

Profoxydim in Focus: A Structural Examination of Herbicide Behavior in Gas and Aqueous Phases

 , , and
, , and 
Abstract

1. Introduction
2. Material and Methods
2.1. Reagents and Solutions
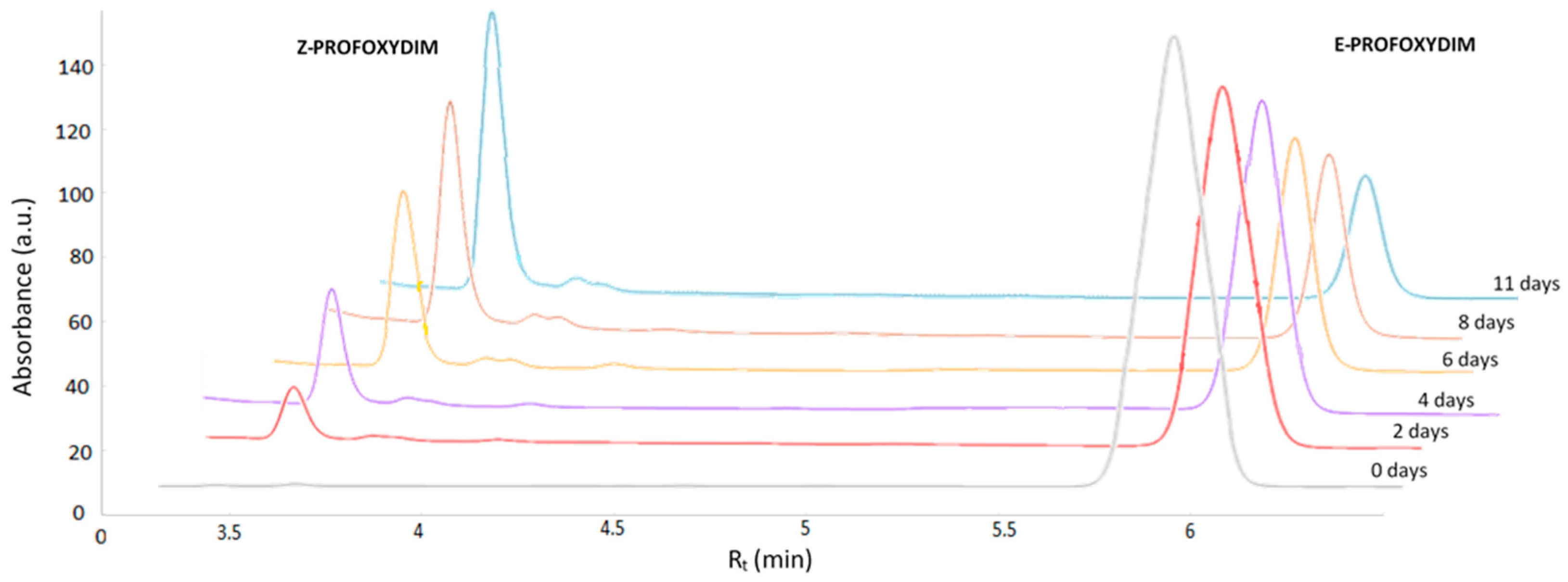
2.2. Isomerizacion Experiments
2.3. Chromatographic Analysis
2.4. NMR Experiments
2.5. Computational Details
2.6. QSAR Models
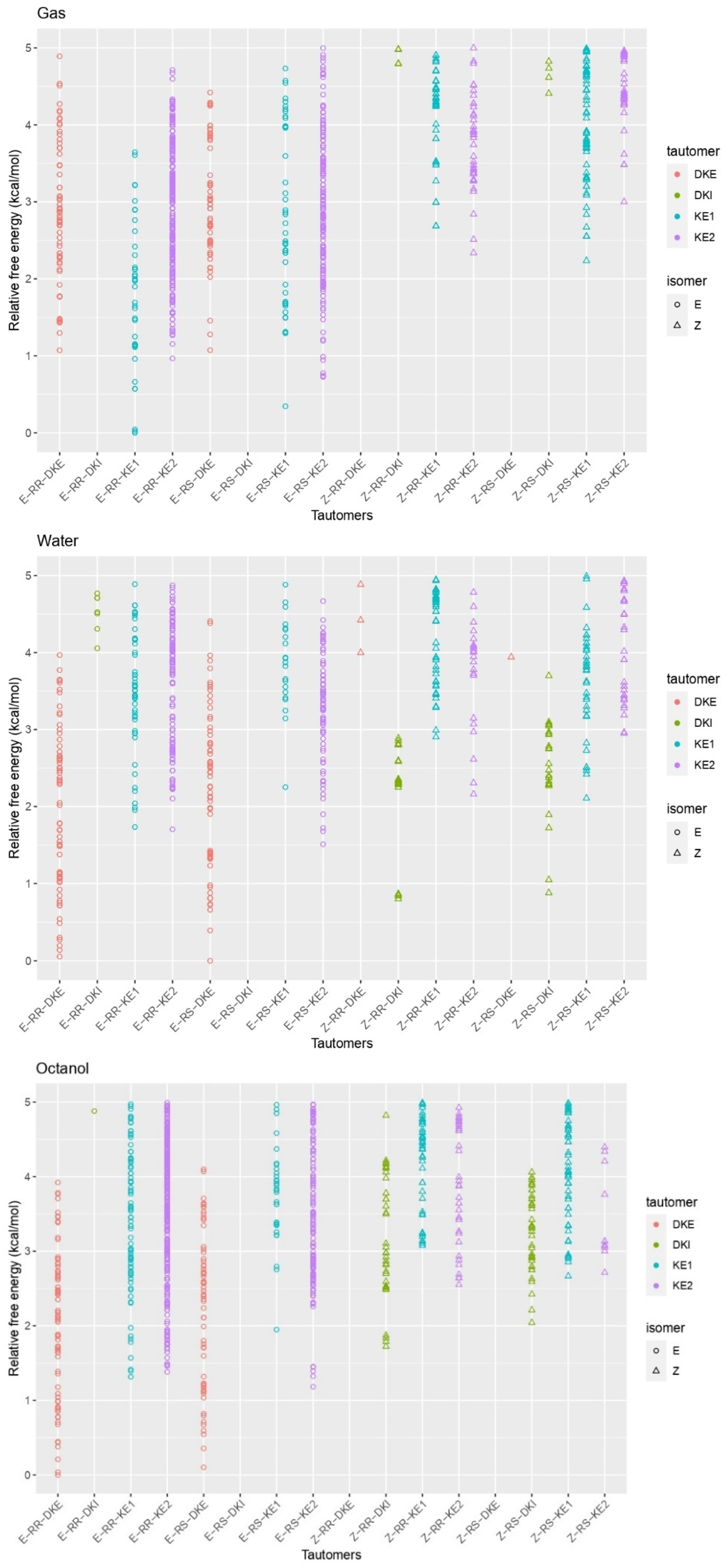
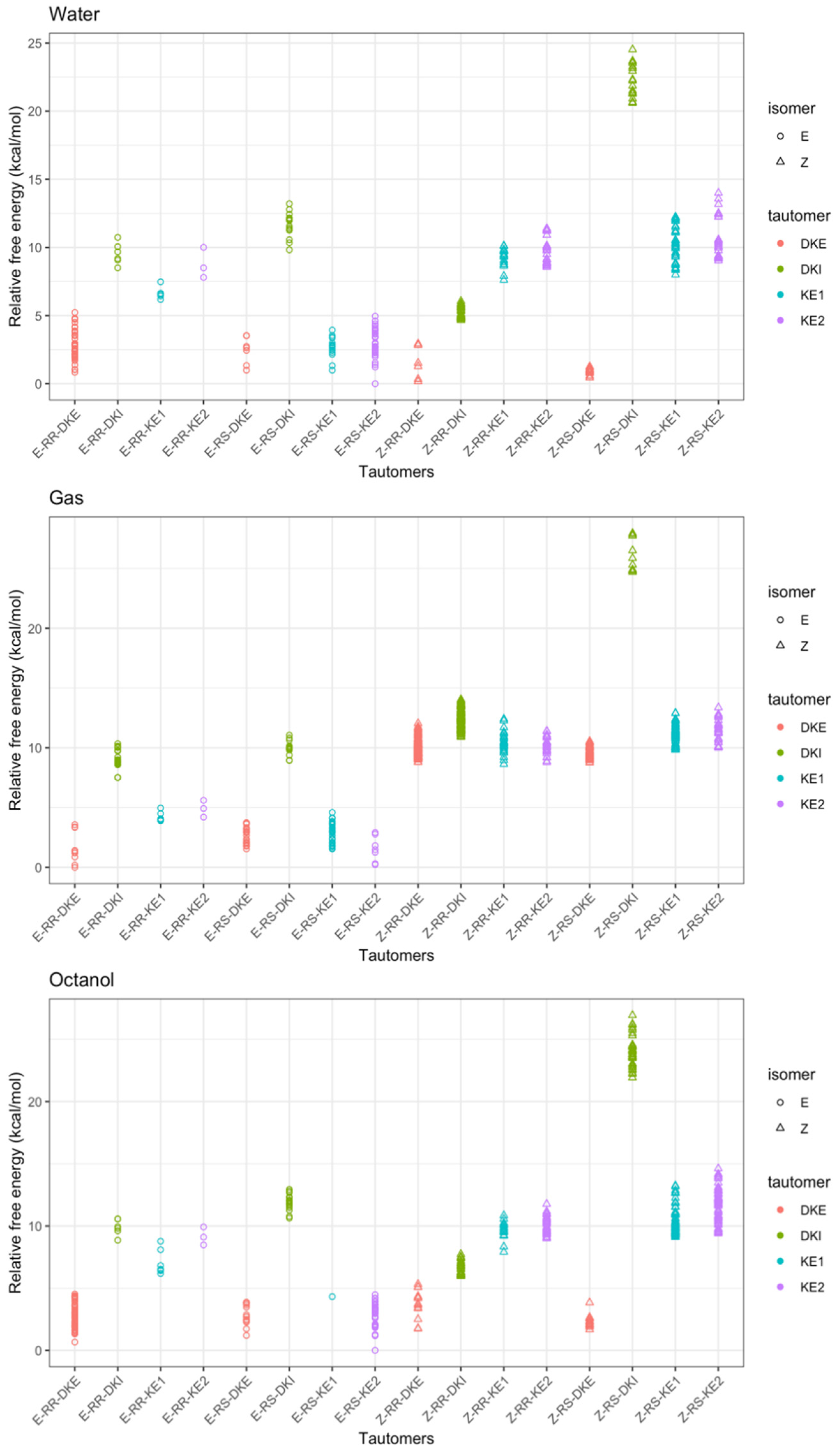
3. Results and Discussion
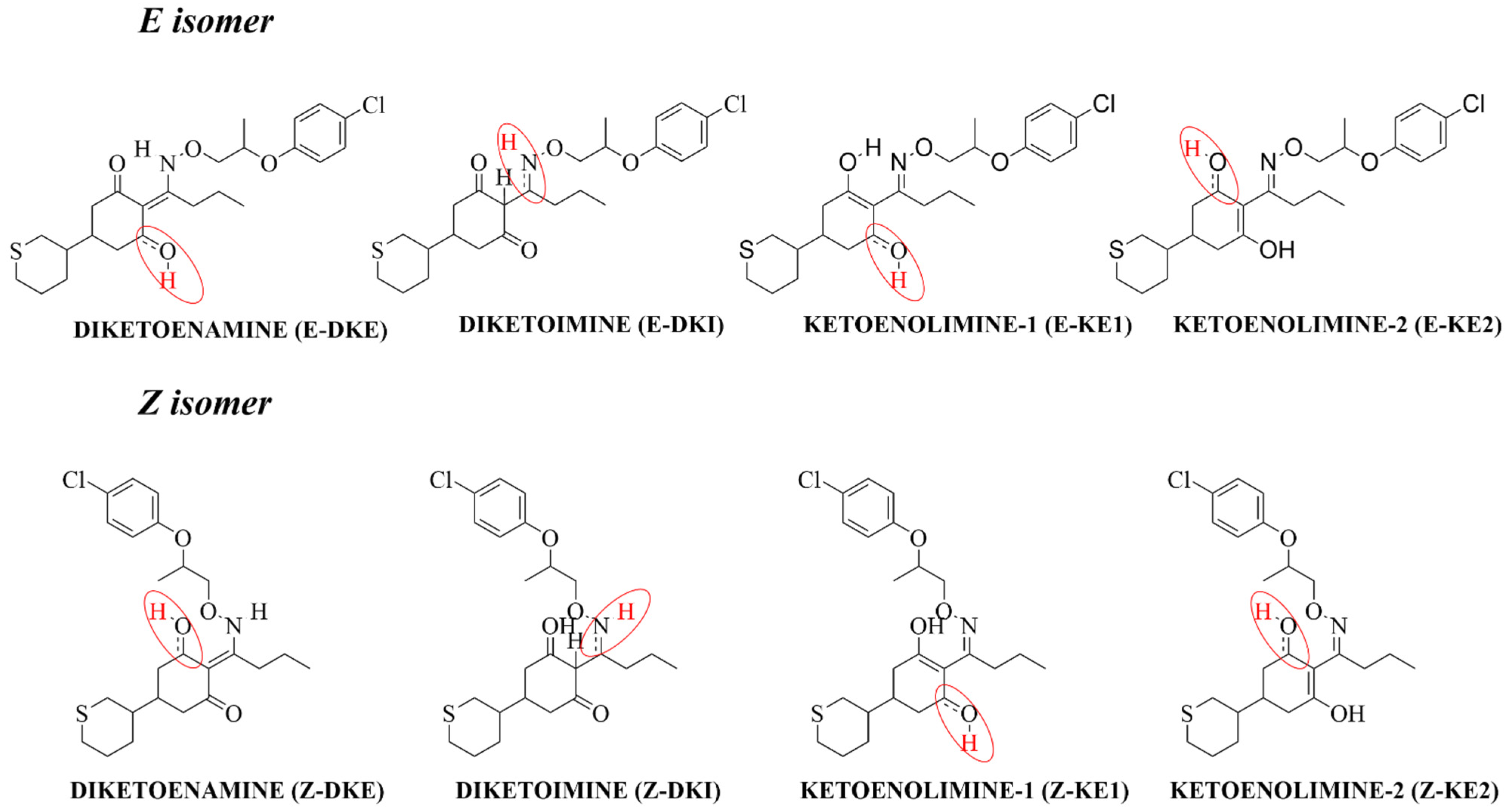
3.1. Structural Analysis
3.2. NMR Analysis
3.3. Effect of pH
3.4. QSAR Models
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Watson, K.G. Cyclohexane–1,3–dione Oxime Ether Grass–Specific Herbicides and the Discovery of Butroxydim. Aust. J. Chem. 2011, 64, 367–372. [Google Scholar] [CrossRef]
- Walter, H. Profoxydim: Development of a Herbicide from Laboratory to Field. In Uso De Herbicidas En La Agricultura Del Siglo XXI; Servicio Publicaciones Universidad de Córdoba: Córdoba, Spain, 2001; pp. 19–30. [Google Scholar]
- Iwataki, I. Cyclohexanedione Herbicides: Their Activities and Properties; CRC Press: Boca Raton, FL, USA, 1992; pp. 397–426. [Google Scholar]
- Torra, J.; Montull, J.M.; Calha, I.M.; Osuna, M.D.; Portugal, J.; de Prado, R. Current status of herbicide resistance in the Iberian Peninsula: Future trends and challenges. Agronomy 2022, 12, 929. [Google Scholar] [CrossRef]
- Travlos, I.; Kanatas, P.; Tsekoura, A.; Gazoulis, I.; Papastylianou, P.; Kakabouki, I.; Antonopoulos, N. Efficacy of different herbicides on Echinochloa colona (L.) link control and the first case of its glyphosate resistance in Greece. Agronomy 2020, 10, 1056. [Google Scholar] [CrossRef]
- European Food Safety Authority (EFSA); Brancato, A.; Brocca, D.; Carrasco Cabrera, L.; De Lentdecker, C.; Ferreira, L.; Greco, L.; Jarrah, S.; Kardassi, D.; Leuschner, R.; et al. Review of the existing maximum residue levels for profoxydim according to Article 12 of Regulation (EC) No 396/2005. EFSA J. 2018, 16, e05282. [Google Scholar] [CrossRef]
- Kacan, K.; Tursun, N.; Ullah, H.; Datta, A. Barnyardgrass (Echinochloa crus–galli (L.) P. Beauv.) resistance to acetolactate synthase–inhibiting and other herbicides in rice in Turkey. Plant Soil Environ. 2020, 66, 357–365. [Google Scholar] [CrossRef]
- Panwar, R.S.; Malik, R.K.; Rathi, S.S. Effect of tralkoxydim and its combination with other new herbicides on the control of weeds in wheat (Triticum aestivum). Indian, J. Agron. 1996, 41, 401–405. [Google Scholar]
- Caputo, M.; Colasurdo, D.D.; Allegretti, P.E.; Laurella, S.L. Tautomeric and E- Zequilibria of the herbicide clethodim in water and organic solvents: A nuclear magnetic resonance and theoretical study. J. Phys. Org. Chem. 2020, 33, e4108. [Google Scholar] [CrossRef]
- McMullan, P.M. Grass Herbicide Efficacy as Influenced by Adjuvant, Spray Solution pH, and Ultraviolet Light. Weed Technol. 1996, 10, 72–77. [Google Scholar] [CrossRef]
- Bridges, D.C.; Falb, L.N.; Smith, A.E. Stability and activity of clethodim as influenced by pH, UV light, and adjuvant. In Adjuvants for Agrichemicals; CRC Press: Boca Raton, FL, USA, 2018; pp. 215–223. [Google Scholar]
- Royneberg, T.; Balke, N.E.; Lund-Hoie, K. Cycloxydim absorption by suspension–cultured velvetleaf (Abutilon theophrasti Medic.) cells. Weed Res. 1994, 34, 1–9. [Google Scholar] [CrossRef]
- Whitwell, T.; Kalmowitz, K.E.; Stapleton, G.S. Postemergence Grass Herbicide Activity Changes with Adjuvant and pH and Sodium Level in Spray Solutions. J. Environ. Hortic. 1992, 10, 55–58. [Google Scholar] [CrossRef]
- Le Coadou, L.; Le Ménach, K.; Labadie, P.; Dévier, M.-H.; Pardon, P.; Augagneur, S.; Budzinski, H. Quality survey of natural mineral water and spring water sold in France: Monitoring of hormones, pharmaceuticals, pesticides, perfluoroalkyl substances, phthalates, and alkylphenols at the ultra–trace level. Sci. Total Environ. 2017, 603, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Boxall, A.B.A. Transformation Products of Synthetic Chemicals in the Environment; Springer: Berlin/Heidelberg, Germany, 2009; pp. 177–204. [Google Scholar]
- Hildebrandt, A.; Guillamón, M.; Lacorte, S.; Tauler, R.; Barceló, D. Impact of pesticides used in agriculture and vineyards to surface and groundwater quality (North Spain). Water Res. 2008, 42, 3315–3326. [Google Scholar] [CrossRef] [PubMed]
- Boxall, A.B.A.; Sinclair, C.J.; Fenner, K.; Kolpin, D.; Maund, S.J. Peer Reviewed: When Synthetic Chemicals Degrade in the Environment. Environ. Sci. Technol. 2004, 38, 368A–375A. [Google Scholar] [CrossRef] [PubMed]
- Cervantes-Díaz, A.; Alonso-Prados, E.; Alonso-Prados, J.L.; Sandín-España, P. Assessing the effect of organic amendments on the degradation of profoxydim in paddy soils: Kinetic modeling and identification of degradation products. Sci. Total Environ. 2024, 912, 169072. [Google Scholar] [CrossRef] [PubMed]
- Santín-Montanyá, M.I.; Patiño-Ropero, M.J.; Cervantes-Díaz, Á.; López-Goti, C.; Alonso-Prados, J.L.; Sandín-España, P. Impacto del uso de la enmienda orgánica compost de alperujo en la eficacia de herbicidas y en el rendimiento del cultivo de arroz. Braz. J. Anim. Environ. Res. 2024, 7, e71889. [Google Scholar] [CrossRef]
- Sandín-España, P.; Sevilla-Morán, B.; Alonso-Prados, J.L.; Santín-Montanya, I.S. Chemical Behaviour and Herbicidal Activity of Cyclohexanedione Oxime Herbicides. In Herbicides: Properties, Synthesis and Control of Weeds; BoD: Norderstedt Germany, 2012; pp. 75–102. [Google Scholar]
- Cervantes-Díaz, A.; Mateo-Miranda, M.; Torrado-Cubero, N.H.; Alonso-Prados, J.L.; Sandín-España, P. Stereoisomeric separation of the chiral herbicide profoxydim and residue method development in rice by QuEChERS and LC–MS/MS. Food Chem. 2021, 443, 138536. [Google Scholar] [CrossRef]
- Turner, J.A. The Pesticide Manual: A World Compendium, 17th ed.; British Crop Protection Council: Alton, UK, 2015; pp. 924–925. [Google Scholar]
- Bannwarth, C.; Caldeweyher, E.; Ehlert, S.; Hansen, A.; Pracht, P.; Seibert, J.; Spicher, S.; Grimme, S. Extended tight-binding quantum chemistry methods. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2021, 11, e1493. [Google Scholar] [CrossRef]
- Pracht, P.; Bohle, F.; Grimme, S. Automated exploration of the low–energy chemical space with fast quantum chemical methods. Phys. Chem. Chem. Phys. 2020, 22, 7169–7192. [Google Scholar] [CrossRef]
- Grimme, S.; Bohle, F.; Hansen, A.; Pracht, P.; Spicher, S.; Stahn, M. Efficient Quantum Chemical Calculation of Structure Ensembles and Free Energies for Non rigid Molecules. J. Phys. Chem. A 2021, 125, 4039–4054. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Grimme, S.; Brandenburg, J.G.; Bannwarth, C.; Hansen, A. Consistent structures and interactions by density functional theory with small atomic orbital basis sets. J. Chem. Phys. 2015, 143, 054107. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S. Density functional theory with London dispersion corrections. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 211–228. [Google Scholar] [CrossRef]
- Chai, J.–D.; Head-Gordon, M. Systematic optimization of long–range corrected hybrid density functionals. J. Chem. Phys. 2008, 128, 084106–084115. [Google Scholar] [CrossRef] [PubMed]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Wolinski, K.; Hinton, J.F.; Pulay, P. Efficient implementation of the gauge–independent atomic orbital method for NMR chemical shift calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar] [CrossRef]
- Becke, A.D. Density–functional thermochesmistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.T.; Yang, W.T.; Parr, R.G. Development of the Colle–Salvetti correlation–energy formula into a functional of the electron–density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H. Gaussian 16 Rev. C.01; Gaussian: Wallingford, CT, USA, 2016. [Google Scholar]
- US EPA ECOlogical Structure–Activity Relationship Model (ECOSAR) Class Program. Available online: https://www.epa.gov/tsca-screening-tools/ecological-structure-activity-relationships-ecosar-predictive-model (accessed on 2 February 2022).
- Könemann, H. Quantitative structure–activity relationships in fish toxicity studies Part 1: Relationship for 50 industrial pollutants. Toxicology 1981, 19, 209–221. [Google Scholar] [CrossRef]
- Hermens, J.; Leeuwangh, P.; Musch, A. Quantitative structure–activity relationships and mixture toxicity studies of chloro– and alkylanilines at an acute lethal toxicity level to the guppy (Poecilia reticulata). Ecotoxicol. Environ. Saf. 1984, 8, 388–394. [Google Scholar] [CrossRef]
- Meylan, W.M.; Howard, P.H. Atom/fragment contribution method for estimating octanol–water partition coefficients. J. Pharm. Sci. 1995, 84, 83–92. [Google Scholar] [CrossRef]
- Xu, X.; Chen, J.; Qu, R.; Wang, Z. Oxidation of Tris (2–chloroethyl) phosphate in aqueous solution by UV–activated peroxymonosulfate: Kinetics, water matrix effects, degradation products and reaction pathways. Chemosphere 2017, 185, 833–843. [Google Scholar] [CrossRef] [PubMed]
- Grygorovych, O.; Nevskii, O.; Moskalenko, S.; Pivovarenko, V.; Doroshenko, A. Protolytic properties of the structurally rigid analogs of 2,6–distyrylpyridine. Widening the pH sensitivity range by the photochemical E→Z isomerisation and introduction of substituents capable to protolytic interactions. Open Chem. 2010, 8, 766–782. [Google Scholar] [CrossRef]
- Landge, S.M.; Aprahamian, I. A pH Activated Configurational Rotary Switch: Controlling the E/Z Isomerization in Hydrazones. J. Am. Chem. Soc. 2009, 131, 18269–18271. [Google Scholar] [CrossRef] [PubMed]
- Nsikabaka, S.; Harb, W.; Ruiz-López, M.F. The role of water on the acid–promoted E/Z isomerization of oximes in aqueous solution. J. Mol. Struct. THEOCHEM 2006, 764, 161–166. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Exp. δ | Exp. δ | E–DKE | E–DKI | E–KE1 | E–KE2 | |
|---|---|---|---|---|---|---|
| (ppm) CH3OD | (ppm) CDCl3 | |||||
| Diketone ring | ||||||
| H4a | 2.19 | 2.19 | 1.78 | 1.94 | 2.13 | 2.54 |
| H4b | 2.10 | 2.13 | 2.23 | 3.09 | 2.67 | 2.37 |
| H5 | 1.99 | 2.03 | 0.50 | 3.45 | 3.18 | 1.38 |
| H6a | 2.36 | 2.41 | 2.23 | 2.80 | 2.52 | 2.60 |
| H6b | 2.32 | 2.38 | 1.63 | 2.01 | 1.88 | 2.64 |
| C–chain | ||||||
| H8a | 2.36 | 2.4 | 2.90 | 2.06 | 2.13 | 2.77 |
| H8b | 2.42 | 2.43 | 2.47 | 2.65 | 1.65 | 2.72 |
| H9a,b | 1.37 | 1.50 | 1.53 | 1.87 | 1.31 | 1.78 |
| 1.64 | 0.90 | 1.40 | 1.13 | |||
| H10a | 0.85 | 0.95 | 1.11 | –0.07 | 0.81 | 0.15 |
| H10b | 0.82 | 0.93 | 0.97 | –0.96 | 0.75 | –0.72 |
| H10c | 0.80 | 0.92 | 0.92 | –0.02 | 1.00 | 0.23 |
| Aromatic | ||||||
| H12 | 6.99 | 6.89 | 6.94 | 7.09 | 7.23 | 7.07 |
| H13 | 7.21 | 7.24 | 7.43 | 7.45 | 7.26 | 7.37 |
| H15 | 7.21 | 7.24 | 7.31 | 7.54 | 7.81 | 7.42 |
| H16 | 6.99 | 6.89 | 6.95 | 7.32 | 7.15 | 7.10 |
| C–chain (O–O) | ||||||
| H17 | 4.93–4.71 | 4.68–4.57 | 4.87 | 5.25 | 5.15 | 5.19 |
| H18 | 4.20–4.04 | 4.26–4.16 | 4.30 | 4.24 | 4.16 | 4.21 |
| 4.15 | 4.29 | 3.97 | 4.24 | |||
| H19a,b,c | 1.30 | 1.36 | 0.80 | 1.52 | 1.10 | 0.45 |
| 0.96 | 0.82 | 1.48 | 0.95 | |||
| 1.60 | 0.99 | 1.10 | 1.03 | |||
| Thiane ring | ||||||
| H20 | 1.59 | 1.61 | 0.95 | 1.34 | 1.39 | 1.51 |
| H21a | 2.06 | 2.08 | 1.09 | 1.84 | 1.78 | 0.80 |
| H21b | 1.67 | 1.60 | 1.82 | 1.40 | 1.37 | 1.45 |
| H22a | 2.50 | 2.49 | 1.92 | 1.79 | 2.03 | 1.98 |
| H22b | 2.40 | 2.46 | 1.45 | 1.55 | 1.40 | 0.97 |
| H23a | 1.88 | 1.86 | 2.72 | 2.86 | 2.53 | 2.55 |
| H23b | 1.20 | 1.19 | 2.33 | 2.90 | 2.91 | 2.39 |
| H24a | 2.40 | 2.42 | 2.61 | 2.71 | 2.76 | 2.11 |
| H24b | 2.60 | 2.61 | 2.53 | 3.00 | 2.95 | 2.52 |
| Moving H | ||||||
| NH OH H2 | – | 14.2 | 13.43 (N–H) | 3.74 (H2) | 14.57 (O–H) | 13.62 (O–H) |
| Exp. δ | E–DKE | E–DKI | E–KE1 | E–KE2 | |
|---|---|---|---|---|---|
| (ppm) CH3OD | |||||
| Diketone ring | |||||
| C1 | 193.38 | 204.2 | 220.7 | 205.2 | 187.1 |
| C2 | 110.7 | 110.4 | 78.1 | 112.0 | 116.4 |
| C3 | 193.3 | 206.4 | 224.7 | 192.9 | 204.9 |
| C4 | 39.54 | 44.0 | 45.8 | 37.1 | 43.5 |
| C5 | 38.82 | 38.6 | 28.1 | 31.9 | 38.1 |
| C6 | 39.61 | 44.4 | 45.8 | 44.0 | 34.4 |
| C–chain | |||||
| C7 | 161.70 | 176.4 | 167.4 | 168.6 | 168.5 |
| C8 | 32.16 | 33.6 | 30.3 | 33.5 | 28.3 |
| C9 | 18.86 | 23.7 | 19.6 | 22.8 | 20.9 |
| C10 | 13.73 | 13.5 | 11.0 | 13.3 | 14.8 |
| Aromatic | |||||
| C11 | 157.0 | 160.2 | 161.6 | 163.5 | 161.2 |
| C12 | 117.6 | 115.6 | 122.6 | 125.6 | 114.3 |
| C13 | 129.2 | 134.2 | 133.7 | 134.1 | 134.1 |
| C14 | 125.3 | 133.4 | 132.3 | 135.3 | 132.8 |
| C15 | 129.2 | 133.5 | 133.7 | 133.4 | 134.7 |
| C16 | 117.6 | 122.6 | 116.7 | 123.3 | 123.2 |
| C–chain (O–O) | |||||
| C17 | 73.0 | 70.2 | 75.4 | 75.7 | 71.3 |
| C18 | 76.2 | 76.9 | 79.4 | 78.9 | 77.4 |
| C19 | 16.36 | 12.3 | 13.1 | 15.6 | 13.0 |
| Thiane ring | |||||
| C20 | 42.60 | 45.2 | 42.8 | 39.6 | 39.2 |
| C21 | 28.01 | 30.4 | 28.9 | 28.4 | 31.1 |
| C22 | 28.50 | 30.7 | 24.0 | 22.5 | 32.7 |
| C23 | 29.87 | 33.2 | 34.9 | 34.1 | 33.5 |
| C24 | 31.48 | 35.5 | 35.6 | 35.6 | 37.5 |
| Time (Days) | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 11 | 13 | 15 | 30 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| % Z–Profoxydim | 1 | 8 | 13 | 24 | 25 | 30 | 35 | 42 | 45 | 56 | 62 | 70 | 70 |
| % E–Profoxydim | 99 | 92 | 87 | 76 | 75 | 70 | 65 | 58 | 55 | 44 | 38 | 30 | 30 |
| Tautomers | Acute Toxicity [mg/L] | Chronic Toxicity (ChV) [mg/L] | ||||
|---|---|---|---|---|---|---|
| Fish (LC50) | Daphnid (LC50) | Algae (EC50) | Fish | Daphnid | Algae | |
| E/Z–DKE | 0.376 | 0.068 | 0.025 | 0.0061 | 0.0084 | 0.0110 |
| E/Z–DKI | 0.163 | 0.032 | 0.010 | 0.0022 | 0.0041 | 0.0047 |
| E/Z–KE1 | 0.160 | 0.031 | 0.0098 | 0.0021 | 0.0041 | 0.0046 |
| E/Z–KE2 | 0.160 | 0.031 | 0.0098 | 0.0021 | 0.0041 | 0.0046 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cobos-Escudero, M.; Pla, P.; Cervantes-Diaz, Á.; Alonso-Prados, J.L.; Sandín-España, P.; Alcamí, M.; Lamsabhi, A.M. Profoxydim in Focus: A Structural Examination of Herbicide Behavior in Gas and Aqueous Phases. Molecules 2024, 29, 4371. https://doi.org/10.3390/molecules29184371
Cobos-Escudero M, Pla P, Cervantes-Diaz Á, Alonso-Prados JL, Sandín-España P, Alcamí M, Lamsabhi AM. Profoxydim in Focus: A Structural Examination of Herbicide Behavior in Gas and Aqueous Phases. Molecules. 2024; 29(18):4371. https://doi.org/10.3390/molecules29184371
Chicago/Turabian StyleCobos-Escudero, María, Paula Pla, Álvaro Cervantes-Diaz, José Luis Alonso-Prados, Pilar Sandín-España, Manuel Alcamí, and Al Mokhtar Lamsabhi. 2024. "Profoxydim in Focus: A Structural Examination of Herbicide Behavior in Gas and Aqueous Phases" Molecules 29, no. 18: 4371. https://doi.org/10.3390/molecules29184371
APA StyleCobos-Escudero, M., Pla, P., Cervantes-Diaz, Á., Alonso-Prados, J. L., Sandín-España, P., Alcamí, M., & Lamsabhi, A. M. (2024). Profoxydim in Focus: A Structural Examination of Herbicide Behavior in Gas and Aqueous Phases. Molecules, 29(18), 4371. https://doi.org/10.3390/molecules29184371