Abstract
A truly organocatalytic approach to the Darzens reaction affording α,β-epoxy carbonyl compounds in good yields was developed taking advantage of the high basic strength and low nucleophilicity of cyclopropenimine superbases. The catalytic active free base can easily be generated in situ from its hydrochloride salt and maintained in the active deprotonated form by performing the reactions in a heterogeneous reaction system in the presence of excess potassium carbonate as a sacrificial base.
1. Introduction
The Darzens reaction, i.e., the condensation of aldehydes or ketones with α-halo carbonyl compounds in the presence of strong inorganic and/or organic bases is a non-oxidative approach to the preparation of α,β-epoxy esters and other α,β-epoxy carbonyl compounds. At variance with oxidation reactions, either metal catalysed [1,2,3,4,5,6,7,8] or organocatalytic [9,10,11,12,13,14,15], that require the forerunning preparation of α,β-unsaturated compounds, the Darzens reaction enables the formation of a new carbon-carbon bond and closure of the epoxide ring in a single synthetic step. This approach is remarkably advantageous since it may help shorten long synthetic routes and requires low-cost fragments. In view of that, and given the relevance of epoxides and α,β-epoxy carbonyl compounds as synthetic intermediates, refs. [16,17,18,19,20,21]. This reaction recently enjoyed a renewed interest, and some effort has been devoted to improving its synthetic applicability. This resulted in the development of a variety of different reaction conditions among which Phase Transfer Catalysis (PTC), ref. [22] and Lewis acid catalysis [23] are particularly relevant. However, in its base promoted versions—including those performed under PTC—the Darzens reaction leading to α,β-epoxy esters still represents a challenge. The major limitation is represented by the easy hydrolysis of the epoxyester formed in the very same conditions employed for the condensation [24]. Hydrolysis is also selective towards the trans isomer of the epoxyester and is significant even in the case of t-butyl esters [25]. This essentially limits the scope of Darzens reactions to pronucleophiles such as α-haloketones [26,27,28,29,30], α-chloroamides, refs. [31,32] or nitriles [32,33] as well as α-halosulfones [34,35,36].
While the most recent approaches to Darzens and Darzens-like reactions mainly rely on Lewis acid, and PTC catalysis, novel strategies based on exploiting supramolecular catalysis are emerging. As far as Lewis acid catalysis is concerned, Xie, Guo and co-workers recently developed an asymmetric Darzens reaction of isatins that provides access to spiro-epoxyoxindoles using Ni(acac)2 as the Lewis acid and imidazolidine-pyrroloimidazolone pyridine as ligand [23]. Highly enantioselective Darzens-like epoxidation of diazoesters with glyoxal derivatives could be achieved using a chiral boron–Lewis acid catalyst, allowing the asymmetric synthesis of trisubstituted α,β-epoxy esters. Ref. [37] In The field of PCT catalysis for Darzens reactions, the use of chiral phosphonium ions instead of quaternary ammonium ions has also been pursued in recent years. Indeed, Wang ad co-workers developed a highly efficient aza-Darzens cyclization between cyclic imines and α-halogenated ketones by employing a dipeptide-based chiral bifunctional phosphonium salt [38,39]. Catalytic approaches to Darzens reactions by supramolecular hosts used as nanoreactors have recently allowed three-component aza-Darzens reactions leading to aziridines to be carried out in water. The three components are aldehydes, anilines and substituted diazo esters; suitable supramolecular hosts were γ-cyclodextrins or metallacages [40,41,42,43]. Notwithstanding these achievements, the Darzens condensation involving α-halo esters still remains challenging.
We recently developed a base-promoted Darzens reaction in aprotic solvents—under non solvolytic conditions—involving the use of stoichiometric amounts of the charge-neutral Schwesinger bases P1-t-Bu and P4-t-Bu [44]. Although phosphazenes potentially have great utility, both the problems of their stability and difficulties of their preparation make the identification of alternative superbases for Darzens reactions an important goal. Most importantly an organocatalytic approach to this reaction, of which there is no example at present, would be highly desirable. Herein we describe the use of 2,3-bis(dicyclohexylamino)cyclopropenimines to fill this gap, taking advantage of its high basic strength that has been exploited in the development of enantioselective Mannich [45], Michael [46,47,48], and [3+2] cycloadditions reactions [49].
2. Results
The choice of the cyclopropenimine base was guided by its high basic strength, pKBH+ = 26.9, which is similar to that of the Schwesinger base P1-t-Bu [50]. The high basicity of cyclopropenimines is due to the aromatic cyclopropenium ion, which is formed upon protonation at the imino nitrogen [51]. In addition, at variance with phosphazene bases, the cyclopropenimine scaffold can be easily decorated with chiral moieties providing an easy entry to chiral catalysts.
The chiral enantiopure cyclopropenimine I is easily accessible on the multigram scale in a straightforward manner, see Materials and Methods section, alternatively, base I is also commercially available.
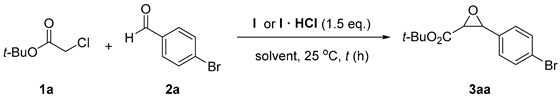
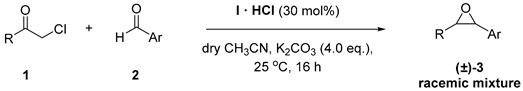
At the outset of our study, we carried out a preliminary investigation of the Darzens reaction between t-butyl chloroacetate (1a) and 4-bromobenzaldehyde (2a), Figure 1, seeking the best solvent system. This screening was performed by using a stoichiometric amount of cyclopropenimine free base I or its hydrochloride salt I·HCl [47], with respect to the α-haloester and the aldehyde, Table 1. When using I·HCl, the reactions were carried out in the presence of concentrated aqueous KOH to ensure the generation of the free base I. For this screening, the reactions were carried out on 0.25 mmol of aldehyde using a 1a/2a/I or 1a/2a/I·HCl molar ratio of 1.5:1:1.5 in 1 mL of solvent at 25 °C. The solvents considered were: dry CH3CN, dry ethyl acetate, which has often been employed in reactions involving cyclopropenimine bases [52], CH2Cl2 or toluene.
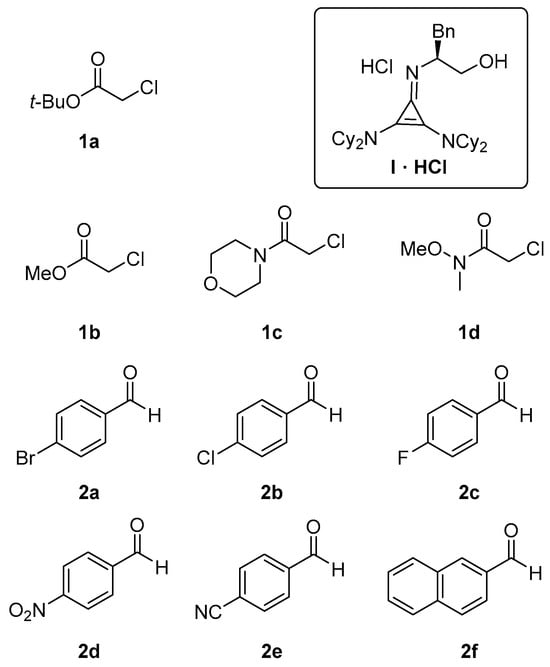
Figure 1.
Structures of pronucleophiles (1a–1d), aldehydes (2a–2f) and catalyst (I) used in the present study.

Table 1.
Darzens reaction of t-butyl chloroacetate (1a) and 4-bromobenzaldehyde (2a) in the presence of stoichiometric cyclopropenimine I or its hydrochloride salt I HCl 1.
The use of CH3CN resulted in 44% conversion after 16 h and 34% isolated yield of epoxide 3aa, Table 1 entry 1, while the reaction performed in dry ethyl acetate gave no conversion to the product after 24 h. Using a stoichiometric amount of the hydrochloride salt I·HCl instead of the free base in solvents such as DCM or toluene in the presence of concentrated, aqueous KOH gave low conversions and yields. However, under these biphasic conditions, lowering the amount of I·HCl to 20 mol% still gave conversion to the product similar to that achieved using a stoichiometric amount of I·HCl, Table 1, entry 4 vs. entry 3. Considering that under the same conditions but in the absence of a catalyst, no reactions occurred, the above observation pinpoints that free base I generated by in situ deprotonation of the hydrochloride salt is catalytically active and the hydroxide anion promoted Darzens condensation is negligible.
This observation prompted us to explore different reaction conditions in order to optimize the conversion, considering systems comprising low polarity solvents, two equivalents of alkali metal carbonates as bases and a 20 mol% of I·HCl as catalyst. When the reaction was performed in acetonitrile, using Cs2CO3 the product was obtained in low yield regardless of the presence of catalyst, Table 2 entry 1 vs. entry 2.
Table 2.
Development of Darzens reaction in the presence of catalytic loading of I·HCl 1.
On the contrary, by using potassium carbonate, the reaction proceeds only in the presence of I·HCl affording the product with 49% conversion and 36% isolated yield, Table 2, entries 3 and 4.
Increasing the amount of potassium carbonate to four equivalents and increasing the amount of I·HCl to 30 mol% yielded almost quantitative conversion to the product with a 67% isolated yield because of product instability under chromatographic conditions, Table 2 entry 6 vs. entry 5; without the addition of I·HCl the reaction did not proceed despite the increased amount of potassium carbonate. Under these conditions, the condensation of methyl chloroacetate was less efficient than that of t-butyl chloroacetate, Table 2, entry 7 vs. entry 4.
Other solvents such as THF or toluene, in combination with K2CO3 or Cs2CO3 proved unsuitable for this reaction since no conversion could be observed in 16 h, Table 2, entries 8–9. Organic bases such as pyridine, N,N-Diisopropylethylamine (DIPEA) and N,N,N′,N′-1,8-bis(dimethylamino)naphthalene (Proton Sponge) used in dichlorometane, or acetonitrile were also considered in the screening but did not provide measurable conversion to the product, Table 2, entries 10–14.
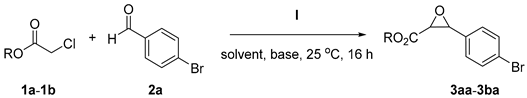
This survey allowed us to pinpoint that the best conditions require the use of acetonitrile as a solvent in the presence of 30 mol% of I·HCl and the use of 4 molar equivalents of potassium carbonate. Under these optimized conditions the performance of I·HCl (30 mol %) was assessed against various aromatic aldehydes and pronucleophiles, Figure 1 and Table 3.
Table 3.
Catalytic Darzens reactions of chloroacetate esters and amides with aromatic aldehydes in the presence of catalyst I·HCl 1.
The reactions proceed smoothly, generally scoring high conversion within 16 h. With p-chlorobenzaldehyde (2b) and p-fluorobenzaldehyde (2c) the epoxides derived from the pronucleophile 1a were obtained with 65% and 32% isolated yield, respectively. With the 4-cyanobenzaldehyde (2e), Table 3 entry 4, the reaction displayed a full conversion and the epoxide 3ae was obtained with 78% yield and 1/0.7 cis/trans ratio. Also, 2-naphthaldehyde 2f displayed good reactivity under these conditions.
Aldehyde 2e was then selected to test the reactivity of different pronucleofiles, including N-(Chloroacetyl)morpholine 1c, Table 3 entry 6, and the Weinreb amide of chloroacetic acid 1d, Table 3 entry 7. In the first case the product was obtained with low conversion and yield, while in the latter, the product was obtained with quantitative conversion enabling the preparation of epoxide 3de with an excellent isolated yield. In particular, the preparation of Weinreb amides of aryl glycidic acids was achieved so far only by catalytic oxidation of the cinnamic acid amides [53,54], amidation of the free acids which are known to be unstable, or by reaction of sulfur yilides obtained from diazo acetamides [55].
3. Discussion
Our preliminary experiments using t-butyl-chloroacetate (1a) as pronucleophile and p-bromobenzaldehyde (2a) as the carbonyl component displayed that the basicity of cyclopropenimine I was sufficient to carry out smooth stoichiometric deprotonation of the α-haloester in low polarity solvents such as acetonitrile, dichloromethane and toluene. The cyclopropenimine I can be used as a free base or, alternatively, the hydrochloride salt I·HCl can be also used provided that a sacrificial base such as KOH is introduced in the reaction system as a concentrated aqueous solution. The use of I·HCl represents an advantage since it has been reported that free base I is unstable and rearranges to the corresponding oxazoline while I·HCl is an essentially indefinitely stable compound [52]. Control experiments performed in order to assess any participation of KOH in the reaction displayed that under these heterogeneous conditions the background Darzens condensation not involving I was negligible, in line with literature reports that highlight the necessity of phase transfer catalysis to achieve this transformation [32,56,57,58,59]. Moreover, by lowering the amount of I·HCl to substechiometric, we could still observe measurable conversion to the products, pinpointing that base I can be used to devise a catalytic approach to this transformation. To this end, using 30 mol% of I·HCl, we addressed a sacrificial base screening primarily exploring heterogeneous systems in which the base was introduced as a solid phase. Alkali metal carbonates such as K2CO3 or Cs2CO3 proved to be effective only in combination with acetonitrile as a solvent; however, the reactions performed using Cs2CO3 suffered a significant contribution of the background, uncatalysed, reaction, likely because of the higher solubility of this salt in acetonitrile due to the softer nature of the Cs+ cation respect to K+ [60]. Under optimized conditions, the Darzens reaction involving t-butyl-chloroacetate (1a) and p-bromobenzaldehyde (2a) required 4 molar equivalents of K2CO3 and a 30% of I·HCl with respect to the substrates. The conversion of the reagents, assessed after 16 h, was 93% and the product could be isolated in 67% yield. Control experiments confirmed that in the absence of I·HCl, the transformation is ineffective also under these conditions. In all of these reactions the diastereoisomeric products are formed with only limited selectivity for the cis isomer, both the cis and the trans isomers were however found to be racemic, despite the enantiopure nature of base I, see the Supplementary Materials. The cis selectivity observed, despite limited, shares some similarities with the general outcome of the Darzens reactions carried out under PCT conditions [61]. We speculate that, by analogy to those conditions, the cis selectivity might be partly accounted for considering the steric bulk of base I. Indeed, under PCT, the most favorable ion pair formed upon nucleophilic addition of the ester enolate to the aldehyde is the one that better accommodates the sterically demanding quaternary ammonium ions; this intermediate leads to the cis epoxy ester [61].
A plausible mechanism for the reaction explaining the catalytic role of base I can therefore be sketched based on our observations and the available information on the base-promoted Darzens reactions [32,56,57,58,59,61], Figure 2. We consider that I·HCl will be first deprotonated by the sacrificial heterogeneous base generating the free base in the organic phase. Base I will enter a deprotonation equilibrium leading to the ester enolate that will provide nucleophilic addition to the aldehyde carbonyl group. As mentioned above, the little cis-diastereoelectivity observed in our experiments led us to assume the almost equimolar formation of the two diastereomeric haloidrin anion intermediates Int′ and Int″, with a slight preference for Int′ in which the protonated base can occupy the least hindered side of the intermediate. Finally, by epoxide ring closure, I·HCl is reformed closing the catalytic cycle.
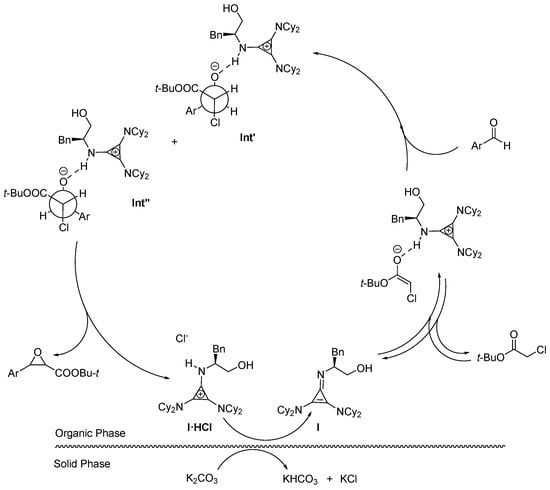
Figure 2.
Proposed catalytic cycle involving I·HCl.
Other organic bases such as DIPEA and Proton Sponge were used in order to extend our study to homogeneous systems. Not surprisingly, in the presence of these species, no conversion to the product could be observed because of their weaker basicity with respect to cyclopropenimine I, making it impossible to achieve significant concentrations of free base I starting from the hydrochloride salt.
The catalytic system developed proved to be effective in promoting the Darzens condensations of aromatic aldehydes with t-butyl chloroacetate achieving good to excellent conversions of the reagents. Other pronucleophiles such as N-(Chloroacetyl)morpholine and the Weinreb amide of chloroacetic acid were also tested providing the condensation products in moderate to good yields.
4. Materials and Methods
4.1. General Information
Unless otherwise noted, all reactions were performed in oven-dried or flame-dried glassware. Air-sensitive reagents and solutions were transferred via a syringe and were introduced to the apparatus through rubber septa. All reagents were purchased from Sigma-Aldrich srl (Milan, Italy). or Alfa Aesar GmbH (Karlsruhe, Germany) and used as received. All solvents were purchased from Sigma-Aldrich Co. LLC or Alfa Aesar GmbH. Dry dichloromethane, dry acetonitrile, dry ethyl acetate, dry THF, toluene (ACS grade) were used as received. Solvents for chromatography and filtration including ethyl acetate, dichloromethane, petroleum ether and methanol were used as received; hexane and 2-propanol were HPLC grade. Analytical thin layer chromatography (TLC) was performed on silica gel 60 F254 pre-coated plates with visualization under short-wavelength UV light. Additionally, spots were visualized by dipping the plates with potassium permanganate (aqueous H2SO4 solution of potassium permanganate) and ninhydrin reagent (n-butanol solution of ninhydrin and acetic acid) followed by heating. Flash column chromatography was performed using Biotage® SNAP Cartridge KP-Sil 10 g, Biotage apparatus and the indicated solvent mixtures. Analytical chiral HPLC analyses were carried out using the indicated columns, solvents and conditions.
Proton NMR spectra were recorded at 400 MHz (Bruker 400 MHz). Carbon NMR spectra were recorded at 100 MHz (Bruker 400 MHz). The proton chemical shifts were referenced to the residual non deuterated solvent (δ = 7.26 for CDCl3; δ = 2.49 for DMSO-d6). Chemical shifts (δ) are reported in parts per million (ppm), and multiplicities are indicated as s (singlet), d (doublet), t (triplet), q (quartet), dd (double doublet), m (multiplet), and b (broad). Coupling constants, J, are quoted in Hertz. 1H and 13C NMR assignments were supported by 2D experiments (gCOSY, gHSQC, ROESY experiments).
ESI-mass spectra were recorded on AcquityTM Ultra Performance LC apparatus and are reported in the form of (m/z). LC runs were performed using an Acquity UPLC CSH C18 column (50 mm × 2.1 mm i.d. 1.7 μm particle size) at 40 °C; solvents: A = 0.1% v/v solution of HCOOH in water B = 0.1% v/v solution of HCOOH in acetonitrile; gradient: from 3% to 99.9% of solvent B; flow rate: 1 mL/min; acquisition stop time: 2.0 min.
4.2. Preparation of Catalyst I·HCl
Catalyst I was prepared according to reported procedures, and its spectral data perfectly matched those reported in the literature [40,43].
Dicyclohexylamine (33.5 mL, 168.66 mmol) was slowly added to a solution of tetrachlorocyclopropene (5 g, 28.11 mmol) in CH2Cl2 (280 mL) in a 1L round bottom flask. A white precipitate was formed. The reaction mixture was stirred for 4 h at 25 °C. Next, (S)-2-amino-3-phenylpropan-1-ol (4.67 g, 30.92 mmol) was added in one portion and the reaction mixture was stirred for an additional 10 h. The crude reaction mixture was filtered through a celite plug, then washed with 1.0 M HCl (3 × 130 mL), dried over anhydrous Na2SO4, filtered and concentrated under vacuum to yield pure cyclopropenimine hydrochloride salt I·HCl (16.3 g, >99% yield) as yellow solid.
4.3. Preparation of Compound 3 in the Presence of Catalyst I·HCl
To a solution of aldehyde 2 (0.25 mmol, 1.0 equiv.) and α-halo carbonyl compound 1 (0.375 mmol, 1.5 equiv.) in anhydrous acetonitrile (1 mL), catalyst I·HCl (43 mg, 0.075 mmol) and K2CO3 (138 mg, 1.0 mmol) were added at 25 °C. The resulting mixture was stirred at 25 °C for 16 h. Then, a saturated aqueous solution of ammonium chloride (1 mL) was added. The resulting mixture was extracted with CH2Cl2 (3 × 2 mL). The organic layers were combined, dried over Na2SO4 anhydrous, filtered and concentrated in vacuum to yield a crude compound. The crude compounds were purified by silica gel flash chromatography (90/10 cyclohexane/ethyl acetate) to yield compound 3, spectroscopic data match those reported in the literature [32,61,62].
5. Conclusions
In summary, in this work, we explored the feasibility of Darzens condensation reactions between α-chloroesters and substituted aromatic aldehydes promoted by a series of organic bases. Low basicity amines such as DIPEA and pyridine used in a stoichiometric amount proved to be ineffective but also the higher basicity Proton Sponge did not allow to observe measurable conversion to the products. On the contrary, the use of cyclopropenimine superbase I allowed us to achieve smooth conversion of the reagents. Furthermore, the cyclopropenimine superbase I was proved to be active even at substoichiometric levels, in the presence of excess K2CO3 as a sacrificial base, thus enabling the set-up of a heterogeneous catalytic system for Darzens Reactions. Under our optimised conditions, the reaction of α-haloesters and amides with a series of aromatic aldehydes afforded α,β-epoxyesters and α,β-epoxyamides in high conversions and acceptable to excellent yields. To the best of our knowledge, this is the first report in which a cyclopropenimine superbase is used either stoichiometrically or catalytically for this kind of transformations. The low nucleophilicity of these bases makes this method a potentially valuable alternative to other base promoted/catalysed Darzens reactions. A plausible mechanistic hypothesis is provided to account for the limited diastereoselectivity of the reactions that displayed only a slight preference for the formation of the cis-epoxide. We consider that the steric bulk of cyclopropenimine I is primarily responsible for the limited, but consistent throughout the study, selectivity for the cis-epoxide products in analogy with the Darzens reaction performed under PTC conditions. However, since the cyclo-propenimine scaffold is amenable to decoration with various fragments at the imino nitrogen, we envision that substituents capable of providing further interactions with the aldehyde carbonyl group will possibly improve the strereoselectivity of our method.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules29184350/s1, characterization data for catalyst I·HCl, characterization data for compounds 3ab, 3ac, 3ad, 3ae, 3af, 3ce, 3de. References are cited in [32,61,62].
Author Contributions
Conceptualization, C.L., P.P. and L.P.; investigation, C.L.; data curation, C.L, P.P.; writing—original draft preparation, P.P.; writing—review and editing, C.L., P.P. and L.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original data not present in the article main text are reported in the Supplementary Materials.
Acknowledgments
C.L. wishes to acknowledge generous support from Aptuit, in particular for the access to the facilities of the analytical department. In this context the assistance of Alexa Lorenzon is gratefully acknowledged.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Liu, H.; Liu, W.; Xue, G.; Tan, T.; Yang, C.; An, P.; Chen, W.; Zhao, W.; Fan, T.; Cui, C.; et al. Modulating Charges of Dual Sites in Multivariate Metal–Organic Frameworks for Boosting Selective Aerobic Epoxidation of Alkenes. J. Am. Chem. Soc. 2023, 145, 11085–11096. [Google Scholar] [CrossRef] [PubMed]
- Cussó, O.; Garcia-Bosch, I.; Ribas, X.; Lloret-Fillol, J.; Costas, M. Asymmetric Epoxidation with H2O2 by Manipulating the Electronic Properties of Non-heme Iron Catalysts. J. Am. Chem. Soc. 2013, 135, 14871–14878. [Google Scholar] [CrossRef]
- Chang, S.; Galvin, J.M.; Jacobsen, E.N. Effect of Chiral Quaternary Ammonium Salts on (salen)Mn-Catalyzed Epoxidation of cis-Olefins. A Highly Enantioselective, Catalytic Route to Trans-Epoxides. J. Am. Chem. Soc. 1994, 116, 6937–6938. [Google Scholar] [CrossRef]
- Luo, L.; Yamamoto, H. Iron(II)-Catalyzed Asymmetric Epoxidation of Trisubstituted α,β-Unsaturated Esters. Eur. J. Org. Chem. 2014, 35, 7803–7805. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Sun, Q. Bioinspired Manganese and Iron Complexes for Enantioselective Oxidation Reactions: Ligand Design, Catalytic Activity, and Beyond. Acc. Chem. Res. 2019, 52, 2370–2381. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Miao, C.; Xia, C.; Lee, Y.-M.; Nam, W.; Sun, W. Mechanistic Insights into the Enantioselective Epoxidation of Olefins by Bioinspired Manganese Complexes: Role of Carboxylic Acid and Nature of Active Oxidant. ACS Catal. 2018, 8, 4528–4538. [Google Scholar] [CrossRef]
- Deng, L.; Jacobsen, E.N. A practical, highly enantioselective synthesis of the taxol side chain via asymmetric catalysis. J. Org. Chem. 1992, 57, 4320–4323. [Google Scholar] [CrossRef]
- Sawano, T.; Yamamoto, H. Regio- and Enantioselective Substrate-Directed Epoxidation. Eur. J. Org. Chem. 2020, 2020, 2369–2378. [Google Scholar] [CrossRef]
- Wang, Z.X.; Shi, Y. A New Type of Ketone Catalyst for Asymmetric Epoxidation. J. Org. Chem. 1997, 62, 8622–8623. [Google Scholar] [CrossRef]
- Takayuki Ohyoshi, T.; Iizumi, H.; Hosono, S.; Tano, H.; Kigoshi, H. Total Synthesis of Aplysiasecosterols A and B, Two Marine 9,11-Secosteroids. Org. Lett. 2023, 25, 4725–4729. [Google Scholar] [CrossRef]
- Höthker, S.; Plato, A.; Grimme, S.; Qu, A.-W.; Gansäuer, A. Stereoconvergent Approach to the Enantioselective Construction of α-Quaternary Alcohols by Radical Epoxide Allylation. Angew. Chem. Int. Ed. 2024, 63, e202405911. [Google Scholar] [CrossRef] [PubMed]
- Furutani, T.; Imashiro, R.; Hatsuda, M.; Seki, M. A Practical Procedure for the Large-Scale Preparation of Methyl (2R,3S)-3-(4-Methoxyphenyl)glycidate, a Key Intermediate for Diltiazem. J. Org. Chem. 2002, 67, 4599–4601. [Google Scholar] [CrossRef]
- Solladié-Cavallo, A.; Bouérat, L. Epoxidation of p-Methoxycinnamates Using Chiral Dioxiranes Derived from New Trisubstituted Halogenated Cyclohexanones: Enhanced Efficiency of Ketones Having an Axial Halogen. Org. Lett. 2000, 2, 3531–3534. [Google Scholar] [CrossRef] [PubMed]
- Imperio, D.; Valloni, F.; Caprioglio, D.; Minassi, A.; Casali, E.; Panza, L. Stereoselective Shi-type epoxidation with 3-oxo-4,6-O-benzylidene pyranoside catalysts: Unveiling the role of carbohydrate skeletons. RSC Adv. 2024, 14, 16778–16783. [Google Scholar] [CrossRef] [PubMed]
- Höthker, S.; Mika, R.; Goli, H.; Gansäuer, A. Converging Stereodivergent Reactions: Highly Stereoselective Formal anti-Markovnikov Addition of H2O to Mixtures of Olefins. Chem. Eur. J. 2023, 29, e202301031. [Google Scholar] [CrossRef] [PubMed]
- Yudin, A.K. Aziridines and Epoxides in Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2006. [Google Scholar]
- Moschona, F.; Savvopoulou, I.; Tsitopoulou, M.; Tataraki, D.; Rassias, G. Epoxide Syntheses and Ring-Opening Reactions in Drug Development. Catalysts 2020, 10, 1117. [Google Scholar] [CrossRef]
- Hanif, M.; Zahoor, A.F.; Saif, M.J.; Nazeer, U.; Ali, K.G.; Parveen, B.; Mansha, A.; Chaudhry, A.R.; Irfan, A. Exploring the synthetic potential of epoxide ring opening reactions toward the synthesis of alkaloids and terpenoids: A review. RSC Adv. 2024, 14, 13100–13128. [Google Scholar] [CrossRef]
- Meninno, S.; Alessandra Lattanzi, A. Epoxides: Small Rings to Play with under Asymmetric Organocatalysis. ACS Org. Inorg. Au 2022, 2, 289–305. [Google Scholar] [CrossRef]
- Jat, J.L.; Kumar, G. Isomerization of Epoxides. Adv. Synth. Catal. 2019, 361, 4426–4441. [Google Scholar] [CrossRef]
- López, I.; Rodríguez, S.; Izquierdo, J.; González, F.V. Highly Stereoselective Epoxidation of α-Methyl-γ-hydroxy-α,β-unsaturated Esters: Rationalization and Synthetic Applications. J. Org. Chem. 2007, 72, 6614–6617. [Google Scholar] [CrossRef]
- de los Santos, J.M.; Ochoa de Retana, A.M.; Martı´nez de Marigorta, E.; Vicario, J.; Palacios, F. Catalytic Asymmetric Darzens and Aza-Darzens Reactions for the Synthesis of Chiral Epoxides and Aziridines. ChemCatChem 2018, 10, 5092–5114. [Google Scholar] [CrossRef]
- Wang, H.L.; Wei, T.; Bi, R.-Y.; Xie, M.-S.; Guo, H.-M. Imidazolidine-Pyrroloimidazolone Pyridine-Ni(acac)2 Complex Catalyzed Asymmetric Darzens Reaction of Isatins with Phenacyl Chlorides: A Chiral Amplification Effect. Adv. Synth. Catal. 2024, 366, 3188–3193. [Google Scholar] [CrossRef]
- Kimura, C.; Kashiwaya, K.; Murai, K.; Katada, H. Darzens glycidic ester condensation of benzaldehyde in solid-liquid two-phase system. Ind. Eng. Chem. Prod. Res. Dev. 1983, 22, 118–120. [Google Scholar] [CrossRef]
- Jonczyk, A.; Zomerfeld, T. Convenient synthesis of t-butyl Z-3-substituted glycidates under conditions of phase-transfer catalysis. Tetrahedron Lett. 2003, 44, 2359–2361. [Google Scholar] [CrossRef]
- Liu, Y.; Provencher, B.A.; Bartelson, K.J.; Deng, L. Highly enantioselective asymmetric Darzens reactions with a phase transfer catalyst. Chem. Sci. 2011, 2, 1301–1304. [Google Scholar] [CrossRef]
- Zha, Q.; Wu, Y. Enantioselective Total Synthesis of 10-Desoxy Analogue of a Previously Reported Natural Peroxyguaidiol. J. Org. Chem. 2022, 87, 10114–10137. [Google Scholar] [CrossRef]
- Delost, M.D.; Njardarson, J.T. Mild Darzens Annulations for the Assembly of Trifluoromethylthiolated (SCF3) Aziridine and Cyclopropane Structures. Org. Lett. 2021, 23, 6121–6125. [Google Scholar] [CrossRef]
- Luo, J.; Hu, L.; Zhang, M.; Tang, Q. An efficient Darzens reaction promoted by 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU). Tetrahedron Lett. 2019, 60, 1949–1951. [Google Scholar] [CrossRef]
- Ashokkumar, V.; Siva, A.; Chidamdaram, R.R. A highly enantioselective asymmetric Darzens reaction catalysed by proline based efficient organocatalysts for the synthesis of di- and tri-substituted epoxides. Chem. Commun. 2017, 53, 10926–10929. [Google Scholar] [CrossRef]
- Mamedov, V.A.; Mamedova, V.L.; Syakaev, V.V.; Khikmatova, G.Z.; Korshin, D.E.; Kushatov, T.A.; Latypov, S.K. A new and efficient method for the synthesis of 3-(2-nitrophenyl) pyruvic acid derivatives and indoles based on the Reissert reaction. Tetrahedron Lett. 2018, 59, 3923–3925. [Google Scholar] [CrossRef]
- Arai, S.; Suzuki, Y.; Tokumaru, K.; Shioiri, T. Diastereoselective Darzens reactions of α-chloroesters, amides and nitriles with aromatic aldehydes under phase-transfer catalyzed conditions. Tetrahedron Lett. 2002, 43, 833–836. [Google Scholar] [CrossRef]
- Dong, Z.; Chen, C.; Wang, J.; Xu, J.; Yang, Z. Dual roles of bisphosphines and epoxides: Rh-catalyzed highly chemoselective and diastereoselective (3 + 2) transannulations of 1,2,3-thiadiazoles with cyanoepoxides. Org. Chem. Front. 2021, 8, 6687–6698. [Google Scholar] [CrossRef]
- Uritis, A.; Phillips, H.; Phillips, H.; Coombs, T.C. House-Meinwald rearrangement of aryl-substituted epoxysulfones in hexafluoroisopropanol (HFIP). Tetrahedron Lett. 2023, 128, 154716. [Google Scholar] [CrossRef]
- Ku, J.-M.; Yoo, M.-S.; Park, H.-g.; Jew, S.-s.; Jeong, B.-S. Asymmetric synthesis of α,β-epoxysulfones via phase-transfer catalytic Darzens reaction. Tetrahedron 2007, 63, 8099–8103. [Google Scholar] [CrossRef]
- Delost, M.D.; Njardarson, J.T. Strategic Vinyl Sulfone Nucleophile β-Substitution Significantly Impacts Selectivity in Vinylogous Darzens and Aza-Darzens Reactions. Org. Lett. 2020, 22, 6917–6921. [Google Scholar] [CrossRef]
- Nam, D.G.; Shim, S.Y.; Jeong, H.-M.; Ryu, D.H. Catalytic Asymmetric Darzens-Type Epoxidation of Diazoesters: Highly Enantioselective Synthesis of Trisubstituted Epoxides. Angew. Chem. Int. Ed. 2021, 60, 22236–22240. [Google Scholar] [CrossRef]
- Pan, J.; Wu, J.-H.; Zhang, H.; Ren, X.; Tan, J.-P.; Zhu, L.; Zhang, H.-S.; Jiang, C.; Wang, T. Highly Enantioselective Synthesis of Fused Tri- and Tetrasubstituted Aziridines: Aza-Darzens Reaction of Cyclic Imines with a-Halogenated Ketones Catalyzed by Bifunc-tional Phosphonium Salt. Angew. Chem. Int. Ed. 2019, 58, 7425–7430. [Google Scholar] [CrossRef]
- Wu, J.-H.; Pan, J.; Wang, T. Dipeptide-Based Phosphonium Salt Catalysis: Application to Enantioselective Synthesis of Fused Tri- and Tetrasubstituted Aziridines. Synlett 2019, 30, 2101–2106. [Google Scholar] [CrossRef]
- Patamia, V.; Saccullo, E.; Zagni, C.; Tomarchio, R.; Quattrocchi, G.; Floresta, G.; Rescifina, A. γ-Cyclodextrins as Supramolecular Reactors for the Three-component Aza-Darzens Reaction in Water. Chem. Eur. J. 2024, 30, e202303984. [Google Scholar] [CrossRef]
- Bierschenk, S.M.; Pan, J.Y.; Settineri, N.S.; Warzok, U.; Bergman, R.G.; Raymond, K.N.; Toste, D.F. Impact of Host Flexibility on Selectivity in a Supramolecular Host-Catalyzed Enantioselective aza-Darzens Reaction. J. Am. Chem. Soc. 2022, 144, 11425–11433. [Google Scholar] [CrossRef]
- Zhou, S.; Zhang, Z.; Bai, D.; Li, J.; Cui, X.; Xu, Z.J.; Tang, Y.; Tang, X.; Liu, W. A Discrete 3d–4f Metallacage as an Efficient Catalytic Nanoreactor for a Three-Component Aza-Darzens Reaction. Inorg. Chem. 2022, 61, 4009–4017. [Google Scholar] [CrossRef] [PubMed]
- Bierschenk, S.M.; Bergman, R.G.; Raymond, K.N.; Toste, D.F. A Nanovessel-Catalyzed Three-Component Aza-Darzens Reaction. J. Am. Chem. Soc. 2020, 142, 733–737. [Google Scholar] [CrossRef] [PubMed]
- Lops, C.A.; Pengo, P.; Pasquato, L. Highly Efficient Darzens Reactions Mediated by Phosphazene Bases under Mild Conditions. Chem. Open 2022, 11, e202200179. [Google Scholar] [CrossRef] [PubMed]
- Bandar, J.S.; Lambert, T.H. Cyclopropenimine-Catalyzed Enantioselective Mannich Reactions of tert-Butyl Glycinates with N-Boc-Imines. J. Am. Chem. Soc. 2013, 135, 11799–11802. [Google Scholar] [CrossRef] [PubMed]
- Bandar, J.S.; Sauer, G.S.; Wulff, W.D.; Lambert, T.H.; Vetticatt, M.J. Transition State Analysis of Enantioselective Brønsted Base Catalysis by Chiral Cyclopropenimines. J. Am. Chem. Soc. 2014, 136, 10700–10707. [Google Scholar] [CrossRef]
- Bandar, J.S.; Lambert, T.H. Enantioselective Brønsted Base Catalysis with Chiral Cyclopropenimines. J. Am. Chem. Soc. 2012, 134, 5552–5555. [Google Scholar] [CrossRef]
- Leonardi, C.; Brandolese, A.; Preti, L.; Bortolini, O.; Polo, E.; Dambruoso, P.; Ragno, D.; Di Carmine, G.; Massia, A. Expanding the Toolbox of Heterogeneous Asymmetric Organocatalysts: Bifunctional Cyclopropenimine Superbases for Enantioselective Catalysis in Batch and Continuous Flow. Adv. Synth. Catal. 2021, 363, 5473–5485. [Google Scholar] [CrossRef]
- Lauridsen, V.H.; Ibsen, L.; Blom, J.; Jørgensen, K.A. Asymmetric Brønsted Base Catalyzed and Directed [3+2] Cycloaddition of 2-Acyl Cycloheptatrienes with Azomethine Ylides. Chem. Eur. J. 2016, 22, 3259–3263. [Google Scholar] [CrossRef]
- Nacsa, E.D.; Lambert, T.H. Higher-Order Cyclopropenimine Superbases: Direct Neutral Brønsted Base Catalyzed Michael Reactions with α-Aryl Esters. J. Am. Chem. Soc. 2015, 137, 10246–10253. [Google Scholar] [CrossRef]
- Komatsu, K.; Kitagawa, T. Cyclopropenylium Cations, Cyclopropenones, and Heteroanalogues-Recent Advances. Chem. Rev. 2003, 103, 1371–1428. [Google Scholar] [CrossRef]
- Bandar, J.S.; Barthelme, A.; Mazori, A.Y.; Lambert, T.H. Structure–activity relationship studies of cyclopropenimines as enantioselective Brønsted base catalysts. Chem. Sci. 2015, 6, 1537–1547. [Google Scholar] [CrossRef] [PubMed]
- Cussó, O.; Garcia-Bosch, I.; Font, D.; Ribas, X.; Lloret-Fillol, J.; Costas, M. Highly Stereoselective Epoxidation with H2O2 Catalyzed by Electron-Rich Aminopyridine Manganese Catalysts. Org. Lett. 2013, 15, 6158–6161. [Google Scholar] [CrossRef] [PubMed]
- Borrell, M.; Costas, M. Mechanistically Driven Development of an Iron Catalyst for Selective Syn-Dihydroxylation of Alkenes with Aqueous Hydrogen Peroxide. J. Am. Chem. Soc. 2017, 139, 12821–12829. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, V.K.; Blackburn, P.; Fieldhouse, R.; Jones, R.V.H. Catalytic sulfur ylide reactions: Use of diazoacetamides for the diastereoselective synthesis of glycidic amides. Tetrahedron Lett. 1998, 39, 8517–8520. [Google Scholar] [CrossRef]
- Ooi, T.; Maruoka, K. Recent Advances in Asymmetric Phase-Transfer Catalysis. Angew. Chem. Int. Ed. 2007, 46, 4222–4266. [Google Scholar] [CrossRef]
- Bakó, P.; Rapi, Z.; Keglevich, G.; Szabó, T.; Sóti, P.L.; Vígh, T.; Grűn, A.; Holczbauer, T. Asymmetric C–C bond formation via Darzens condensation and Michael addition using monosaccharide-based chiral crown ethers. Tetrahedron Lett. 2011, 52, 1473–1476. [Google Scholar] [CrossRef]
- Rapi, Z.; Szabó, T.; Keglevich, G.; Szöllősy, Á.; Drahos, L.; Bakó, P. Enantioselective synthesis of heteroaromatic epoxyketones under phase-transfer catalysis using D-glucose- and D-mannose-based crown ethers. Tetrahedron Asymmetry 2011, 22, 1189–1196. [Google Scholar] [CrossRef]
- Rapi, Z.; Bakó, P.; Keglevich, G.; Szöllősy, Á.; Drahos, L.; Botyánszki, A.; Holczbauer, T. Asymmetric phase transfer Darzens reactions catalyzed by D-glucose- and D-mannose-based chiral crown ethers. Tetrahedron Asymmetry 2012, 23, 489–496. [Google Scholar] [CrossRef]
- Lehmann, F. Cesium Carbonate (Cs2CO3). Synlett 2004, 15, 2447–2448. [Google Scholar] [CrossRef]
- Kowalkowska, A.; Jończyk, A. Effect of Phase-Transfer Catalyst on Stereochemistry of tert-Butyl-3-aryl(alkyl)-Substituted Glycidates. Org. Process Res. Dev. 2010, 14, 728–731. [Google Scholar] [CrossRef]
- Sharifi, A.; Abaee, M.S.; Mirzaei, M.; Salimi, R. Ionic Liquid-Mediated Darzens Condensation: An Environmentally-Friendly Procedure for the Room-Temperature Synthesis of α,β-Epoxy Ketones. J. Iran. Chem. Soc. 2008, 5, 135–139. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).