
Metal-Based Drug–DNA Interactions and Analytical Determination Methods

 ,
, 
 ,
,  and
and .jpg)
Abstract
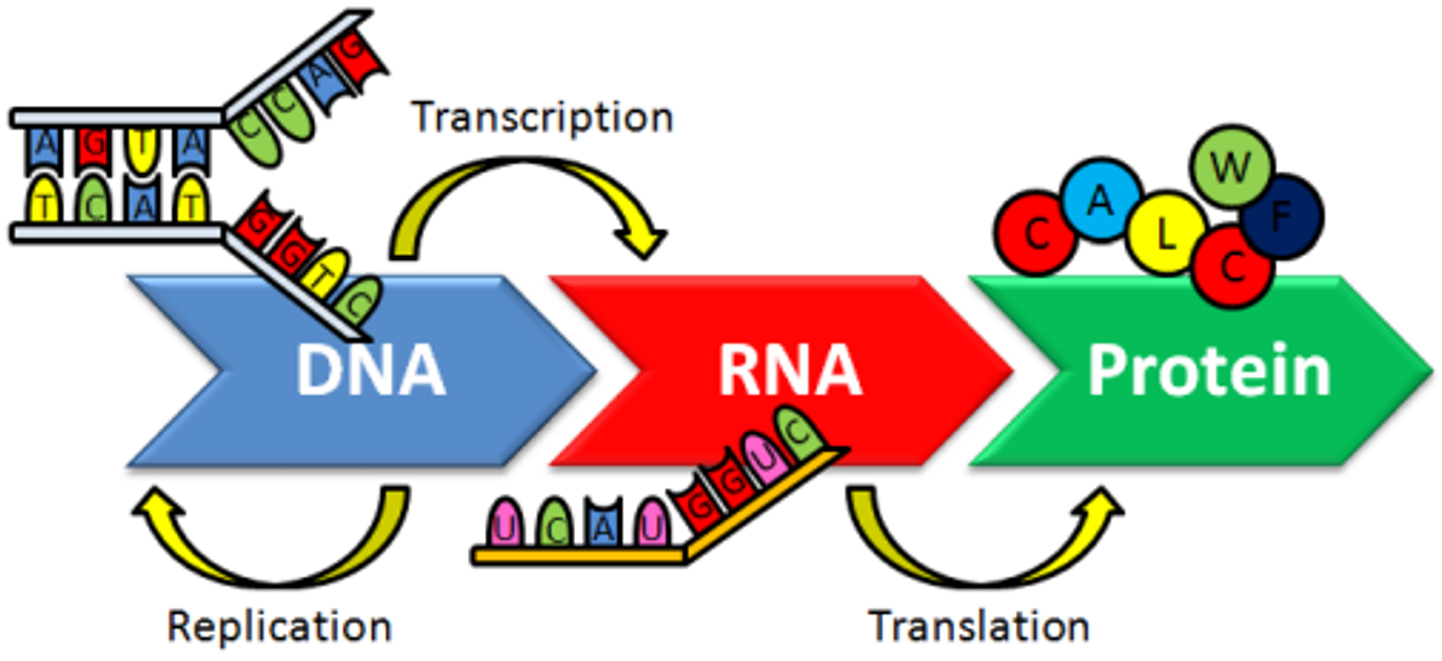
:1. Introduction
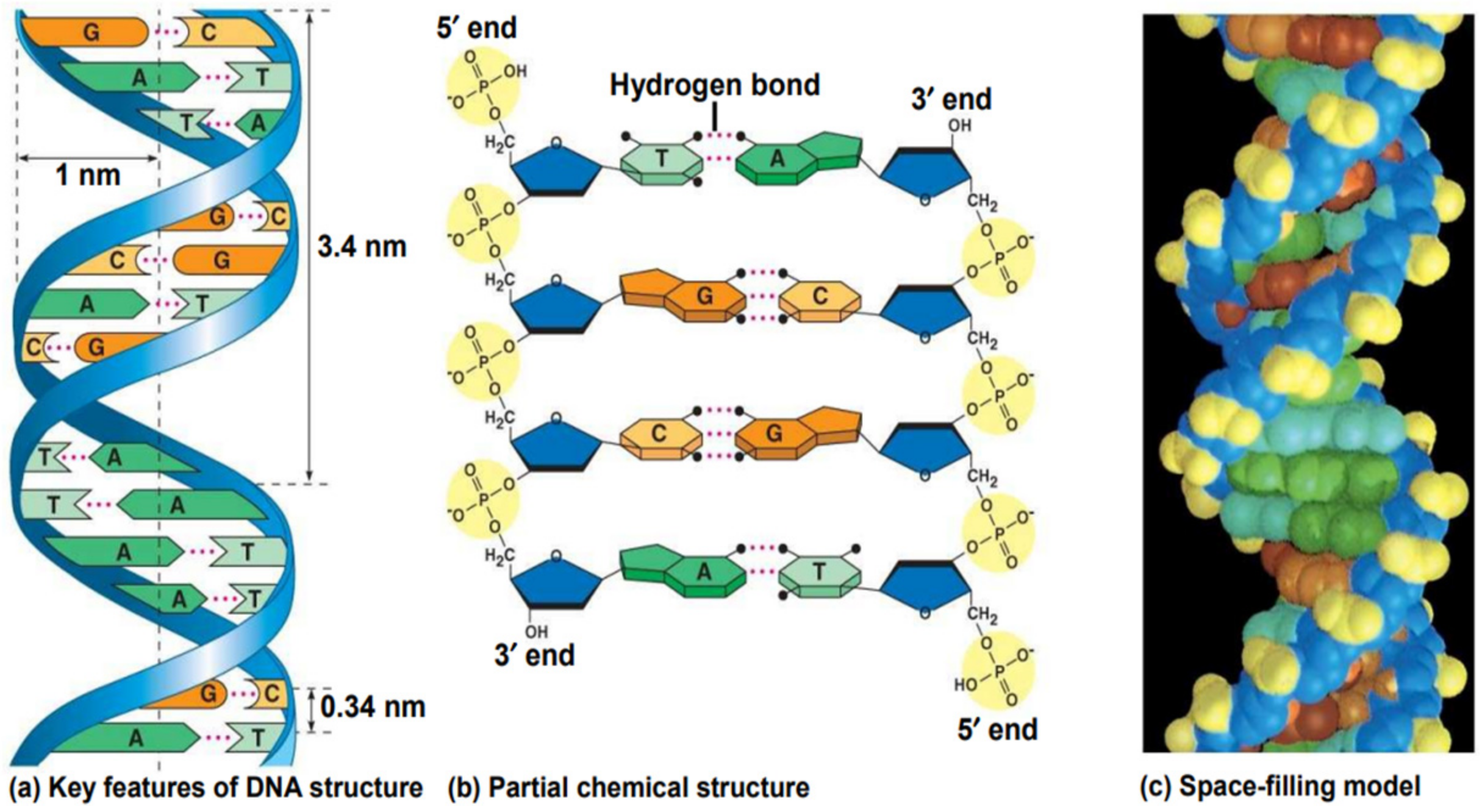
2. DNA Structure
3. Types of DNA–Metal Complex Interactions
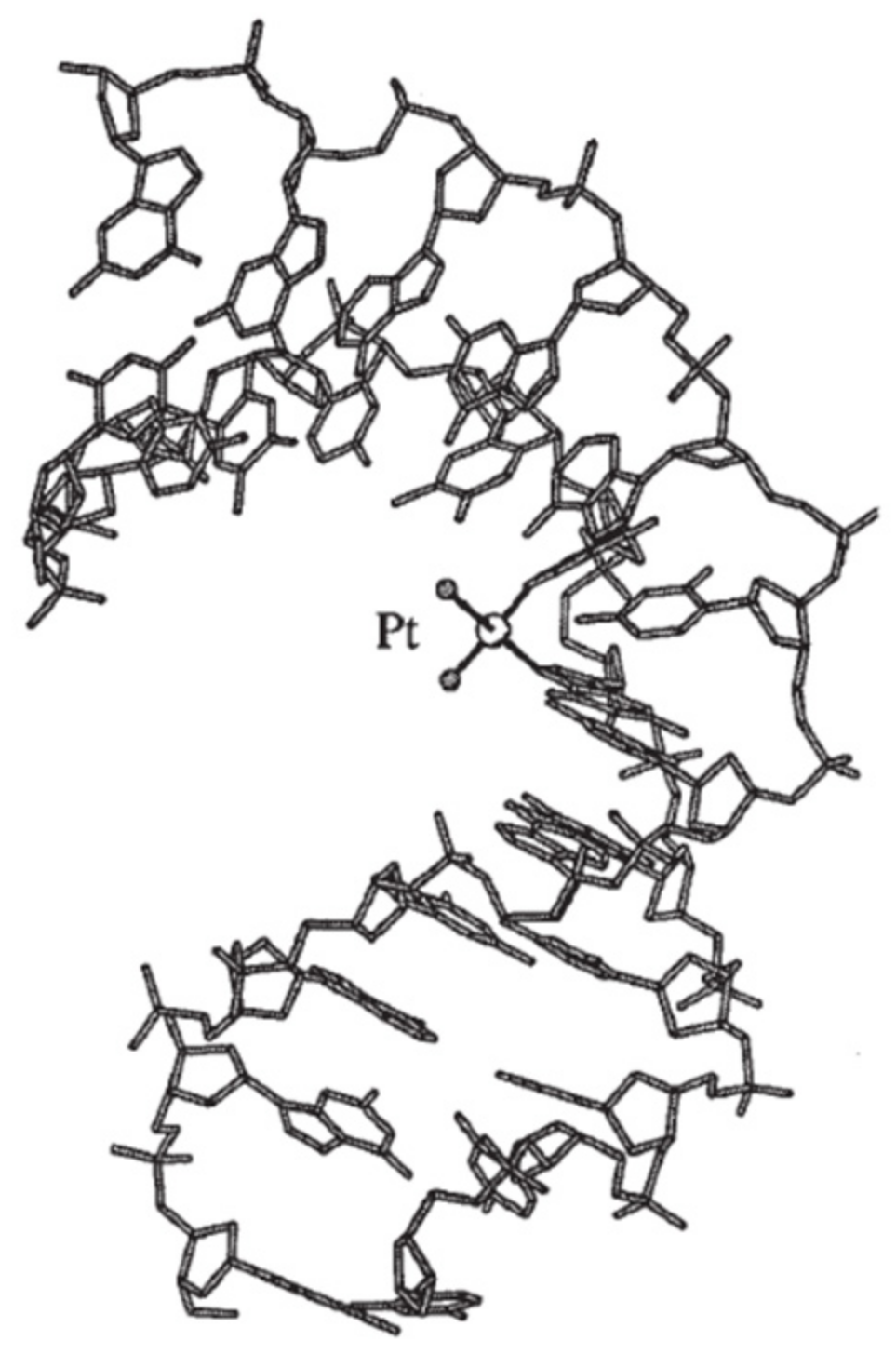
3.1. Intra-/Interstrand Cross-Link
3.2. Intercalators
3.3. Insertors
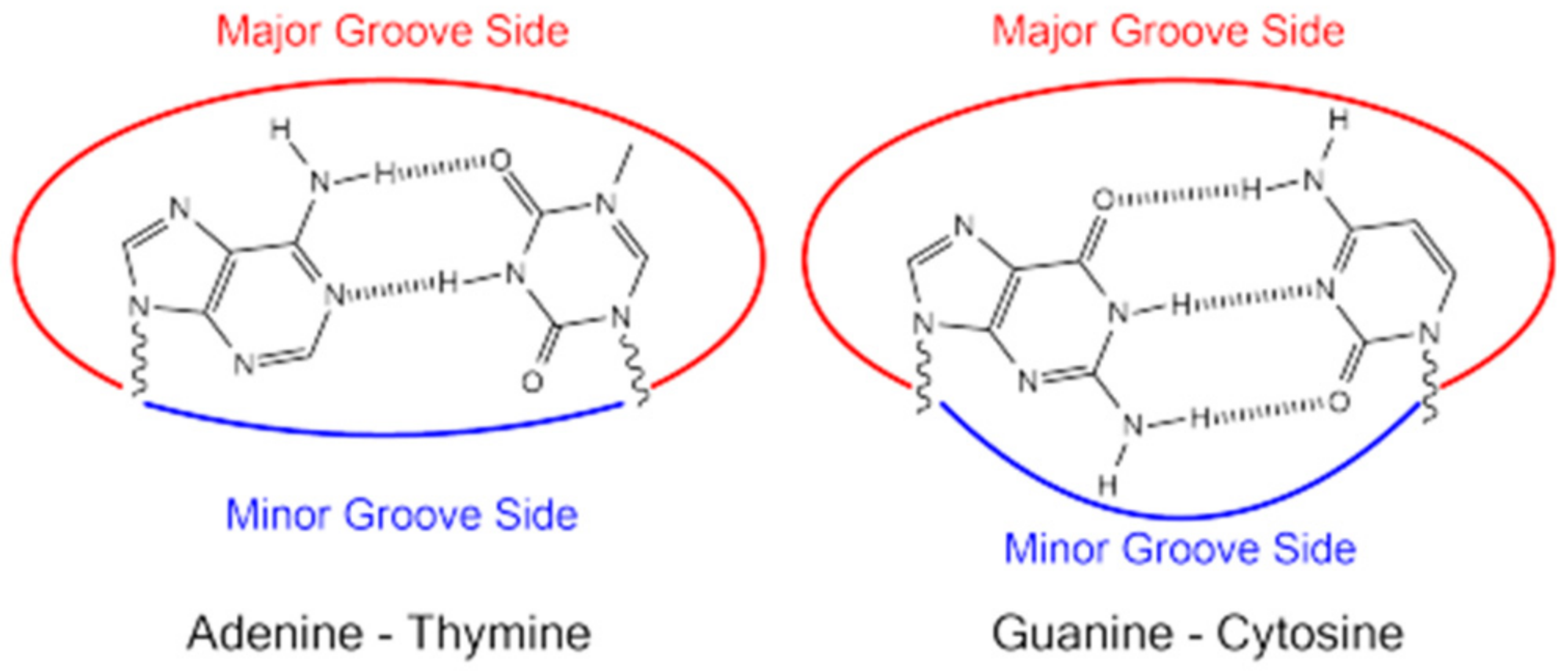
3.4. Major and Minor Groove Binders
3.5. Electrostatic Interactions with the Sugar Phosphate DNA Backbone
4. Methods for Assessment of Metallodrug–DNA Interaction
4.1. Molecular Spectroscopy
Fluorescence Spectroscopic Studies
4.2. Electrochemical Methods
4.3. Atomic Spectroscopy
4.3.1. X-ray Crystallography
4.3.2. NMR Spectrometry
4.3.3. Mass Spectrometry
4.4. Electrophoretic Method
Agarose Gel Electrophoresis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Minchin, S.; Lodge, J. Understanding biochemistry: Structure and function of nucleic acids. Essays Biochem. 2019, 63, 433–456. [Google Scholar] [CrossRef]
- Odani, A. Inorganic biochemistry. In Metal Ions and Complexes in Solution; Yamaguchi, T., Persson, I., Eds.; Royal Society of Chemistry: London, UK, 2023; Volume 2. [Google Scholar]
- Hadjiliadis, N.; Sletten, E. Metal Complexes—DNA Interactions; Wiley: Hoboken, NJ, USA, 2009. [Google Scholar]
- Mehrdad, S.A.; Cucchiarini, A.; Mergny, J.L.; Noureini, S.K. Heavy metal ions interactions with G-quadruplex-prone DNA sequences. Biochemie 2024, 225, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Muthaiah, S.; Bhatia, A.; Kannan, M. Stability of metal complexes. In Stability and Applications of Coordination Copounds; Srivastva, A.N., Ed.; IntechOpen: London, UK, 2020. [Google Scholar]
- Stankovic, M.; Kljun, J.; Stevanovic, N.L.J.; Lazic, J.; Bogojevic, S.S.; Vojnovic, S.; Zlatar, M.; Nikodinovic-Runic, J.; Turel, I.; Djuran, M.I.; et al. Silver (I) complexes containing antifungal azoles: Significant improvement of the anti-Candida potential of the azole drug after its coordination to the silver (I) ion. Dalton Trans. 2024, 53, 2218–2230. [Google Scholar] [CrossRef] [PubMed]
- Stevanovic, N.L.J.; Aleksic, I.; Kljun, J.; Bogojevic, S.S.; Veselinovic, A.; Nikodinovic-Runic, J.; Turel, I.; Djuran, M.I.; Glisic, B.D. Copper(II) and Zinc(II) complexes with the clinically used Fluconazole: Comparison of antifungal activity and therapeutic potential. Pharmaceuticals 2021, 14, 24. [Google Scholar] [CrossRef] [PubMed]
- Alshater, H.; Al-Sulami, A.I.; Aly, S.A.; Abdalla, E.M.; Sakr, M.A.; Hassan, S.S. Antitumor and antibacterial activity of Ni(II), Cu(II), Ag(I), and Hg(II) complexes with ligand derived from thiosemicarbazones: Characterization and theoretical studies. Molecules 2023, 28, 2590. [Google Scholar] [CrossRef] [PubMed]
- de la Mata Moratilla, S.; Casado Angulo, S.; Gómez-Casanova, N.; Copa-Patiño, J.L.; Heredero-Bermejo, I.; de la Mata, F.J.; García-Gallego, S. Zinc(II) Iminopyridine complexes as antibacterial agents: A structure-to-activity study. Int. J. Mol. Sci. 2024, 25, 4011. [Google Scholar] [CrossRef]
- Hangan, A.; Turza, A.; Lucaciu, R.L.; Sevastre, B.; Pall, E.; Oprean, L.S.; Borodi, G. New Cu+2 complexes with N-sulfonamide ligands: Potential antitumor, antibacterial and antioxidant agents. Molecules 2022, 27, 3338. [Google Scholar] [CrossRef]
- Rusu, D.; Stănilă, A.; Marian, I.O.; Marian, C.O.; Rusu, M.; Lucaciu, R. Synthesis and caracterization of some cobalt (II) complexes with amino acids having biological activities. Rev. Chim. 2009, 60, 939–943. [Google Scholar]
- Hubin, T.J.; Amoyaw, P.N.; Roewe, K.D.; Simpson, N.C.; Maples, R.D.; Carder Freeman, T.N.; Cain, A.N.; Le, J.G.; Archibald, S.J.; Khan, S.I.; et al. Synthesis and antimalarial activity of metal complexes of cross-bridged tetraazamacrocyclic ligands. Bioorg. Med. Chem. 2014, 22, 3239–3244. [Google Scholar] [CrossRef]
- Bortolamiol, E.; Visentin, F.; Scattolin, T. Recent advances in bioconjugated transition metal complexes for cancer therapy. Appl. Sci. 2023, 13, 5561. [Google Scholar] [CrossRef]
- Kostova, I. Anticancer metallocenes and metal complexes of transition elements from groups 4 to 7. Molecules 2024, 29, 824. [Google Scholar] [CrossRef] [PubMed]
- Hangan, A.C.; Lucaciu, R.L.; Turza, A.; Dican, L.; Sevastre, B.; Páll, E.; Oprean, L.S.; Borodi, G. New Copper complexes with antibacterial and cytotoxic activity. Int. J. Mol. Sci. 2023, 24, 13819. [Google Scholar] [CrossRef] [PubMed]
- Hangan, A.C.; Stan, R.L.; Turza, A.; Oprean, L.S.; Pall, E.; Gheorghe-Cetean, S.; Sevastre, B. Synthesis, crystal structures, characterization and antitumor activities of two copper(II) complexes of a sulfonamide ligand. Transit. Met. Chem. 2017, 42, 153–164. [Google Scholar] [CrossRef]
- Tsoupras, A.; Pafli, S.; Stylianoudakis, C.; Ladomenou, K.; Demopoulos, C.A.; Philippopoulos, A. Anti-inflammatory and antithrombotic potential of metal-based complexes and porphyrins. Compounds 2024, 4, 376–400. [Google Scholar] [CrossRef]
- Wlodarczyk, J.; Krajewska, J.; Szeleszczuk, L.; Szalwinska, P.; Gurba, A.; Lipiec, S.; Taciak, P.; Szczepaniak, R.; Mlynarzuk-Bialy, I.; Fichna, J. A new Gold(III) complex, TGS 703, shows potent anti-inflammatory activity in colitis via the enzymatic and non-enzymatic antioxidant system—An in vitro, in silico, and in vivo study. Int. J. Mol. Sci. 2024, 24, 7025. [Google Scholar] [CrossRef]
- Abate, C.; Carnamucio, F.; Giuffre, O.; Foti, C. Metal-based compounds in antiviral therapy. Biomolecules 2022, 12, 933. [Google Scholar] [CrossRef]
- Chuong, C.; DuChane, C.M.; Webb, E.M.; Rai, P.; Marano, J.M.; Bernier, C.M.; Merola, J.S.; Weger-Lucarelli, J. Noble metal organometallic complexes display antiviral activity against SARS-CoV-2. Viruses 2021, 13, 980. [Google Scholar] [CrossRef]
- Hangan, A.C.; Stan, R.L.; Sevastre, B.; Gheorghe-Cetean, S.; Oprean, L. DNA cleavage study and SOD-mimetic activity of a new Cu (II) complex. Farmacia 2017, 65, 368–373. [Google Scholar]
- Kumar, P.; Gorai, S.; Santra, M.K.; Mondal, B.; Manna, D. DNA binding, nuclease activity and cytotoxicity studies of Cu(II) complexes of tridentate ligands. Dalton Trans. 2012, 41, 7573–7581. [Google Scholar] [CrossRef]
- Sirajuddin, M.; Ali, S.; Badshah, A. Drug-DNA interactions and their study by UV-Visible, fluorescence and cyclic voltametry. J. Photochem. Photobiol. B Biol. 2013, 124, 1–19. [Google Scholar] [CrossRef]
- Available online: https://muhammad-asif88.medium.com/central-dogma-of-molecular-biology-294d2600a484 (accessed on 9 September 2024).
- Watson, J.D.; Crick, F.H.C. Molecular structure of nucleic acids: A structure for deoxyribose nucleic acid. Nature 1953, 171, 737–738. [Google Scholar] [CrossRef] [PubMed]
- Maddox, B. The Double Helix and the “wronged heroine”. Nature 2003, 421, 407–408. [Google Scholar] [CrossRef] [PubMed]
- Kennelly, P.J.; Botham, K.M.; McGuinness, O.; Rodwell, V.W.; Weil, P.A. Nucleotides and nucleic acid structure & function. In Harper’s Illustrated Biochemistry, 32nd ed.; McGraw: Lange, NY, USA, 2023; pp. 329–337, 348–360. [Google Scholar]
- Harvey, R.A.; Ferrier, D.R. Lippincott’s Illustrated Reviews: Biochemistry, 5th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA; Wolters Kluwer: Alphen aan den Rijn, The Netherlands, 2011. [Google Scholar]
- Berg, J.M.; Tymoczko, J.L.; Gatto, G.J., Jr.; Stryer, L. Biochemistry, 8th ed.; W. H. Freeman and Company: New York, NY, USA, 2015. [Google Scholar]
- Available online: https://discover.hubpages.com/education/DNA-What-is-it-The-replication-process-Consequences-if-it-is-not-carried-out-correctly (accessed on 9 September 2024).
- Cowan, J.A. Bioinorganic Chemistry. An Introduction, 2nd ed.; Wiley-VCH: New York, NY, USA, 1997. [Google Scholar]
- Yakovchuk, P.; Protozanova, E.; Frank-Kamenetskii, M.D. Base-stacking and base-pairing contributions into the thermal stability of the DNA double helix. Nucleic Acids Res. 2006, 34, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Hannon, M.J. Supramolecular DNA recognition. Chem. Soc. Rev. 2007, 36, 280–295. [Google Scholar] [CrossRef] [PubMed]
- Bertini, I.; Gray, H.B.; Lippard, S.J.; Valentine, J.S. Bioinorganic Chemistry; University Science Books: Mill Valley, CA, USA, 1994; ISBN 0-935702-57-1. Available online: http://resolver.caltech.edu/CaltechBOOK:1994.002 (accessed on 9 September 2024).
- Shakked, Z.; Guerstein-Guzikevich, G.; Eisenstein, M.; Frolow, F.; Rabinovich, D. The conformation of the DNA double helix in the crystal is dependent on its environment. Nature 1989, 342, 456–460. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Ramos, J.C.; Galindo-Murillo, R.; Cortez-Guzman, F.; Ruiz-Azuara, L. Metal-based drug-DNA interactions. J. Mex. Chem. Soc. 2013, 57, 245–259. [Google Scholar] [CrossRef]
- Neidle, S. Oxford Handbook of Nucleic Acid Structure; Oxford University Press: New York, NY, USA, 1999. [Google Scholar]
- Hägerlöf, M.; Papsai, P.; Chow, C.S.; Elmroth, S.K.C. More pronounced salt dependence and higher reactivity for platination of the hairpin r(CGCGUUGUUCGCG) compared with d(CGCGTTGTTCGCG). J. Biol. Inorg. Chem. 2006, 11, 974–990. [Google Scholar] [CrossRef]
- François, J.; Thuong, N.T.; Hélène, C. Recognition and cleavage of hairpin structures in nucleic acids by oligodeoxynucleotides. Nucl. Acids Res. 1994, 22, 3943–3950. [Google Scholar] [CrossRef]
- Huppert, J.L. Structure, location and interactions of G-quadruplexes. FEBS J. 2010, 277, 3452–3458. [Google Scholar] [CrossRef]
- Brooks, T.A.; Kendrick, S.; Hurley, L. Making sense of G-quadruplex and i-motif functions in oncogene promoters. FEBS J. 2010, 277, 3459–3469. [Google Scholar] [CrossRef]
- Li, X.; Peng, Y.; Ren, J.; Qu, X. Carboxyl-modified singlewalled carbon nanotubes selectively induce human telomeric imotif formation. Proc. Natl. Acad. Sci. USA 2006, 103, 19658–19663. [Google Scholar] [CrossRef] [PubMed]
- Kendrick, S.; Akiyama, Y.; Hecht, S.M.; Hurley, L.H. The i-motif in the bcl-2 P1 promoter forms an unexpectedly stable structure with a unique 8:5:7 loop folding pattern. J. Am. Chem. Soc. 2009, 131, 17667–17676. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wei, C.; Jia, G.; Wang, X.; Feng, Z.; Li, C. Formation of i-motif structure at neutral and slightly alkaline pH. Mol. Biosyst. 2010, 6, 580–586. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Vasquez, K.M. Triplex technology in studies of DNA damage, DNA repair, and mutagenesis. Biochimie 2011, 93, 1197–1208. [Google Scholar] [CrossRef]
- Marian, E.; Vicas, L.G.; Tunde, J.; Muresan, M.; Stan, R.L.; Sevastre, B.; Diaconeasa, Z.; Ionescu, C.; Hangan, A.C. A comparative study on the biologic activity of Centaurea cyanus versus Calendula officinalis. Farmacia 2017, 65, 940–946. [Google Scholar]
- Pearson, R.G. Acids and bases. Science 1966, 15, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Barone, G.; Terenzi, A.; Lauria, A.; Almerico, A.M.; Leal, J.M.; Busto, N.; Garcia, B. DNA-binding of nickel(II), copper(II) and zinc(II) complexes: Structure–affinity relationships. Coord. Chem. Rev. 2013, 257, 2848–2862. [Google Scholar] [CrossRef]
- Vinje, J.; Parkinson, J.A.; Sadler, P.J.; Brown, T.; Sletten, E. Sequence selective metalation of double-helical oligodeoxyribonucleotides with Pt(II), Mn(II) and Zn(II) ions. Chem. Eur. J. 2003, 9, 1620–1630. [Google Scholar] [CrossRef]
- Almaqwashi, A.A.; Paramanathan, T.; Rouzina, I.; Williams, M.C. Mechanisms of small molecule–DNA interactions probed by single-molecule force spectroscopy. Nucleic Acids Res. 2016, 44, 3971–3988. [Google Scholar] [CrossRef]
- Keene, F.R.; Smith, J.A.; Collins, J.G. Metal complexes as structure-selective binding agents for nucleic acids. Coord. Chem. Rev. 2009, 253, 2021–2035. [Google Scholar] [CrossRef]
- Rilak, A.; Masnikosa, R.; Bratsos, I.; Alessio, E. Chemistry and reactivity of ruthenium(II) complexes: DNA/protein binding mode and anticancer activity are related to the complex structure. Coord. Chem. Rev. 2019, 398, 113011. [Google Scholar] [CrossRef]
- Srivastava, S.K. Transitional metal based anticancer drug: A review on current cancer chemotherapy drug. JETIR 2018, 5, 943–959. [Google Scholar]
- Johnstone, T.C.; Suntharalingam, K.; Lippard, S.J. The next generation of platinum drugs: Targeted Pt(II) agents, nanoparticle delivery and Pt(IV) prodrugs. Chem. Rev. 2016, 116, 3436–3486. [Google Scholar] [CrossRef] [PubMed]
- Cetean, S.; Ciuleanu, T.; Leucuta, D.C.; Cainap, C.; Constantin, A.M.; Cazacu, I.; Cainap, S.; Gherman, A.; Oprean, L.; Hangan, A.; et al. Hypersensitivity reactions to platinum derivatives: Findings of new predictive markers. J. BUON 2015, 20, 1617–1623. [Google Scholar] [PubMed]
- He, Y.; Ding, Y.; Wang, D.; Zhang, W.; Chen, W.; Liu, X.; Qin, W.; Qian, X.; Chen, H.; Guo, Z. HMGB1 bound to cisplatin-DNA adducts undergoes extensive acetylation and phosphorylation in vivo. Chem. Sci. 2015, 6, 2074–2078. [Google Scholar] [CrossRef]
- Stefàno, E.; De Castro, F.; Ciccarese, A.; Muscella, A.; Marsigliante, S.; Benedetti, M.; Fanizzi, F.P. An overview of altered pathways associated with sensitivity to platinum-based chemotherapy in neuroendocrine tumors: Strengths and prospects. Int. J. Mol. Sci. 2024, 25, 8568. [Google Scholar] [CrossRef]
- Takahara, P.M.; Rosenzweig, A.C.; Frederick, C.A.; Lippard, S.J. Crystal structure of double-stranded DNA containing the major adduct of the anticancer drug cisplatin. Nature 1995, 377, 649–652. [Google Scholar] [CrossRef]
- Demeunynck, M.; Bailly, C.; Wilson, W.D. Small Molecule DNA and RNA Binders: From Synthesis to Nucleic Acid Complexes; Wiley-VCH Verlag: Weinheim, Germany, 2003. [Google Scholar]
- Nakamoto, K.; Tsuboi, M.; Strahan, G.D. Drug-DNA Interactions. Structures and Spectra; John Wiley and Sons Ltd.: New York, NY, USA, 2008. [Google Scholar]
- Zeglis, B.M.; Pierre, V.C.; Barton, J.K. Metallo-intercalators and metallo-insertors. Chem. Comm. 2007, 44, 4565–4579. [Google Scholar] [CrossRef]
- Shobha Devi, C.; Thulasiram, B.; Aerva, R.R.; Nagababu, P. Recent advances in copper intercalators as anticancer agents. J. Fluoresc. 2018, 28, 1195–1205. [Google Scholar] [CrossRef]
- Biver, T.; Secco, F.; Venturini, M. Mechanistic aspects of the interaction of intercalating metal complexes with nucleic acids. Coord. Chem. Rev. 2008, 252, 1163–1177. [Google Scholar] [CrossRef]
- Maciel-Flores, C.E.; Lozano-Alvarez, J.A.; Bivián-Castro, E.Y. Recently reported biological activities and action targets of Pt(II)- and Cu(II)-based complexes. Molecules 2024, 29, 1066. [Google Scholar] [CrossRef] [PubMed]
- Jia, F.; Wang, S.; Man, Y.; Kumar, P.; Liu, B. Recent developments in the interactions of classic intercalated Ruthenium compounds: [Ru(bpy)2dppz]2+ and [Ru(phen)2dppz]2+ with a DNA Molecule. Molecules 2019, 24, 769. [Google Scholar] [CrossRef]
- Modrich, P. Mechanisms in eukaryotic mismatch repair. J. Biol. Chem. 2006, 281, 30305–30309. [Google Scholar] [CrossRef] [PubMed]
- Erxleben, A. Investigation of non-covalent interactions of metal complexes with DNA in cell-free systems. Chimia 2017, 71, 102. [Google Scholar] [CrossRef] [PubMed]
- Dumont, E.; Monari, A. Understanding DNA under oxidative stress and sensitization: The role of molecular modeling. Front. Chem. 2015, 3, 43. [Google Scholar] [CrossRef]
- Morris, D.L. DNA-bound metal ions: Recent developments. Biomol. Concepts 2014, 5, 397–407. [Google Scholar] [CrossRef]
- Khan, G.S.; Shah, A.; Zia-ur-Rehman; Baker, D. Chemistry of DNA minor groove binding agents. J. Photochem. Photobiol. B Biol. 2012, 115, 105–118. [Google Scholar] [CrossRef]
- Pages, B.J.; Ang, D.L.; Wright, E.P.; Aldrich-Wright, J.R. Metal complex interactions with DNA. Dalton Trans. 2015, 44, 3505–3526. [Google Scholar] [CrossRef]
- Nelson, S.M.; Ferguson, L.R.; Denny, W.A. Non-covalent ligand/DNA interactions: Minor groove binding agents. Mutat. Res. 2007, 623, 24–40. [Google Scholar] [CrossRef]
- Erxleben, A. Interactions of copper complexes with nucleic acids. Coord. Chem. Rev. 2018, 360, 92–121. [Google Scholar] [CrossRef]
- Galindo-Murillo, R.; Winkler, L.; Garcia-Ramos, J.C.; Ruiz-Azuara, L.; Cortes-Guzman, F.; Cheatham, T.E. Ancillary ligand on thernary Cu(II) complexes guides binding selectivity toward minor-grove DNA. J. Phys. Chem. B 2020, 124, 11648–11658. [Google Scholar] [CrossRef] [PubMed]
- Galindo-Murillo, R.; Garcia-Ramos, J.C.; Ruiz-Azuara, L.; Cheatham, T.E.; Cortes-Guzman, F. Intercalation processes of copper complexes in DNA. Nucleic Acids Res. 2015, 43, 5364–5376. [Google Scholar] [CrossRef] [PubMed]
- Sevastre, B.; Sarpataki, O.; Olah, N.K.; Stan, R.L.; Taulescu, M.; Marcus, I.; Cătoi, C. Antitumor effect of Euonymus Europaeus on Ehrlich tumor cells in vivo. Farmacia 2014, 62, 907–917. [Google Scholar]
- González-Ballesteros, M.M.; Sánchez-Sánchez, L.; Espinoza-Guillén, A.; Espinal-Enríquez, J.; Mejía, C.; Hernández-Lemus, E.; Ruiz-Azuara, L. Antitumoral and antimetastatic activity by mixed chelate Copper(II) compounds (Casiopeínas®) on triple-negative breast cancer, in vitro and in vivo models. Int. J. Mol. Sci. 2024, 25, 8803. [Google Scholar] [CrossRef] [PubMed]
- von Hippel, P.H.; Marcus, A.H. The many roles of binding cooperativity in the control of DNA replication. Biophys. J. 2019, 117, 2143–2146. [Google Scholar] [CrossRef]
- Komeda, S.; Moulaei, T.; Kruger Woods, K.; Chikuma, M.; Farrell, N.P.; Williams, L.D. A third mode of DNA binding: Phosphate clamps by a polynuclear platinum complex. J. Am. Chem. Soc. 2006, 128, 16092–16103. [Google Scholar] [CrossRef]
- Jany, T.; Moreth, A.; Gruschka, C.; Sischka, A.; Spiering, A.; Dieding, M.; Wang, Y.; Haji Samo, S.; Stammler, A.; Bögge, H.; et al. Rational design of a cytotoxic dinuclear Cu2+ complex that binds by molecular recognition at two neighboring phosphates of the DNA backbone. Inorg. Chem. 2015, 54, 2679–2690. [Google Scholar] [CrossRef]
- Li, S.; Yuan, B.; Wang, X.; Zhang, J.; Yue, L.; Hou, H.; Hu, J.; Chen, S. Crystal structure, DNA interaction and in vitro anticancer activity of Cu(II) and Pt(II) compounds based on benzimidazole-quinoline derivative. Polyhedron 2020, 179, 114369. [Google Scholar] [CrossRef]
- Kirthan, B.R.; Prabhakara, M.C.; Bhojya Naik, H.S.; Nayak, P.H.A.; Naik, E.I. Synthesis, characterization, DNA interaction and anti-bacterial studies of Cu(II), Co(II) and Ni(II) metal complexes containing azo-dye ligand. Chem. Data Collect. 2020, 29, 100506. [Google Scholar] [CrossRef]
- Saha, U.; Chatterjee, S.; Dolai, M.; Kumar, G.S. Biophysical and Thermodynamic Investigations on the Differentiation of Fluorescence Response towards Interaction of DNA: A Pyrene-Based Receptor versus Its Fe(III) Complex. Bio Mater. 2020, 3, 7810. [Google Scholar] [CrossRef]
- Abu-Dief, A.M.; Alotaibi, N.H.; Al-Farraj, E.S.; Qasem, H.A.; Alzahrani, S.; Mahfouz, M.K.; Abdou, A. Fabrication, structural elucidation, theoretical, TD-DFT, vibrational calculation and molecular docking studies of some novel adenine imine. J. Molec. Liquids 2021, 326, 115277. [Google Scholar] [CrossRef]
- Kurt, B.; Temel, H.; Atlan, M.; Kaya, S. Synthesis, characterization, DNA interaction and docking studies of novel Schiff base ligand derived from 2,6-diaminopyridine and its complexes. J. Mol. Struct. 2020, 1209, 127928. [Google Scholar] [CrossRef]
- Shinde RG, Khan AA, Barik A, Exploring the interaction of copper-esculetin complex with ct-DNA: Insight from spectroscopic and docking studies. J. Mol. Struct. 2020, 1208, 127901. [CrossRef]
- Kiwaan, H.A.; El-Mowafy, A.S.; El-Bindary, A.A. Synthesis, spectral characterization, DNA binding, catalytic and in vitro cytotoxicity of some metal complexes. J. Molec. Liquids 2023, 326, 115381. [Google Scholar] [CrossRef]
- Baskaran, S.; Krishnan, M.N.; Arumugham, M.; Kumar, R. Synthesis, crystal structure, DNA interaction, DFT analysis and molecular docking studies of copper(ii) complexes with 1-methyl-l -tryptophan and phenanthroline units. J. Mol. Struct. 2022, 1224, 129236. [Google Scholar] [CrossRef]
- Kumar, N.; Kaushal, R.; Awasthi, P. Non-covalent binding studies of transition metal complexes with DNA: A review. J. Mol. Struct. 2023, 1288, 135751. [Google Scholar] [CrossRef]
- Kellett, A.; Molphy, Z.; Slator, C.; McKee, V.; Farrell, N.P. Molecular methods for assessment of non-covalent metallodrug–DNA interactions. Chem. Soc. Rev. 2019, 48, 971–988. [Google Scholar] [CrossRef]
- Vaidyanathan, V.G.; Nair, B.U. Photooxidation of DNA by a cobalt(II) tridentate complex. J. Inorg. Biochem. 2003, 94, 121–126. [Google Scholar] [CrossRef]
- Vijayalakshmi, R. Interaction of DNA with [Cr(Schiff base)(H2O)2]ClO4. Biochim. Biophys. Acta 2000, 1475, 157–162. [Google Scholar] [CrossRef]
- Karacan, P.; Okay, O. Ethidium bromide binding to DNA cryogels. React. Funct. Polym. 2013, 73, 442–450. [Google Scholar] [CrossRef]
- Phukan, S.; Mitra, S. Fluorescence behavior of ethidium bromide in homogeneous solvents and in presence of bile acid hosts. J. Photochem. Photobiol. A Chem. 2012, 244, 9–17. [Google Scholar] [CrossRef]
- Nafisi, S.; Saboury, A.A.; Keramat, N.; Neault, J.F.; Tajmir-Riahi, H.A. Stability and structural features of DNA intercalation with ethidium bromide, acridine orange and methylene blue. J. Mol. Struct. 2007, 827, 35–43. [Google Scholar] [CrossRef]
- Sathyadevi, P.; Krishnamoorthy, P.; Butorac, R.R.; Cowley, A.H.; Bhuvanesh, N.S.P.; Dharmaraj, N. Effect of substitution and planarity of the ligand on DNA/BSA interaction, free radical scavenging and cytotoxicity of diamagnetic Ni(II) complexes: A systematic investigation. Dalton Trans. 2011, 40, 9690–9702. [Google Scholar] [CrossRef] [PubMed]
- Arjmand, F.; Parveen, S.; Afzal, M.; Shahid, M. Synthesis, characterization, biological studies (DNA binding, cleavage, antibacterial and topoisomerase I) and molecular docking of copper(II) benzimidazole complexes. J. Photochem. Photobiol. B Biol. 2012, 114, 15–26. [Google Scholar] [CrossRef]
- Lepecq, J.B.; Paoletti, C. A fluorescent complex between ethidium bromide and nucleic acids. J. Mol. Biol. 1967, 27, 87–106. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gimenez, J.L.; Gonzalez-Alvarez, M.; Liu-Gonzalez, M.; Macias, B.; Borras, J.; Alzuet, G. Toward the development of metal-based synthetic nucleases: DNA binding and oxidative DNA cleavage of a mixed copper(II) complex with N-(9H-purin-6-yl)benzenesulfonamide and 1,10-phenantroline. Antitumor activity in human Caco-2 cells and Jurkat T lymphocytes. Evaluation of p53 and Bcl-2 proteins in the apoptotic mechanism. J. Inorg. Biochem. 2009, 103, 923–934. [Google Scholar] [CrossRef]
- Aslanoglu, M. Electrochemical and spectroscopic studies of the interaction of proflavine with DNA. Anal. Sci. 2006, 22, 439–443. [Google Scholar] [CrossRef]
- Shah, A.; Zaheer, M.; Qureshi, R.; Akhter, Z.; Nazar, M.F. Voltammetric and spectroscopic investigations of 4-nitrophenylferrocene interacting with DNA. Spectrochim. Acta A 2010, 75, 1082–1087. [Google Scholar] [CrossRef]
- Ngoepe, M.; Clayton, H. Metal complexes as DNA synthesis and/or repair inhibitors: Anticancer and antimicrobial agents. Pharm. Fronts 2021, 03, e164–e182. [Google Scholar] [CrossRef]
- Mucha, P.; Hikisz, P.; Gwoździński, K.; Krajewska, U.; Leniart, A.; Budzisz, E. Cytotoxic effect, generation of reactive oxygen/nitrogen species and electrochemical properties of Cu(II) complexes in comparison to half-sandwich complexes of Ru(II) with aminochromone derivatives. SC Adv. 2019, 9, 31943–31952. [Google Scholar] [CrossRef]
- Garman, E.F. Developments in x-ray crystallographic structure determination of biological macromolecules. Science 2014, 343, 1102–1108. [Google Scholar] [CrossRef] [PubMed]
- Helliwell, J.R.; Mitchell, E.P. Synchrotron radiation macromolecular crystallography: Science and spin-offs. IUCrJ 2015, 2, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Hangan, A.; Borras, J.; Liu-Gonzalez, M.; Oprean, L. Synthesis, crystal structures and properties of [Cu (L1)2(py)2(H2O)](H2O) [HL1 = N-(5-ethyl-[1,3,4]—Thiadiazole-2-yl)-toluenesulfonamidate] and [Cu (L2)2(py)2(H2O)] [HL2 = N-(5-ethyl-[1,3,4]—Thiadiazole-2-yl)-benzenesulfonamidate]. Z. Anorg. Allg. Chem. 2007, 633, 1837–1841. [Google Scholar] [CrossRef]
- Barton, J.K.; Boynton, A.N.; Boyle, K.M. Targeting DNA mismatches with coordination complexes. In DNA-Targeting Molecules as Therapeutic Agents; Waring, M.J., Ed.; Royal Society of Chemistry: Cambridge, UK, 2018; pp. 367–390. [Google Scholar] [CrossRef]
- Da Vela, S.; Svergun, D.I. Methods, development and applications of small-angle X-ray scattering to characterize biological macromolecules in solution. Curr. Res. Struct. Biol. 2020, 2, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Feigin, L.A.; Svergun, D.I. Structure Analysis by Small-Angle X-ray and Neutron Scattering; Plenum Press: New York, NY, USA, 1987. [Google Scholar]
- Kumar, P.P.P.; Lim, D.K. Gold-Polymer Nanocomposites for future therapeutic and tissue engineering applications. Pharmaceutics 2022, 14, 70. [Google Scholar] [CrossRef]
- Jensen, T.H.; Bech, M.; Bunk, O.; Thomsen, M.; Menzel, A.; Bouchet, A.; Le Duc, G.; Feidenhans, R.; Pfeiffer, F. Brain tumor imaging using small-angle X-ray scattering tomography. Phys. Med. Biol. 2011, 56, 1717. [Google Scholar] [CrossRef]
- Sidhu, S.; Falzon, G.; Hart, S.A.; Fox, J.G.; Lewis, R.A.; Siu, K.K.W. Classification of breast tissue using a laboratory system for small-angle x-ray scattering. Phys. Med. Biol. 2011, 56, 6779. [Google Scholar] [CrossRef]
- Jacques, D.A.; Trewhella, J. Small-angle scattering for structural biology expanding the frontier while avoiding the pitfalls. Protein Sci. 2010, 19, 642–657. [Google Scholar] [CrossRef]
- Allec, N.; Choi, M.; Yesupriya, N.; Szychowski, B.; White, M.R.; Kann, M.G.; Garcin, E.D.; Daniel, M.C.; Badano, A. Small-angle X-ray scattering method to characterize molecular interactions: Proof of concept. Sci. Rep. 2015, 5, 12085. [Google Scholar] [CrossRef]
- Komeda, S.; Qu, Y.; Mangrum, J.B.; Hegmans, A.; Williams, L.D.; Farrell, N.P. The phosphate clamp as recognition motif in platinum. Inorg. Chim. Acta 2016, 452, 25–33. [Google Scholar] [CrossRef]
- Berners-Price, S.J.; Ronconi, L.; Sadler, P.J. Insights into the mechanism of action of platinum anticancer drugs from multinuclear NMR spectroscopy. Prog. Nucl. Magn. Reson. Spectrosc. 2006, 49, 65–98. [Google Scholar] [CrossRef]
- Hangan, A.; Borodi, G.; Filip, X.; Tripon, C.; Morari, C.; Oprean, L.; Filip, C. Structure of N-(5-ethyl-[1,3,4]-thiadiazole-2-yl)toluenesulfonamide by combined X-ray powder diffraction, 13C solid-state NMR and molecular modelling. Acta Crystallogr. B 2010, 66, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Urathamakul, T.; Waller, D.J.; Beck, J.L.; Aldrich-Wright, J.R.; Ralph, S.F. Comparison of mass spectrometry and other techniques for probing interactions between metal complexes and DNA interactions between metal complexes and DNA. Inorg. Chem. 2008, 47, 6621–6632. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Wang, J.; Zhang, X.; Yang, Z.; Zhang, J.C.; Zhao, L.; Peng, H.; Lei, J.; Wang, H.W. Single particle cryo-EM reconstruction of 52 kDa streptavidin at 3.2 Angstrom resolution. Nat. Commun. 2019, 10, 2386. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein, J.L. Cryo-EM captures the dynamics of ion channel opening. Cell 2017, 168, 341–343. [Google Scholar] [CrossRef]
- Benjin, X.; Ling, L. Developments, applications, and prospects of cryo-electron microscopy. Protein Sci. 2020, 29, 872–882. [Google Scholar] [CrossRef]
- Punjani, A.; Rubinstein, J.L.; Fleet, D.J.; Brubaker, M.A. CryoSPARC: Algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 2017, 14, 290–296. [Google Scholar] [CrossRef]
- Goldstein, A.; Soroka, Y.; Frušic-Zlotkin, M.; Popov, I.; Kohen, R. High resolution SEM imaging of gold nanoparticles in cells and tissues. J. Microsc. 2014, 256, 237–247. [Google Scholar] [CrossRef]
- Havrdova, M.; Polakova, K.; Skopalik, J.; Vujtek, M.; Mokdad, A.; Homolkova, M.; Tucek, J.; Nebesarova, J.; Zboril, R. Field emission scanning electron microscopy (FE-SEM) as an approach for nanoparticle detection inside cells. Micron 2014, 67, 149–154. [Google Scholar] [CrossRef]
- Malatesta, M. Transmission electron microscopy for nanomedicine: Novel applications for long-established techniques. Eur. J. Histochem. 2016, 60, 2751. [Google Scholar] [CrossRef]
- Malatesta, M. Transmission electron microscopy as a powerful tool to investigate the interaction of nanoparticles with subcellular structures. Int. J. Mol. Sci. 2021, 22, 12789. [Google Scholar] [CrossRef]
- Rodríguez, M.R.; Lavecchia, M.J.; Parajón-Costa, B.Z.; González-Baró, A.C.; González-Baró, M.R.; Cattáneo, E. DNA cleavage mechanism by metal complexes of Cu(II), Zn(II) and VO(IV) with a schiff-base ligand. Biochimie 2021, 186, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Alaghaz, A.N.; Aldulmani, S. Preparation, structural characterization and DNA binding/cleavage affinity of new bioactive nano-sized metal (II/IV) complexes with oxazon-Schiff’s base ligand. Appl. Organomet. Chem. 2019, 33, e5135. [Google Scholar] [CrossRef]
- Hangan, A.C.; Turza, A.; Stan, R.L.; Sevastre, B.; Páll, E.; Cetean, S.; Oprean, L.S. Synthesis, crystal structure and characterization of new biologically active Cu(II) complexes with ligand derived from N-substituted sulfonamides. J. Chem. Sci. 2016, 128, 815–824. [Google Scholar] [CrossRef]
- Yadav, A.; Poonia, K. Recent advances on the DNA interaction properties of Schiff bases and their metal complexes. Appl. Organomet. Chem. 2024, 38, e7496. [Google Scholar] [CrossRef]
- Hangan, A.C.; Turza, A.; Stan, R.L.; Ștefan, R.; Oprean, L.S. Synthesis, crystal structure, properties and nuclease activity of a new Cu(II) complex [Cu(L)2(py)2(H2O)]. (HL = N-(5-(4-methylphenyl)-[1,3,4]–thiadiazole–2-yl)-toluenesulfonamide). Russ. J. Coord. Chem. 2015, 41, 395–404. [Google Scholar] [CrossRef]
- Fox, K.R. Methods in Molecular Biology. Drug-DNA Interaction Protocols; Humana Press: Totowa, NJ, USA, 2007. [Google Scholar]
- Macias, B.; Villa, M.V.; Lapresa, R.; Alzuet, G.; Hernandez-Gil, J.; Sanz, F. Mn(II) complexes with sulfonamides as ligands. DNA interaction studies and nuclease activity. J. Inorg. Biochem. 2012, 115, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Chaires, J.B. Thermal denaturation of drug–DNA complexes. In DNA-Targeting Molecules as Therapeutic Agents; Waring, M.J., Ed.; Royal Society of Chemistry: Cambridge, UK, 2018; pp. 74–95. [Google Scholar] [CrossRef]
- Mudasir, M.; Wahyuni, E.T.; Tjahjono, D.H.; Yoshioka, N.; Inoue, H. Spectroscopic studies on the thermodynamic and thermal denaturation of the ct-DNA binding of methylene blue. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2010, 77, 528–534. [Google Scholar] [CrossRef]
- Jaividhya, P.; Dhivya, R.; Akbarsha, M.A.; Palaniandavar, M. Efficient DNA cleavage mediated by mononuclear mixed ligand copper(II) phenolate complexes: The role of co-ligand planarity on DNA binding and cleavage and anticancer activity. J. Inorg. Biochem. 2012, 114, 94–105. [Google Scholar] [CrossRef]
- Raman, N.; Jeyamurugan, R.; Sakthivel, A.; Mitu, L. Novel metal-based pharmacologically dynamic agents of transition metal(II) complexes: Designing, synthesis, structural elucidation, DNA binding and photo-induced DNA cleavage activity. Spectrochim. Acta A 2010, 75, 88–97. [Google Scholar] [CrossRef]
- Prisecaru, A.; Molphy, Z.; Kipping, R.G.; Peterson, E.J.; Qu, Y.; Kellett, A.; Farrell, N.P. The phosphate clamp: Sequence selective nucleic acid binding profiles and conformational induction of endonuclease inhibition by cationic Triplatin complexes. Nucleic Acids Res. 2014, 42, 13474–13487. [Google Scholar] [CrossRef] [PubMed]
- García-Giménez, J.L.; Hernández-Gil, J.; Martínez-Ruíz, A.; Castiñeiras, A.; Liu-Gonzáles, M.; Pallardó, F.V.; Borrás, J.; Alzuet Piña, G. DNA binding, nuclease activity, DNA photocleavage and cytotoxic properties of Cu(II) complexes of N-substituted sulfonamides. J. Inorg. Biochem. 2013, 121, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Swathi, M.; Shankar, D.S.; Daravath, S.; Ganji, N.; Lakshmi, P.V.A.; Shivaraj, R. Computational studies, cytotoxicity, DNA inter actions of bioactive novel 2-methoxy 5-trifluoromethyl benzenamine Schiff base metal complexes. Inorg. Chem. Comm. 2023, 153, 110826. [Google Scholar] [CrossRef]
- Pérez, A.; Luque, F.J.; Orozco, M. Dynamics of B-DNA on the microsecond time scale. J. Am. Chem. Soc. 2007, 129, 14739–14745. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, N.M.; Davis, D.R.; Cheatham, T.E., III. Molecular dynamics re-refinement of two different small RNA loop structures using the original NMR data suggest a common structure. J. Biomol. NMR 2012, 53, 321–339. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E.I.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Computat Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef]
- Peters, M.B.; Yang, Y.; Wang, B.; Füsti-Molnár, L.; Weaver, M.N.; Merz, K.M. Structural survey of Zinc containing proteins and the development of the Zinc AMBER force field (ZAFF). J. Chem. Theory Comput. 2010, 6, 2935–2947. [Google Scholar] [CrossRef]
- Sahadevan, M.; Sundaram, M.; Subramanian, K. Quantum mechanical approaches and molecular docking studies of metal based anticancer drugs cis-Diammine glycolato platinum and Diaminocyclohexane oxalatoplatinum structures. Comput. Biol. Chem. 2023, 106, 107940. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metal Complexes Investigated for Interaction with DNA | DNA Type | Identified Interaction | Reference | |
|---|---|---|---|---|
| Metal Ion | Ligand Type | |||
Copper (II) Platinum (II) | 2-((2-(pyridin-2-yl)-1H-benzo[d]imidazol- 1-yl)methyl)quinolone | ct-DNA and pBR322 plasmid DNA | Intercalation | Li et al. [81] |
Cobalt (II) Nickel (II) Copper (II) | 5-methyl-2-phenyl-1,2-dihydro-3Hpyrazole-3-one and 3-methyl-1-phenyl4-[(E)- phenyldiazenyl]−4,5-dihydro-1H-pyrazole5-ol | ct-DNA and pUC-19 DNA | Intercalation | Kirthan et al. [82] |
| Iron (III) | 1-amino pyrene and 2- hydroxy-1-napthaldehyde | ct-DNA | Intercalation | Saha et al. [83] |
| Palladium (II) Vanadium (II) Silver (I) | 1-(Pyridin3-yliminomethyl)-naphthalen-2-ol (HNAP) | ct-DNA | Intercalation | Abu-Dief et al. [84] |
| Rh (III) | 2,2-bypiridine, 5,6-chrysenequinone diimine 2,2-bipyridine, dipyridophenazine | pUC-19 DNA | Insertion | Exleben [67] |
| Copper (II) Iron (III) Palladium (II) | Schiff bases of 2-hydroxy-1-naphthaldehyde and Schiff bases of 4-amino-acetophenone | PcDNA3.1 (-) plasmid DNA | Groove binding | Kurt et al. [85] |
| Copper (II) | Esculetin | ct-DNA | Minor groove binding | Shinde et al. [86] |
| Cobalt (II) Nickel (II) Copper (II) | 2-(4-sulfametazin)hidrazono-5,5dimetilsikloheksan-1,3-dion | ct-DNA | Electrostatic interaction or groove binding | Kiwaan et al. [87] |
| Copper (II) | 1-methyl-l-tryptophan | pBR322 plasmid DNA and ct-DNA | Intercalation or electrostatic interaction | Baskaran et al. [88] |
| Type of Method | Main Advantages | Main Limitations |
|---|---|---|
| Fluorescence spectroscopy |
|
|
| Cyclic voltammetry |
|
|
| X-ray crystallography |
|
|
| Small-angle X-ray scattering |
|
|
| NMR spectrometry |
|
|
| Mass spectrometry |
|
|
| Cryo-electron microscopy |
|
|
| Scanning electron microscopy |
|
|
| Transmission electron microscopy |
|
|
| Agarose gel electrophoresis |
|
|
| Thermal denaturation |
|
|
| Viscosity measurement |
|
|
| Molecular dynamics studies | Studies the structure of a metal compound and determines the way it binds to nucleic acids. | It requires the information obtained by NMR spectroscopy. |
| Quantum mechanical methods |
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hangan, A.C.; Oprean, L.S.; Dican, L.; Procopciuc, L.M.; Sevastre, B.; Lucaciu, R.L. Metal-Based Drug–DNA Interactions and Analytical Determination Methods. Molecules 2024, 29, 4361. https://doi.org/10.3390/molecules29184361
Hangan AC, Oprean LS, Dican L, Procopciuc LM, Sevastre B, Lucaciu RL. Metal-Based Drug–DNA Interactions and Analytical Determination Methods. Molecules. 2024; 29(18):4361. https://doi.org/10.3390/molecules29184361
Chicago/Turabian StyleHangan, Adriana Corina, Luminița Simona Oprean, Lucia Dican, Lucia Maria Procopciuc, Bogdan Sevastre, and Roxana Liana Lucaciu. 2024. "Metal-Based Drug–DNA Interactions and Analytical Determination Methods" Molecules 29, no. 18: 4361. https://doi.org/10.3390/molecules29184361
APA StyleHangan, A. C., Oprean, L. S., Dican, L., Procopciuc, L. M., Sevastre, B., & Lucaciu, R. L. (2024). Metal-Based Drug–DNA Interactions and Analytical Determination Methods. Molecules, 29(18), 4361. https://doi.org/10.3390/molecules29184361