’s disease. Lancet 1993, 342, 338-e2. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.-W.; Cheah, K.-P.; Lin, C.-W.; Li, J.-S.; Yu, W.-Y.; Chang, M.L.; Yeh, G.-C.; Chen, S.-H.; Choy, C.-S.; Hu, C.-M. Myrrh mediates haem oxygenase-1 expression to suppress the lipopolysaccharide-induced inflammatory response in RAW264.7 macrophages. J. Pharm. Pharmacol. 2011, 63, 1211–1218. [Google Scholar] [CrossRef]
- Teng, F.; Zhong, H.-M.; Chen, C.-X.; Liu, H.-Y. Four new eudesmane sesquiterpenoid lactones from Chloranthus serratus. HCA 2009, 92, 1298–1303. [Google Scholar] [CrossRef]
- El-Gamal, A.A. Sesquiterpene lactones from Smyrnium olusatrum. Phytochemistry 2001, 57, 1197–1200. [Google Scholar] [CrossRef]
- Chang, F.-R.; Hsieh, T.-J.; Huang, T.-L.; Chen, C.-Y.; Kuo, R.-Y.; Chang, Y.-C.; Chiu, H.-F.; Wu, Y.-C. Cytotoxic constituents of the stem bark of Neolitsea acuminatissima. J. Nat. Prod. 2002, 65, 255–258. [Google Scholar] [CrossRef]
- Ohno, T.; Nagatsu, A.; Nakagawa, M.; Inoue, M.; Li, Y.-M.; Minatoguchi, S.; Mizukami, H.; Fujiwara, H. New sesquiterpene lactones from water extract of the root of Lindera strychnifolia with cytotoxicity against the human small cell lung cancer cell, SBC-3. Tetrahedron Lett. 2005, 46, 8657–8660. [Google Scholar] [CrossRef]
- Jang, H.-J.; Oh, H.-M.; Hwang, J.T.; Kim, M.-H.; Lee, S.; Jung, K.; Kim, Y.-H.; Lee, S.W.; Rho, M.-C. Eudesmane-type sesquiterpenoids from Salvia plebeia inhibit IL-6-induced STAT3 activation. Phytochemistry 2016, 130, 335–342. [Google Scholar] [CrossRef]
- Liu, Q.; Ahn, J.H.; Kim, S.B.; Lee, C.; Hwang, B.Y.; Lee, M.K. Sesquiterpene lactones from the roots of Lindera strychnifolia. Phytochemistry 2013, 87, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ma, J.; Wang, Y.; Donkor, P.O.; Li, Q.; Gao, S.; Hou, Y.; Xu, Y.; Cui, J.; Ding, L. Eudesmane-type sesquiterpenes from Curcuma phaeocaulis and their inhibitory activities on nitric oxide production in RAW 264.7 cells. Eur. J. Org. Chem. 2014, 2014, 5540–5548. [Google Scholar] [CrossRef]
- Buděšínský, M.; Holub, M.; Šaman, D.; Smítalová, Z.; Ulubelen, A.; Öksüz, S. Structure of istanbulin A and istanbulin B—Two sesquiterpenic lactones from Smyrnium olusatrum L. Collect. Czech. Chem. Commun. 1984, 49, 1311–1317. [Google Scholar] [CrossRef]
- Xu, Y.-J.; Tang, C.-P.; Tan, M.-J.; Ke, C.-Q.; Wu, T.; Ye, Y. Sesquiterpenoids and diterpenoids from Chloranthus anhuiensis. Chem. Biodivers. 2010, 7, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, G.; Rohmer, M.; Beck, J.P.; Anton, R. 7-Oxo-, 7α-hydroxy- and 7β-hydroxysterols from Euphorbia fischeriana. Phytochemistry 1980, 19, 2213–2215. [Google Scholar] [CrossRef]
- Shen, T.; Zhang, L.; Wang, Y.-Y.; Fan, P.-H.; Wang, X.-N.; Lin, Z.-M.; Lou, H.-X. Steroids from Commiphora mukul display antiproliferative effect against human prostate cancer PC3 cells via induction of apoptosis. Bioorg. Med. Chem. Lett. 2012, 22, 4801–4806. [Google Scholar] [CrossRef]
- Brieskorn, C.H.; Noble, P. Drei neue Furanogermacrene aus Myrrhe. Tetrahedron Lett. 1980, 21, 1511–1514. [Google Scholar] [CrossRef]
- Santoro, E.; Messina, F.; Marcotullio, M.C.; Superchi, S. Absolute configuration of bioactive furanogermacrenones from Commiphora erythraea (Ehrenb) Engl. by computational analysis of their chiroptical properties. Tetrahedron 2014, 70, 8033–8039. [Google Scholar] [CrossRef]
- Greve, H.L.; Kaiser, M.; Schmidt, T.J. Investigation of antiplasmodial effects of terpenoid compounds isolated from myrrh. Planta Med. 2020, 86, 643–654. [Google Scholar] [CrossRef]
- Takeda, K.; Horibe, I.; Minato, H. Components of the root of Lindera strychnifolia Vill. Part XIV. Sesquiterpene lactones from the root of Lindera strychnifolia Vill. J. Chem. Soc. 1968, 569–572. [Google Scholar] [CrossRef]
- Tada, H.; Minato, H.; Takeda, K. Components of the root of Lindera strychnifolia Vill. Part XVIII. Neosericenyl acetate and dehydrolindestrenolide. J. Chem. Soc. C 1971, 1070–1073. [Google Scholar] [CrossRef]
- Oshima, Y.; Iwakawa, T.; Hikino, H. Alismol and alismoxide, sesquiterpenoids of Alisma rhizomes. Phytochemistry 1983, 22, 183–185. [Google Scholar] [CrossRef]
- Hikino, H.; Konno, C.; Agatsuma, K.; Takemoto, T.; Horibe, I.; Tori, K.; Ueyama, M.; Takeda, K. Sesquiterpenoids. Part XLVII. Structure, configuration, conformation, and thermal rearrangement of furanodienone, isofuranodienone, curzerenone, epicurzerenone, and pyrocurzerenone, sesquiterpenoids of Curcuma zedoaria. J. Chem. Soc. Perkin Trans. 1 1975, 478–484. [Google Scholar] [CrossRef]
- Brieskorn, C.H.; Noble, P. Furanosesquiterpenes from the essential oil of myrrh. Phytochemistry 1983, 22, 1207–1211. [Google Scholar] [CrossRef]
- Zhu, N.; Sheng, S.; Sang, S.; Rosen, R.T.; Ho, C.-T. Isolation and characterization of several aromatic sesquiterpenes from Commiphora myrrha. Flavour Fragr. J. 2003, 18, 282–285. [Google Scholar] [CrossRef]
- Zhu, N.; Kikuzaki, H.; Sheng, S.; Sang, S.; Rafi, M.M.; Wang, M.; Nakatani, N.; DiPaola, R.S.; Rosen, R.T.; Ho, C.-T. Furanosesquiterpenoids of Commiphora myrrha. J. Nat. Prod. 2001, 64, 1460–1462. [Google Scholar] [CrossRef]
- Stahl, E.; Datta, S.N. Neue sesquiterpenoide Inhaltsstoffe der Gundelrebe (Glechoma hederacea L.). Liebigs Ann. Chem. 1972, 757, 23–32. [Google Scholar] [CrossRef]
- Tashkhodzhaev, B.; Abduazimov, B.K. Stereochemistry of sesquiterpenes of the germacrane type. Chem. Nat. Compd. 1997, 33, 382–388. [Google Scholar] [CrossRef]
- Chaturvedula, V.P.; Schilling, J.K.; Miller, J.S.; Andriantsiferana, R.; Rasamison, V.E.; Kingston, D.G.I. New cytotoxic terpenoids from the wood of Vepris punctata from the Madagascar Rainforest. J. Nat. Prod. 2004, 67, 895–898. [Google Scholar] [CrossRef]
- Yamakawa, K.; Nishitani, K.; Murakami, A.; Yamamoto, A. Studies on the terpenoids and related alicyclic compounds. XXVIII. Chemical transformations of α-santonin into C-8 lactonized eudesmanolides: Yomogin and diastereoisomers of dihydrograveolide. Chem. Pharm. Bull. 1983, 31, 3397–3410. [Google Scholar] [CrossRef]
- Liu, L.-L.; Yang, J.-L.; Shi, Y.-P. Sesquiterpenoids and other constituents from the flower buds of Tussilago farfara. J. Asian Nat. Prod. Res. 2011, 13, 920–929. [Google Scholar] [CrossRef] [PubMed]
- Provan, G.J.; Waterman, P.G. Major triterpenes from the resins of Commiphora incisa and C. kua and their potential chemotaxonomic significance. Phytochemistry 1988, 27, 3841–3843. [Google Scholar] [CrossRef]
- Shen, T.; Yuan, H.-Q.; Wan, W.-Z.; Wang, X.-L.; Wang, X.-N.; Ji, M.; Lou, H.-X. Cycloartane-type triterpenoids from the resinous exudates of Commiphora opobalsamum. J. Nat. Prod. 2008, 71, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, R.; Rashid, R.B.; Sohrab, M.H.; Hasan, C.M. 12α-Hydroxystigmast-4-en-3-one: A new bioactive steroid from Toona ciliata (Meliaceae). Pharmazie 2003, 58, 272–273. [Google Scholar] [CrossRef] [PubMed]
- Dhal, R.; Lalami, K.; Brown, E. Racemic total syntheses of isoaustrobailignan-1 and of some related aryltetralin lactones. Org. Prep. Proced. Int. 1989, 21, 109–118. [Google Scholar] [CrossRef]
- Baser, K.H.C.; Demirci, B.; Dekebo, A.; Dagne, E. Essential oils of some Boswellia spp., myrrh and opopanax. Flavour Fragr. J. 2003, 18, 153–156. [Google Scholar] [CrossRef]
- Tsui, W.-Y.; Brown, G.D. Cycloeudesmanolides from Sarcandra glabra. Phytochemistry 1996, 43, 819–821. [Google Scholar] [CrossRef]
- Guella, G.; Skropeta, D.; Mancini, I.; Pietra, F. The first 6,8-cycloeudesmane sesquiterpene from a marine organism: The red seaweed Laurencia microcladia from the Baia di Calenzana, Elba Island. Z. Naturforsch. B 2002, 57, 1147–1151. [Google Scholar] [CrossRef]
- Ahmed, F.; Ali, M.; Singh, O. New compounds from Commiphora myrrha (Nees) Engl. Pharmazie 2006, 61, 728–731. [Google Scholar] [CrossRef]
- Woyengo, T.A.; Ramprasath, V.R.; Jones, P.J.H. Anticancer effects of phytosterols. Eur. J. Clin. Nutr. 2009, 63, 813–820. [Google Scholar] [CrossRef]
- Provan, G.J.; Waterman, P.G. Picropolygamain: A new lignan from Commiphora incisa resin1,2. Planta Med. 1985, 51, 271–272. [Google Scholar] [CrossRef]
- Dekebo, A.; Lang, M.; Polborn, K.; Dagne, E.; Steglich, W. Four lignans from Commiphora erlangeriana. J. Nat. Prod. 2002, 65, 1252–1257. [Google Scholar] [CrossRef] [PubMed]
- Habtemariam, S. Cytotoxic and cytostatic activity of erlangerins from Commiphora erlangeriana. Toxicon 2003, 41, 723–727. [Google Scholar] [CrossRef]
- Vissiennon, C.; Hammoud, D.; Goos, K.-H.; Nieber, K.; Arnhold, J. Synergistic interactions of chamomile flower, myrrh and coffee charcoal in inhibiting pro-inflammatory chemokine release from activated human macrophages. Planta Med. 2017, 4, 13–18. [Google Scholar] [CrossRef]
- Jang, H.-J.; Lee, S.; Lee, S.-J.; Lim, H.-J.; Jung, K.; Kim, Y.H.; Lee, S.W.; Rho, M.-C. Anti-inflammatory activity of eudesmane-type sesquiterpenoids from Salvia plebeia. J. Nat. Prod. 2017, 80, 2666–2676. [Google Scholar] [CrossRef]
- Xu, J.; Jin, D.; Shi, D.; Ma, Y.; Yang, B.; Zhao, P.; Guo, Y. Sesquiterpenes from Vladimiria souliei and their inhibitory effects on NO production. Fitoterapia 2011, 82, 508–511. [Google Scholar] [CrossRef] [PubMed]
- Dekebo, A.; Dagne, E.; Sterner, O. Furanosesquiterpenes from Commiphora sphaerocarpa and related adulterants of true myrrh. Fitoterapia 2002, 73, 48–55. [Google Scholar] [CrossRef]
- Morteza-Semnani, K.; Saeedi, M. Constituents of the essential oil of Commiphora myrrha (Nees) Engl. var. molmol. J. Essent. Oil Res. 2003, 15, 50–51. [Google Scholar] [CrossRef]
- Dolara, P.; Moneti, G.; Pieraccini, G.; Romanelli, N. Characterization of the action on central opioid receptors of furaneudesma-1,3-diene, a sesquiterpene extracted from myrrh. Phytother. Res. 1996, 1996, 81–83. [Google Scholar]
- Paço, A.; Brás, T.; Santos, J.O.; Sampaio, P.; Gomes, A.C.; Duarte, M.F. Anti-inflammatory and immunoregulatory action of sesquiterpene lactones. Molecules 2022, 27, 1142. [Google Scholar] [CrossRef]
- Dirsch, V.M.; Stuppner, H.; Ellmerer-Müller, E.P.; Vollmar, A.M. Structural requirements of sesquiterpene lactones to inhibit LPS-induced nitric oxide synthesis in RAW 264.7 macrophages. Bioorg. Med. Chem. 2000, 8, 2747–2753. [Google Scholar] [CrossRef] [PubMed]
- Marston, A.; Borel, C.; Hostettmann, K. Separation of natural products by centrifugal partition chromatography. J. Chromatogr. A 1988, 450, 91–99. [Google Scholar] [CrossRef]
- Chen, M.; Lou, Y.; Wu, Y.; Meng, Z.; Li, L.; Yu, L.; Zeng, S.; Zhou, H.; Jiang, H. Characterization of in vivo and in vitro metabolites of furanodiene in rats by high performance liquid chromatography–electrospray ionization mass spectrometry and nuclear magnetic resonance spectra. J. Pharm. Biomed. Anal. 2013, 86, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Glauco, M.B.; Jorge, B.R.; Arlett, M.P.; Samuel, P.T.; Luis, A.M. An eremophilanolide from Senecio rosmarinus. Phytochemistry 1986, 25, 2412–2414. [Google Scholar] [CrossRef]
- Thinh, N.S.; Thu, N.T.B.; Trang, D.T.; van Kiem, P.; Tai, B.H.; Nhiem, N.X. Sesquiterpenes from Fissistigma pallens (Fin. & Gagn.) Merr. Vietnam J. Chem. 2019, 57, 552–557. [Google Scholar] [CrossRef]
- Li, R.-J.; Guo, D.-X.; Lou, H.-X. A new guaiane-type sesquiterpene lactone from the Chinese liverwort Porella acutifolia subsp. tosana. Chin. J. Nat. Med. 2014, 11, 74–76. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
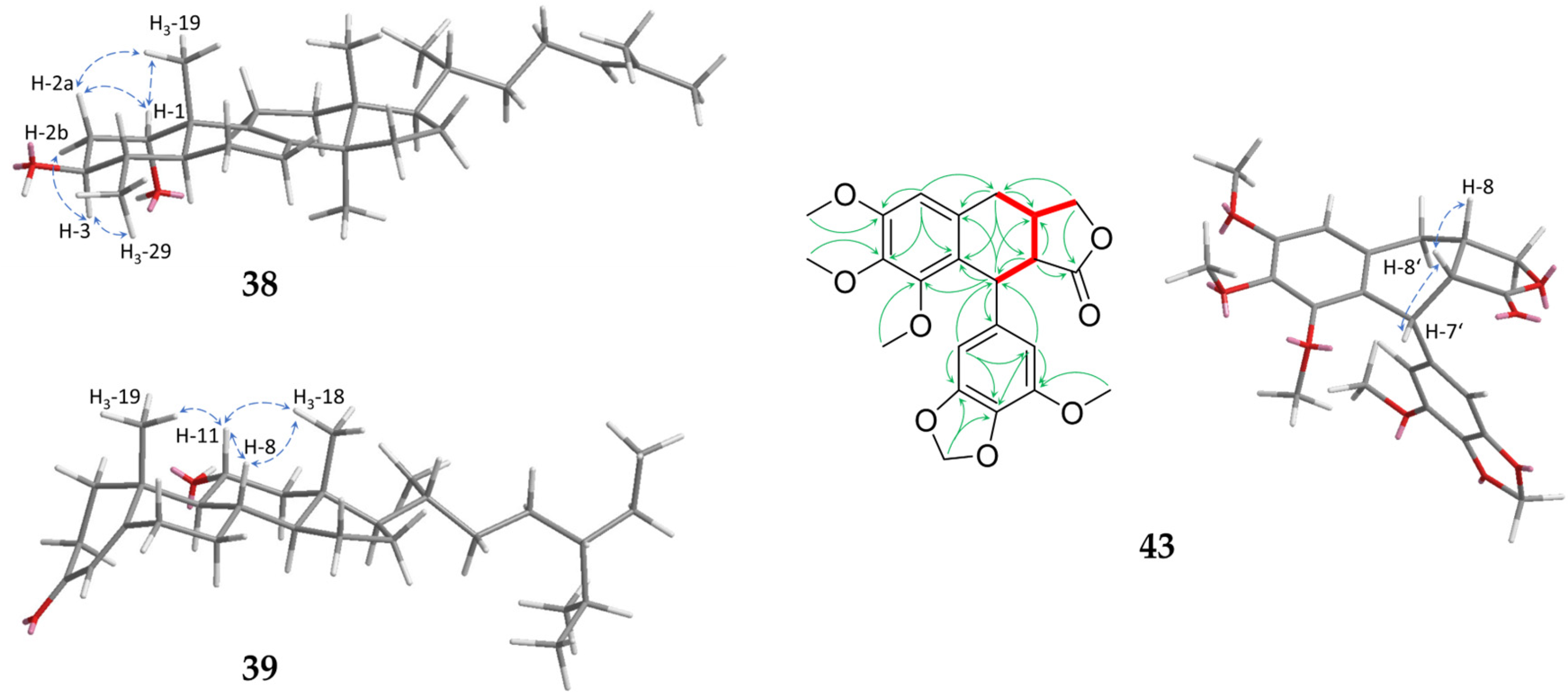


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | 1 | 2 | 3 | 4 | ||||
|---|---|---|---|---|---|---|---|---|
| δH | δC | δH | δC | δH | δC | δH | δC | |
| 1 | 3.61 (1H, d, 4.5) | 76.6 | 5.00 (1H, d, 10.3) | 132.2 | 4.87 (1H, brd, 11.6) | 129.3 | 5.35 (1H, d, 10.4) | 128.4 |
| 2 | 5.97 (1H, dd, 4.5, 9.8) | 130.5 | 5.40 (1H, ddd, 5.0, 10.5, 10.5) | 72.0 | 2.08 a (1H, m) 2.36 a (1H, m) | 24.6 | 5.54 (1H, ddd, 5.0, 10.6, 11.2) | 69.0 |
| 3 | 5.45 (1H, d, 9.7) | 133.2 | 2.15 (1H, dd, 11.0, 11.0) 2.48 (1H, dd, 4.9, 11.3) | 45.4 | 1.14 a (1H, m) 2.01 a (1H, m) | 37.3 | 1.28 a (1H, m) 2.43 a (1H, m) | 42.6 |
| 4 | 55.5 | 134.7 | 60.1 | 59.4 | ||||
| 5 | 79.6 | 5.20 (1H, d, 7.3) | 134.7 | 2.94 a (1H, m) | 63.2 | 3.16 (1H, dd, 3.9, 8.4) | 61.5 | |
| 6 | 2.43 (1H, d, 18.7) 2.75 (1H, d, 18.7) | 38.2 | 5.53 (1H, d, 7.3) | 65.8 | 2.22 (1H, dd, 11.9, 14.9) 2.95 a (1H, m) | 22.0 | 2.56 a (1H, m) 3.01 a (1H, m) | 26.6 |
| 7 | 165.4 | 163.9 | 162.5 | 159.5 | ||||
| 8 | 88.6 | 5.42 a (1H, m) | 83.4 | 5.11 (1H, d, 6.0) | 83.1 | 5.39 (1H, dd, 5.4, 6.2) | 83.5 | |
| 9 | 1.54 (1H, d, 12.5) 1.68 a (1H, d, 12.7) | 47.8 | 2.79 (1H, dd, 6.1, 14.9) 2.91 (1H, dd, 2.3, 14.8) | 42.7 | 2.54 a (1H, m) 2.64 a (1H, m) | 38.6 | 2.40 a (1H, m) 3.00 a (1H, m) | 40.5 |
| 10 | 43.2 | 137.1 | 127.4 | 133.5 | ||||
| 11 | 131.3 | 127.5 | 124.4 | 126.5 | ||||
| 12 | 10.68 (1H, s) | 195.7 | 175.8 | 174.4 | 174.2 | |||
| 13 | 1.67 a (3H, s) | 11.1 | 1.94 (3H, d, 1.7) | 9.7 | 2.17 (3H, s) | 12.4 | 1.80 (3H, brs) | 8.0 |
| 14 | 1.20 (3H, s) | 18.9 | 1.36 (3H, s) | 17.1 | 1.67 (3H, brs) | 18.6 | 1.69 (3H, brs) | 17.9 |
| 15 | 1.02 (3H, s) | 15.0 | 1.59 (3H, s) | 18.7 | 1.16 (3H, s) | 14.8 | 1.23 (3H, s) | 16.8 |
| 16 | 172.4 | 170.6 | ||||||
| 17 | 2.01 (3H, s) | 21.0 | 1.99 (3H, s) | 19.5 | ||||
| No. | 5 | 6 | 7 | |||
|---|---|---|---|---|---|---|
| δH | δC | δH | δC | δH | δC | |
| 1 | 5.21 (1H, d, 9.7) | 129.5 | 3.58 (1H, dd, 6.0, 10.4) | 74.8 | 3.44 (1H, s) | 74.9 |
| 2 | 5.64 (1H, m) | 69.1 | 1.94–1.88 (1H, m) 2.20–2.12 (1H, m) | 31.3 | 3.58 (1H, m) | 80.6 |
| 3 | 1.27 (1H, dd, 10.7, 10.7) 2.46 (1H, dd, 5.2, 11.7) | 41.7 | 5.36 (1H, dd, 1.0, 2.3) | 120.5 | 5.50 (1H, m) | 121.9 |
| 4 | 59.7 | 133.3 | 137.7 | |||
| 5 | 2.74 a (1H, m) | 63.7 | 2.60 (1H, m) | 40.8 | 2.26 a (1H, m) | 42.9 |
| 6 | 2.35 (1H, dd, 8.8, 14.6) 2.74 a (1H, m) | 22.8 | 2.49 (1H, dd, 12.9, 17.7) 2.86 (1H, ddq, 1.9, 3.7, 17.7) | 25.0 | 2.31 (1H, dd, 12.5, 12.5) 2.96 (1H, dd, 2.8, 12.2) | 24.6 |
| 7 | 159.0 | 164.4 | 162.9 | |||
| 8 | 106.0 | 5.20 (1H, ddq, 1.9, 8.0, 8.0) | 78.3 | 106.3 | ||
| 9 | 2.40 (1H, d, 13.4) 2.97 (1H, d, 13.3) | 50.4 | 1.40 (1H, dd, 6.6, 14.1) 2.43 (1H, dd, 10.9, 14.0) | 40.5 | 1.96 (1H, d, 13.2) 2.01 (1H, d, 11.5) | 46.3 |
| 10 | 133.8 | 38.0 | 38.5 | |||
| 11 | 129.5 | 120.2 | 122.8 | |||
| 12 | 171.5 | 176.0 | 174.5 | |||
| 13 | 1.89 (3H, s) | 7.5 | 1.79 (3H, brs) | 6.8 | 1.82 (3H, s) | 8.0 |
| 14 | 1.99 (3H, s) | 17.6 | 0.67 (3H, s) | 12.8 | 1.11 (3H, s) | 16.6 |
| 15 | 1.30 (3H, s) | 16.9 | 1.70 (3H, s) | 19.2 | 1.80 (3H, s) | 21.4 |
| 16 | 170.6 | 3.55 (1H, dq, 7.1, 9.4) 3.65 (1H, dq, 7.1, 9.2) | 65.8 | |||
| 17 | 1.99 (3H, s) | 19.5 | 1.17 (3H, dd, 6.9, 6.9) | 15.9 | ||
| No. | 8 | 9 | 10 | 11 | ||||
|---|---|---|---|---|---|---|---|---|
| δH | δC | δH | δC | δH | δC | δH | δC | |
| 1 | 3.60 (1H, dd, 4.3, 11.6) | 79.1 | 3.29 a (1H, m) | 80.9 | 6.97 (1H, d, 9.9) | 157.3 | 6.99 (1H, d, 10.0) | 162.3 |
| 2 | 1.47 a (1H, m) 1.78 a (1H, m) | 32.3 | 4.80 (1H, ddd, 6.0, 10.5, 11.9) | 74.9 | 6.18 (1H, d, 9.9) | 125.8 | 5.96 (1H, d, 9.9) | 127.1 |
| 3 | 2.17 a (1H, m) 2.37 (1H, ddd, 2.4, 4.4, 13.7) | 35.0 | 2.10 (1H, dd, 12.0, 12.0) 2.71 (1H, dd, 5.5, 12.8) | 40.3 | 187.3 | 190.2 | ||
| 4 | 148.1 | 145.6 | 133.9 | 146.1 | ||||
| 5 | 2.52 (1H, dd, 5.5, 12.7) | 42.0 | 2.02 a (1H, m) | 50.4 | 156.6 | 2.69 (1H, dddd, 2.4, 2.4, 2.4, 12.6) | 50.7 | |
| 6 | 2.69 (1H, ddq, 1.7, 5.1, 17.1) 2.81 (1H, dd, 12.7, 18.8) | 25.5 | 2.48 (1H, dd, 12.5, 12.5) 2.73 (1H, dd, 4.2, 13.3) | 24.7 | 3.79 (1H, s) 3.81 (1H, s) | 29.1 | 2.56 (1H, dd, 13.5, 13.5) 2.96 (1H, dd, 2.9, 13.2) | 24.6 |
| 7 | 165.0 | 162.2 | 162.2 | 160.6 | ||||
| 8 | 5.19 (1H, ddq, 1.7, 9.3, 9.3) | 79.6 | 105.4 | 4.93 (1H, m) | 78.0 | 105.0 | ||
| 9 | 1.20 (1H, dd, 9.1, 13.9) 2.60 (1H, dd, 9.9, 13.9) | 42.6 | 1.45 (1H, d, 13.7) 2.66 (1H, d, 13.6) | 48.5 | 1.90 a (1H, m) 2.40 (1H, dd, 10.7, 14.0) | 40.1 | 1.77 (1H, d, 13.3) 2.43 (1H, d, 13.4) | 47.7 |
| 10 | 41.4 | 41.9 | 40.6 | 39.7 | ||||
| 11 | 121.5 | 123.0 | 122.9 | 124.0 | ||||
| 12 | 177.5 | 174.4 | 176.7 | 174.0 | ||||
| 13 | 1.81 (3H, brs) | 8.2 | 1.80 (3H, brs) | 8.1 | 1.90 (3H, brs) | 8.3 | 1.82 (3H, brs) | 8.2 |
| 14 | 0.64 (3H, s) | 15.2 | 1.03 (3H, s) | 12.1 | 1.19 (3H, s) | 27.9 | 1.24 (3H, s) | 19.7 |
| 15 | 4.75 (1H, brs) 4.93 (1H, brs) | 109.3 | 4.82 a (1H, s) 5.00 (1H, s) | 110.7 | 1.94 (3H, s) | 11.0 | 5.45 (1H, d, 2.1) 6.13 (1H, d, 1.7) | 119.8 |
| 16 | 172.7 | |||||||
| 17 | 2.04 (3H, s) | 21.1 | ||||||
| No. | 12 | 13 | 14 | |||
|---|---|---|---|---|---|---|
| δH | δC | δH | δC | δH | δC | |
| 1 | 7.14 (1H, d, 10.1) | 163.2 | 4.00 (1H, s) | 78.8 | 3.58 (1H, d, 5.3) | 74.5 |
| 2 | 6.03 (1H, d, 10.0) | 127.1 | 5.53 (1H, d, 9.9) | 132.8 | 5.88 (1H, dd, 5.2, 10.1) | 128.9 |
| 3 | 195.3 | 6.17 (1H, dd, 9.9, 2.8) | 131.3 | 6.19 (1H, d, 9.8) | 129.6 | |
| 4 | 58.3 | 145.7 | 147.8 | |||
| 5 | 2.57 (1H, dd, 3.5, 13.8) | 46.4 | 2.17 a (1H, m) | 47.3 | 78.2 | |
| 6 | 2.37 (1H, dd, 13.0, 13.0) 2.57 (1H, dd, 3.5, 13.8) | 21.6 | 2.41 (1H, dd, 13.8, 13.8) 2.92 (1H, dd, 3.8, 13.2) | 24.8 | 2.68 (1H, brd, 13.8) 3.06 (1H, d, 13.6) | 32.1 |
| 7 | 160.6 | 162.0 | 160.2 | |||
| 8 | 104.8 | 105.4 | 106.2 | |||
| 9 | 1.77 (1H, d, 13.3) 2.41 (1H, d, 13.9) | 48.0 | 1.48 (1H, d, 13.7) 2.66 (1H, d, 13.6) | 49.0 | 1.93 (1H, d, 13.7) 2.57 (1H, d, 13.7) | 41.5 |
| 10 | 38.8 | 41.6 | 41.8 | |||
| 11 | 124.1 | 122.9 | 125.8 | |||
| 12 | 173.9 | 174.4 | 174.6 | |||
| 13 | 1.81 (3H, brs) | 8.1 | 1.81 (3H, brs) | 8.1 | 1.81 (3H, brs) | 8.2 |
| 14 | 1.45 (3H, s) | 22.7 | 0.95 (3H, s) | 11.9 | 1.07 (3H, s) | 22.0 |
| 15 | 3.09 (1H, d, 5.5) 3.28 (1H, d, 5.6) | 46.9 | 4.99 (1H, s) 5.03 (1H, s) | 112.1 | 5.16 (1H, s) 5.33 (1H, s) | 114.3 |
| No. | 15 | 16 | 17 | 24 | ||||
|---|---|---|---|---|---|---|---|---|
| δH | δC | δH | δC | δH | δC | δH | δC | |
| 1 | 3.93 (1H, d, 5.3) | 72.5 | 7.08 (1H, d, 9.9) | 155.6 | 7.26 (1H, d, 7.7) | 127.5 | 6.81 (1H, d, 9.9) | 158.6 |
| 2 | 5.91 (1H, dd, 5.3, 9.9) | 126.9 | 6.34 (1H, d, 9.9) | 126.4 | 7.19 (1H, dd, 7.4, 7.4) | 128.3 | 5.91 (1H, d, 10.1) | 126.0 |
| 3 | 6.25 (1H, d, 9.9) | 128.7 | 185.6 | 7.15 (1H, d, 7.1) | 130.8 | 201.6 | ||
| 4 | 145.6 | 139.6 | 139.1 | 2.62 a (1H, m) | 44.2 | |||
| 5 | 75.2 | 149.0 | 134.1 | 2.11 (1H, dd, 3.0, 6.0, 13.3) | 46.4 | |||
| 6 | 2.93 (1H, dq, 1.8, 17.3) 3.16 (1H, d, 17.2) | 29.3 | 8.24 (1H, s) | 143.5 | 3.87 (2H, brs) | 27.6 | 2.50 (1H, dd, 3.0, 13.5) 2.68 a (1H, m) | 25.5 |
| 7 | 146.6 | 137.1 | 161.0 | 159.3 | ||||
| 8 | 147.9 | 194.8 | 107.6 | 103.0 | ||||
| 9 | 5.76 (1H, s) | 112.1 | 2.68 (2H, d, 5.7) | 48.5 | 1.48 (3H, s) | 24.3 | 1.70 (1H, d, 13.6) 2.34 (1H, d, 13.4) | 49.6 |
| 10 | 43.5 | 41.2 | 141.3 | 36.9 | ||||
| 11 | 121.8 | 197.7 | 124.8 | 122.7 | ||||
| 12 | 171.8 | 2.49 (3H, s) | 29.3 | 174.3 | 171.1 | |||
| 13 | 1.91 (3H, brs) | 6.9 | 1.32 (3H, s) | 25.9 | 1.21 (3H, s) | 7.7 | 1.84 (3H, brs) | 8.4 |
| 14 | 0.99 (3H, s) | 22.2 | 2.15 (3H, s) | 10.0 | 4.63 (2H, s) | 63.7 | 1.46 (3H, s) | 22.5 |
| 15 | 5.22 (1H, s) 5.39 (1H, brs) | 113.6 | 2.29 (3H, s) | 20.0 | 1.27 (3H, d, 8.2) | 13.4 | ||
| No. | 38 | 39 | 40 | |||
|---|---|---|---|---|---|---|
| δH | δC | δH | δC | δH | δC | |
| 1 | 3.97 (1H, dd, 3.0, 3.0) | 72.7 | 1.98 a (1H, m) 2.67 (1H, ddd, 4.5, 4.5, 14.2) | 37.6 | 2.00 (1H, ddd, 4.4, 13.8, 13.8) 2.67 (1H, ddd, 4.5, 4.5, 14.2) | 37.6 |
| 2 | 1.67 (1H, ddd, 2.6, 11.1, 13.6) 2.08 a (1H, m) | 36.3 | 2.32 (1H, ddd, 4.5, 4.5, 17.2) 2.44 (1H, ddd, 4.8, 14.2, 17.2) | 34.2 | 2.33 (1H, ddd, 4.3, 4.3, 17.2) 2.45 (1H, ddd, 4.7, 13.5, 17.3) | 34.2 |
| 3 | 3.55 (1H, ddd, 5.1, 9.5, 11.4) | 72.3 | 200.2 | 200.2 | ||
| 4 | 1.44 a (1H, m) | 38.6 | 5.73 (1H, s) | 124.5 | 5.73 (1H, s) | 124.5 |
| 5 | 1.45 a (1H, m) | 40.1 | 171.3 | 171.2 | ||
| 6 | 1.32 a (1H, m) 1.80 a (1H, m) | 19.9 | 2.27 a (1H, m) 2.36 (1H, ddd, 5.1, 14.2, 14.2) | 33.7 | 2.27 a (1H, m) 2.37 (1H, ddd, 5.0, 14.3, 14.3) | 33.7 |
| 7 | 2.15 (2H, m) | 21.4 | 1.06 a (1H, m) 1.83 (1H, m) | 31.6 | 1.05 a (1H, m) 1.83 (1H, dddd, 2.8, 2.8, 5.2, 12.9) | 31.6 |
| 8 | 141.3 | 1.51 a (1H, dd, 12.9, 12.9) | 34.9 | 1.52 a (1H, m) | 35.0 | |
| 9 | 129.2 | 1.08 (1H, dd, 10.8, 10.8) | 59.3 | 1.08 a (1H, m) | 59.3 | |
| 10 | 42.2 | 39.9 | 39.9 | |||
| 11 | 2.06 (2H, m) | 26.2 | 4.01 (1H, m) | 69.3 | 4.02 (1H, ddd, 5.2, 10.7, 15.7) | 39.2 |
| 12 | 1.79 a (2H, m) | 30.9 | 1.24 a (1H, m) 2.30 a (1H, m) | 51.9 | 1.26 a (1H, m) 2.28 a (1H, m) | 51.8 |
| 13 | 44.5 | 43.1 | 43.0 | |||
| 14 | 50.3 | 1.13 a (1H, m) | 55.3 | 1.21 a (1H, m) | 55.8 | |
| 15 | 1.21 (1H, ddd, 1.9, 9.6, 11.8) 1.61 a (1H, m) | 30.8 | 1.10 a (1H, m) 1.61 a (1H, m) | 24.1 | 1.09 a (1H, m) 1.58 a (1H, m) | 24.1 |
| 16 | 1.33 a (1H, m) 1.93 a (1H, m) | 28.0 | 1.26 a (2H, m) | 29.7 | 1.30 a (1H, m) 1.76 (1H, dddd, 5.4, 9.0, 9.0, 14.0) | 28.9 |
| 17 | 1.51 a (1H, m) | 50.3 | 1.17 a (1H, m) | 55.9 | 1.15 a (1H, m) | 55.4 |
| 18 | 0.72 (3H, s) | 15.7 | 0.75 (3H, s) | 13.2 | 0.77 (3H, s) | 13.3 |
| 19 | 1.01 (3H, s) | 19.0 | 1.32 (3H, s) | 18.3 | 1.32 (3H, s) | 18.3 |
| 20 | 1.40 a (1H, m) | 36.2 | 1.37 a (1H, m) | 36.1 | 2.04 a (1H, m) | 40.4 |
| 21 | 0.93 (3H, d, 6.6) | 18.7 | 0.94 (3H, d, 6.1) | 18.6 | 1.04 (3H, d, 6.6) | 21.1 |
| 22 | 1.05 a (1H, m) 1.42 a (1H, m) | 36.4 | 1.02 a (1H, m) 1.32 a (1H, m) | 33.7 | 5.13 (1H, dd, 8.5, 15.1) | 137.7 |
| 23 | 1.86 a (1H, m) 2.04 a (1H, m) | 24.9 | 1.16 a (2H, m) | 26.0 | 5.04 (1H, dd, 8.8, 14.9) | 129.5 |
| 24 | 5.10 a (1H, dd, 7.2, 7.2) | 125.2 | 0.93 a (1H, m) | 45.7 | 1.54 a (1H, m) | 51.2 |
| 25 | 131.2 | 1.66 a (1H, m) | 29.1 | 1.54 a (1H, m) | 31.0 | |
| 26 | 1.60 (3H, s) | 17.6 | 0.81 (3H, d, 6.6) | 19.0 | 0.80 a (3H, m) | 19.0 |
| 27 | 1.68 (3H, s) | 25.7 | 0.84 a (3H, m) | 19.8 | 0.85 (3H, d, 6.6) | 21.1 |
| 28 | 0.92 a (3H, s) | 25.1 | 1.23 a (1H, m) 1.27 a (1H, m) | 23.0 | 1.17 a (1H, m) 1.43 a (1H, m) | 25.4 |
| 29 | 1.02 a (3H, d, 5.5) | 115.0 | 0.84 a (3H, m) | 12.0 | 0.80 a (3H, m) | 12.2 |
| No. | 43 | |
|---|---|---|
| δH | δC | |
| 1 | 132.5 | |
| 2 | 124.9 | |
| 3 | 152.6 | |
| 4 | 142.3 | |
| 5 | 154.2 | |
| 6 | 6.67 (1H, s) | 109.7 |
| 7 | 2.51 (1H, dd, 1.9, 15.7) 2.75 (1H, dd, 7.7, 15.4) | 32.9 |
| 8 | 3.18 (1H, m) | 33.4 |
| 9 | 3.92 (1H, dd, 3.3, 9.4) 4.49 (1H, dd, 7.8, 9.1) | 75.1 |
| 10 | 3.76 (3H, s) | 61.6 |
| 11 | 3.82 (3H, s) | 61.3 |
| 12 | 3.85 (3H, s) | 56.5 |
| 1′ | 137.8 | |
| 2′ | 6.35 (1H, brs) | 108.7 |
| 3′ | 144.9 | |
| 4′ | 135.1 | |
| 5′ | 150.6 | |
| 6′ | 6.29 (1H, brs) | 102.4 |
| 7′ | 4.83 a (1H, m) | 39.7 |
| 8′ | 3.62 (1H, dd, 2.2, 9.9) | 46.3 |
| 9′ | 181.3 | |
| 10′ | 3.79 (3H, s) | 57.4 |
| 11′ | 5.86 (1H, s) 5.92 (1H, s) | 102.5 |
| Fraction | Gradient | Retention Time [min], Compound No. | |
|---|---|---|---|
| Time [min] | ACN [%] | ||
| HEP8C4 | 0 11 11.1 13 | 70 87 100 100 | 7.5, 38; 9.5, 42; 9.8, 40; 10.2, 39 |
| HEP8C6 | 0 20 25 | 60 100 100 | 19.7 min, 41 |
| M1.2C4 | 0 15 16 25 | 55 75 100 100 | 10.5, 29; 12.0 min, curzerenone (2.1 mg); 15.0 min, myrrhone (6.4 mg) |
| M1.4R1F1 | 0 15 20 25 | 30 40 90 90 | 9.5, 16; 10.0, 10; 12.3, mixture of 3, 4 and 22; 15.0, 26; 20.8, 43 |
| M1.4R1F4 | 0 20 21 26 | 20 32 90 90 | 11.6, 12; 12.0, 24; 13.6, 11; 14.0, 23; 15.9, 15; 17.3, 5; 17.6, 27; 18.1, 2; 20.5, 8; 21.4, 6; 22.1, mixture of 18 and 20 |
| M1.4R1F6 | 0 15 16 21 | 20 30 90 90 | 12.4, 7; 13.2 13; 15.5, 17 |
| M1.4R1F7.2 | 4.9, 1, 6.6, 14; 14.4, 9 | ||
| M1.4R1F7.1 | 0 25 26 31 | 17 23 95 95 | 24.4, (–)-21; 25.2, 25; 25.6, 19; 28.0, 28 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Unterholzner, A.; Kuck, K.; Weinzierl, A.; Lipowicz, B.; Heilmann, J. An Unprecedented 4,8-Cycloeudesmane, Further New Sesquiterpenoids, a Triterpene, Steroids, and a Lignan from the Resin of Commiphora myrrha and Their Anti-Inflammatory Activity In Vitro. Molecules 2024, 29, 4315. https://doi.org/10.3390/molecules29184315
Unterholzner A, Kuck K, Weinzierl A, Lipowicz B, Heilmann J. An Unprecedented 4,8-Cycloeudesmane, Further New Sesquiterpenoids, a Triterpene, Steroids, and a Lignan from the Resin of Commiphora myrrha and Their Anti-Inflammatory Activity In Vitro. Molecules. 2024; 29(18):4315. https://doi.org/10.3390/molecules29184315
Chicago/Turabian StyleUnterholzner, Anna, Katrin Kuck, Anna Weinzierl, Bartosz Lipowicz, and Jörg Heilmann. 2024. "An Unprecedented 4,8-Cycloeudesmane, Further New Sesquiterpenoids, a Triterpene, Steroids, and a Lignan from the Resin of Commiphora myrrha and Their Anti-Inflammatory Activity In Vitro" Molecules 29, no. 18: 4315. https://doi.org/10.3390/molecules29184315