
Reaction Rate Rules of Intramolecular H-Migration Reaction Class for RIORIIOO·Radicals in Ether Combustion

Abstract
1. Introduction
2. Results and Discussion
2.1. Energy Barriers
2.1.1. Validation of the Energy Barriers by the CBS–QB3 Method
2.1.2. Energy Barriers for RIORIIOO· Radicals
2.2. Reaction Enthalpies
2.3. Rate Constants and Rate Rules at High-Pressure-Limit
2.3.1. Comparison of Rate Constants by Isodesmic Reaction Method with CBS–QB3 Method
2.3.2. Comparison of the Rate Constants with Values in the Literature for Alkanes
2.3.3. Comparison of the Rate Constants in Analogous Reaction Subclass for RIORIIOO· in Ethers with HOROO· in Alcohols and ROO· in Alkanes
2.3.4. The Rate Rules at High-Pressure-Limit for RIORIIOO· Radicals
3. Methods
3.1. Electronic Structure Calculation
3.2. Calculation of the Rate Constant
4. Conclusions
- (1)
- After adjusting the energy barriers and reaction enthalpies for the target reaction at the M06–2X level using the isodesmic reaction method, the calculated values align closely with CBS–QB3 results and data in the literature.
- (2)
- The overall reaction class exhibits a maximum deviation in energy barriers and rate constants that exceeds chemical accuracy, necessitating the subdivision of H–migration reaction into distinct subclasses.
- (3)
- The high-pressure-limit rate constants derived in this work diverge from values in the literature for alkanes, with the ratio ranging from 1.06 to 2.11 × 104 at 500 K. The presence of C-O-C bonds results in a weakening of the C-H bond adjacent to the ether group.
- (4)
- By comparing the rate constants for ROO·, RIORIIOO·, and HOROO· radicals, we determined that the rate constants for the H–migration reaction of HOROO· radicals are greater than those for ROO· and RIORIIOO· radicals. The maximum deviation in rate constants between RIORIIOO· and HOROO· radicals span two orders of magnitude. Consequently, it is imperative to establish separate rate rules for the H–migration reaction of RIORIIOO· radicals, rather than adopting kinetic parameters from alcohols or alkane fuels with analogous reactions. Additionally, the rate constant derived using the reaction rate rule method exhibits a certain degree of deviation. It is essential to calculate the rate constant for the elementary reaction of ether fuels precisely.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pitsch, H.; Goeb, D.; Cai, L.; Willems, W. Potential of oxymethylene ethers as renewable diesel substitute. Prog. Energy Combust. Sci. 2024, 104, 101173. [Google Scholar] [CrossRef]
- Michelbach, C.A.; Tomlin, A.S. Automatic mechanism generation for the combustion of advanced biofuels: A case study for diethyl ether. Int. J. Chem. Kinet. 2024, 56, 233–262. [Google Scholar] [CrossRef]
- Tommaso, S.D.; Rotureau, P.; Adamo, C. Oxidation mechanism of aliphatic ethers: Theoretical insights on the main reaction channels. J. Phys. Chem A 2012, 116, 9010–9019. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Li, J.Q.; Chen, S.Y.; Zhu, Q.; Li, Z.R. Investigating the kinetics of the intramolecular H-migration reaction class of methyl-ester peroxy radicals in low-temperature oxidation mechanisms of biodiesel. Phys. Chem. Chem. Phys. 2023, 25, 32078–32092. [Google Scholar] [CrossRef] [PubMed]
- Van de Vijver, R.; Vandewiele, N.M.; Bhoorasingh, P.L.; Slakman, B.L.; Khanshan, F.S.; Carstensen, H.H.; Reyniers, M.F.; Marin, G.B.; West, R.H.; Geem, K.M.V. Automatic mechanism and kinetic model generation for gas and solution-phase processes: A perspective on best practices, recent advances, and future challenges. Int. J. Chem. Kinet. 2015, 47, 199–231. [Google Scholar] [CrossRef]
- Miyoshi, A. Systematic computational study on the unimolecular reactions of alkylperoxy (RO2), hydroperoxyalkyl (QOOH), and hydroperoxyalkylperoxy (O2QOOH) radicals. J. Phys. Chem. A 2011, 115, 3301–3325. [Google Scholar] [CrossRef]
- Villano, S.M.; Huynh, L.K.; Carstensen, H.H.; Dean, A.M. High-pressure rate rules for alkyl + O2 reactions. 1. The dissociation, concerted elimination, and isomerization channels of the alkyl peroxy radical. J. Phys. Chem. A 2011, 115, 13425–13442. [Google Scholar] [CrossRef]
- Villano, S.M.; Huynh, L.K.; Carstensen, H.H.; Dean, A.M. High-pressure rate rules for alkyl + O2 reactions. 2. the isomerization, cyclic ether formation, and β-scission reactions of hydroperoxy alkyl radicals. J. Phys. Chem. A 2012, 116, 5068–5089. [Google Scholar] [CrossRef]
- Villano, S.M.; Carstensen, H.H.; Dean, A.M. Rate rules, branching ratios, and pressure dependence of the HO2 + olefin addition channels. J. Phys. Chem. A 2013, 117, 6458–6473. [Google Scholar] [CrossRef]
- Yao, X.X.; Pang, W.Q.; Li, T.; Shentu, J.T.; Zhu, Q.; Li, X.X. High-Pressure-Limit and Pressure-Dependent Rate Rules for Unimolecular Reactions Related to Hydroperoxy Alkyl Radicals in Normal-Alkyl Cyclohexane Combustion. 2. Cyclization Reaction Class. J. Phys. Chem. A 2021, 125, 8959–8977. [Google Scholar] [CrossRef]
- Yao, X.X.; Sun, X.L.; Zhu, Y.F. High-pressure limit and pressure-dependent rate rules for β-scission reaction class of hydroperoxyl alkyl hydroperoxyl radicals(•P(OOH)2) in normal-alkyl cyclohexanes combustion. Molecules 2024, 29, 544. [Google Scholar] [CrossRef] [PubMed]
- Battin-Leclerc, F. Detailed chemical kinetic models for the low-temperature combustion of hydrocarbons with application to gasoline and diesel fuel surrogates. Prog. Energy Combust. Sci. 2008, 34, 440–498. [Google Scholar] [CrossRef]
- Ning, H.B.; Gong, C.M.; Tan, N.X.; Li, Z.R.; Li, X.Y. Low- and intermediate-temperature oxidation of ethylcyclohexane: A theoretical study. Combust. Flame 2015, 162, 4167–4182. [Google Scholar] [CrossRef]
- Simmie, J.M. Detailed chemical kinetic models for the combustion of hydrocarbon fuels. Prog. Energy Combust. Sci. 2003, 29, 599–634. [Google Scholar] [CrossRef]
- Orlando, J.J.; Tyndall, G.S.; Wallington, T.J. The atmospheric chemistry of alkoxy radicals. Chem. Rev. 2003, 103, 4657–4689. [Google Scholar] [CrossRef]
- Atkinson, R. Rate constants for the atmospheric reactions of alkoxy radicals: An updated estimation method. Atmos Environ. 2007, 41, 8468–8485. [Google Scholar] [CrossRef]
- Burke, U.; Somers, K.P.; O’Toole, P.; Zinner, C.M.; Marquet, N.; Bourque, G.; Petersen, E.L.; Metcalfe, W.K.; Serinyel, Z.; Curran, H.J. An ignition delay and kinetic modeling study of methane, dimethyl ether, and their mixtures at high pressures. Combust. Flame 2015, 162, 315–330. [Google Scholar] [CrossRef]
- Di Tommaso, S.; Rotureau, P.; Crescenzi, O.; Adamo, C. Oxidation mechanism of diethyl ether: A complex process for a simple molecule. Phys. Chem. Chem. Phys. 2011, 13, 14636–14645. [Google Scholar] [CrossRef]
- Yang, B.; Zhang, Y.J.; Li, Y.Y.; Chen, Z.; Huang, Z.H.; Qi, F. Research progress and prospect of combustion reaction kinetics for carbon neutrality. J. Eng. Therm. 2022, 43, 1993–2008. [Google Scholar]
- Vereecken, L.; Nozière, B. H migration in peroxy radicals under atmospheric conditions. Atmos. Chem. Phys. 2020, 20, 7429–7458. [Google Scholar] [CrossRef]
- Otkjær, R.V.; Jakobsen, H.H.; Tram, C.M.; Kjaergaard, H.G. Calculated hydrogen shift rate constants in substituted alkyl peroxy radicals. J. Phys. Chem. A 2018, 122, 8665–8673. [Google Scholar] [CrossRef] [PubMed]
- Curtiss, L.A.; Raghavachari, K.; Trucks, G.W.; Pople, J.A. Gaussian-2 theory for molecular energies of first- and second-row compounds. J. Chem. Phys. 1991, 94, 7221–7230. [Google Scholar] [CrossRef]
- Curtiss, L.A.; Redfern, P.C.; Raghavachari, K. Gn theory. Comput. Mol. Sci. 2011, 1, 810–825. [Google Scholar] [CrossRef]
- Curtiss, L.A.; Raghavachari, K.; Redfern, P.C.; Rassolov, V.; Pople, J.A. Gaussian-3 (G3) theory for molecules containing first and second-row atoms. J. Chem. Phys. 1998, 109, 7764–7776. [Google Scholar] [CrossRef]
- Curtiss, L.A.; Jones, C.; Trucks, G.W.; Raghavachari, K.; Pople, J.A. Gaussian-1 theory of molecular energies for second-row compounds. J. Chem. Phys. 1990, 93, 2537–2545. [Google Scholar] [CrossRef]
- Tajti, A.; Szalay, P.G.; Császár, A.G.; Kállay, M.; Gauss, J.; Valeev, E.F.; Flowers, B.A.; Vázquez, J.; Stanton, J.F. HEAT: High accuracy extrapolated ab initio thermochemistry. J. Chem. Phys. 2004, 121, 11599–11613. [Google Scholar] [CrossRef]
- Ochterski, J.W.; Petersson, G.A.; Montgomery Jr, J.A. A complete basis set model chemistry. V. Extensions to six or more heavy atoms. J. Chem. Phys. 1996, 104, 2598–2619. [Google Scholar] [CrossRef]
- Montgomery Jr, J.A.; Ochterski, J.W.; Petersson, G.A. A complete basis set model chemistry. IV. An improved atomic pair natural orbital method. J. Chem. Phys. 1994, 101, 5900–5909. [Google Scholar] [CrossRef]
- Helgaker, T.; Ruden, T.A.; Jørgensen, P.; Olsen, J.; Klopper, W. A priori calculation of molecular properties to chemical accuracy. J. Phys. Org. Chem. 2004, 17, 913–933. [Google Scholar] [CrossRef]
- Sun, X.H.; Yao, Q.; Li, Z.R.; Wang, J.B.; Li, X.Y. Calculation of the rate constants for concerted elimination reaction class of hydroperoxyl-alkyl-peroxyl radicals. Theor. Chem. Acc. 2017, 136, 64. [Google Scholar] [CrossRef]
- Sana, M.; Nguyen, M.T. Comment on the accurate theoretical determination of heats of formation. Chem. Phys. Lett. 1992, 196, 390–396. [Google Scholar] [CrossRef]
- Curran, H.J.; Pitz, W.J.; Westbrook, C.K.; Dagaut, P.; Boettner, J.C.; Cathonnet, M. A wide range modeling study of dimethyl ether oxidation. Int. J. Chem. Kinet. 1998, 30, 229–241. [Google Scholar] [CrossRef]
- Mokrushin, V.; Tsang, W. Chemrate v.1.5.8. National Institute of Standards and Technology: Gaithersburg, MD, USA, 2009. [Google Scholar]
- Bugler, J.; Somers, K.P.; Silke, E.J.; Curran, H.J. Revisiting the kinetics and thermodynamics of the low-tempersture oxidation pathways of alkanes: A case study of the three pentane isomers. J. Phys. Chem. A 2015, 119, 7510–7527. [Google Scholar] [CrossRef] [PubMed]
- Kerschgens, B.; Cai, L.; Pitsch, H.; Heuser, B. Di-n-buthylether, n-octanol, and n-octane as fuel candidates for diesel engine combustion. Combust. Flame 2016, 163, 66–78. [Google Scholar] [CrossRef]
- Pu, J.F.; Yao, X.X.; Li, Z.R.; Li, X.Y. High-pressure limit rate rules for intramolecular H-migration reactions of α, β-hydroxyalkylperoxy radicals. Theor. Chem. Acc. 2021, 140, 147. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision, C. 01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Gonzalez, C.; Schlegel, H.B. Reaction path following in mass-weighted internal coordinates. J. Phys. Chem. 1990, 94, 5523–5527. [Google Scholar] [CrossRef]
- Vandeputte, A.G.; Sabbe, M.K.; Reyniers, M.F.; Van Speybroeck, V.; Waroquier, M.; Marin, G.B. Theoretical Study of the Thermodynamics and Kinetics of Hydrogen Abstractions from Hydrocarbons. J. Phys. Chem. A 2007, 111, 11771–11786. [Google Scholar] [CrossRef]
- Saeys, M.; Reyniers, M.F.; Marin, G.B.; Van Speybroeck, V.; Waroquier, M. Ab Initio Calculations for Hydrocarbons: Enthalpy of Formation, Transition State Geometry, and Activation Energy for Radical Reactions. J. Phys. Chem. A 2003, 107, 9147–9159. [Google Scholar] [CrossRef]
- Truong, T.N. Reaction class transition state theory: Hydrogen abstraction reactions by hydrogen atoms as test cases. J. Chem. Phys. 2000, 113, 4957–4964. [Google Scholar] [CrossRef]
- Ratkiewicz, A.; Huynh, L.K.; Truong, T.N. Performance of first-principles-based reaction class transition state theory. J. Phys. Chem. B 2016, 120, 1871–1884. [Google Scholar] [CrossRef]

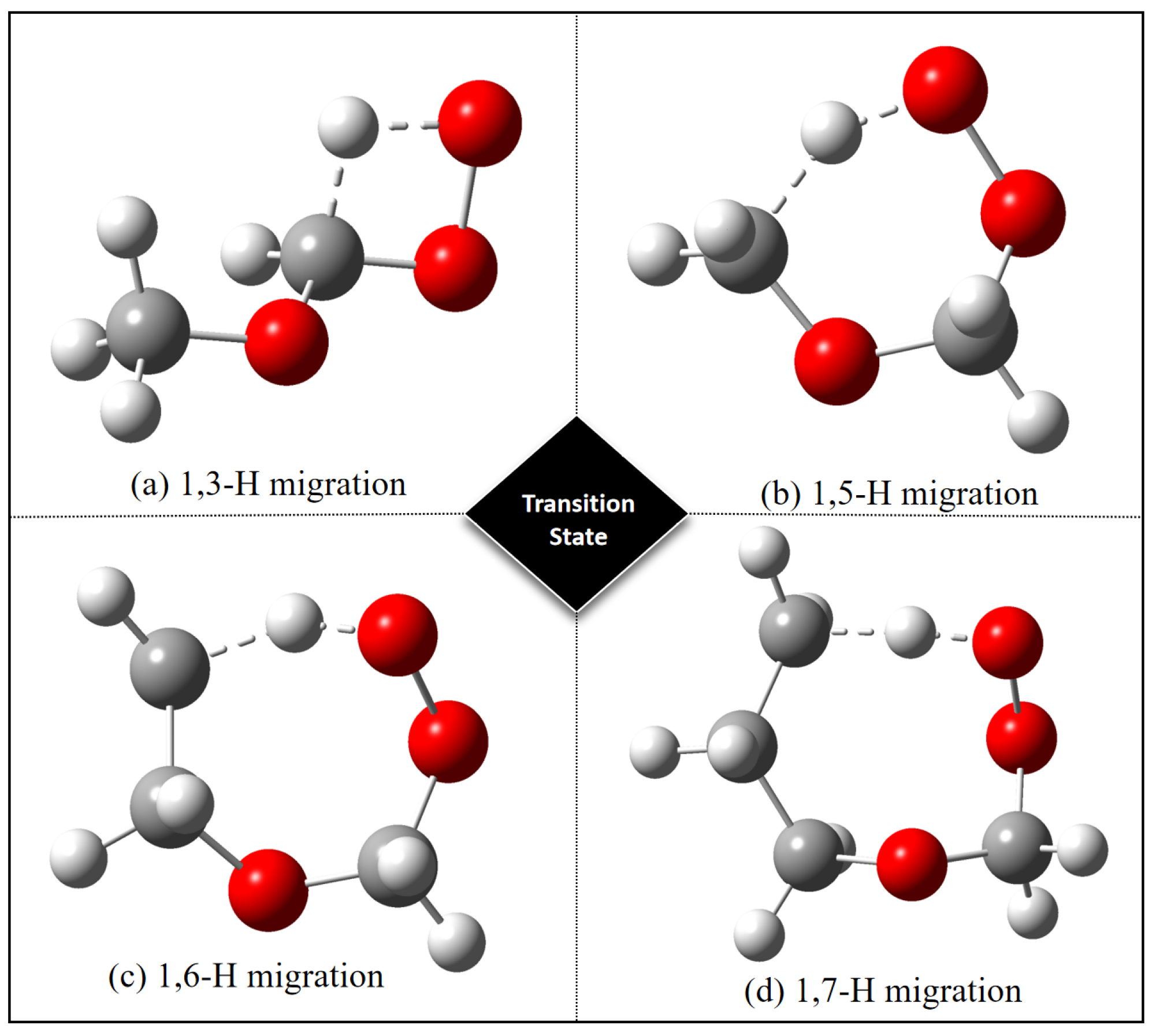

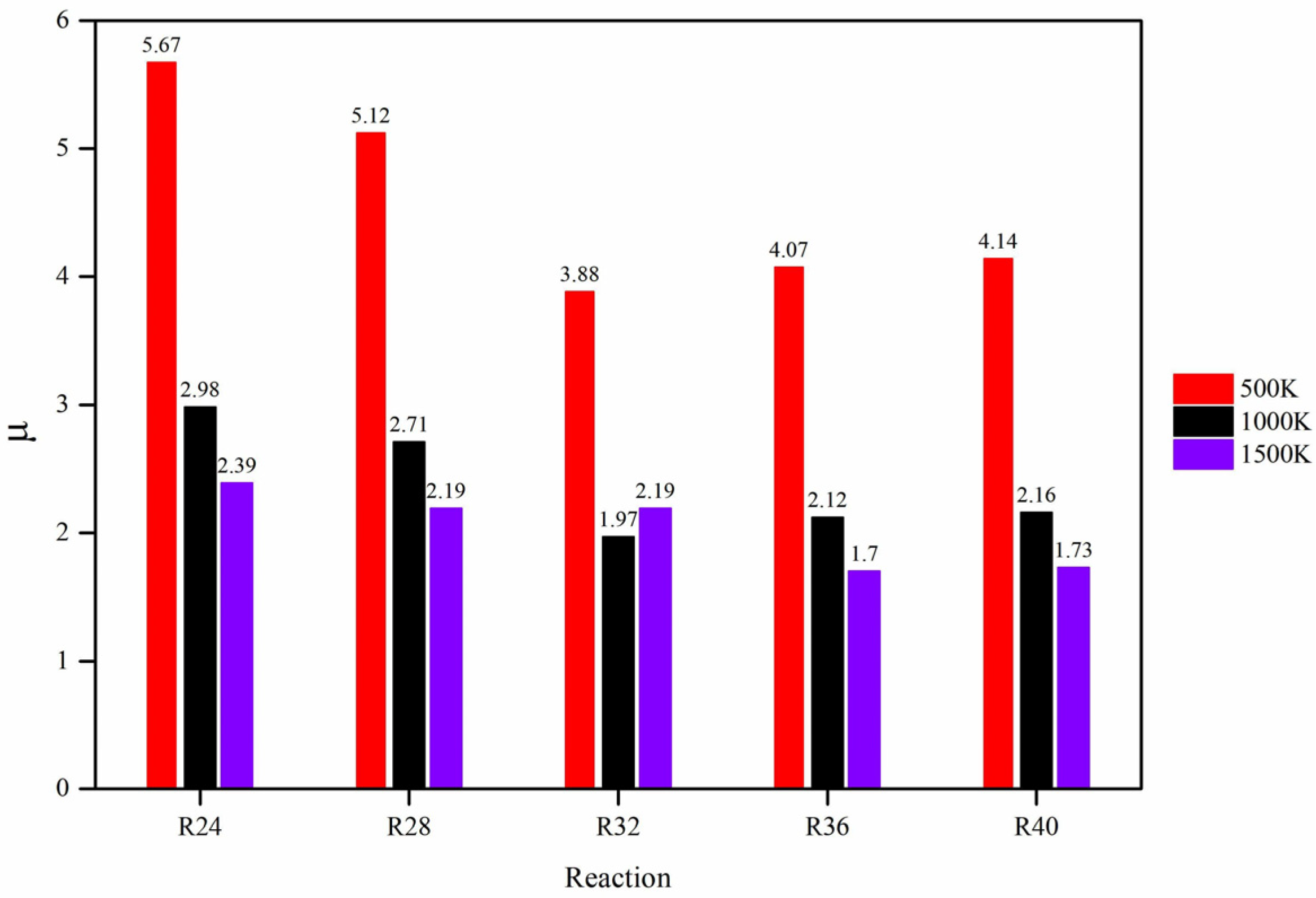
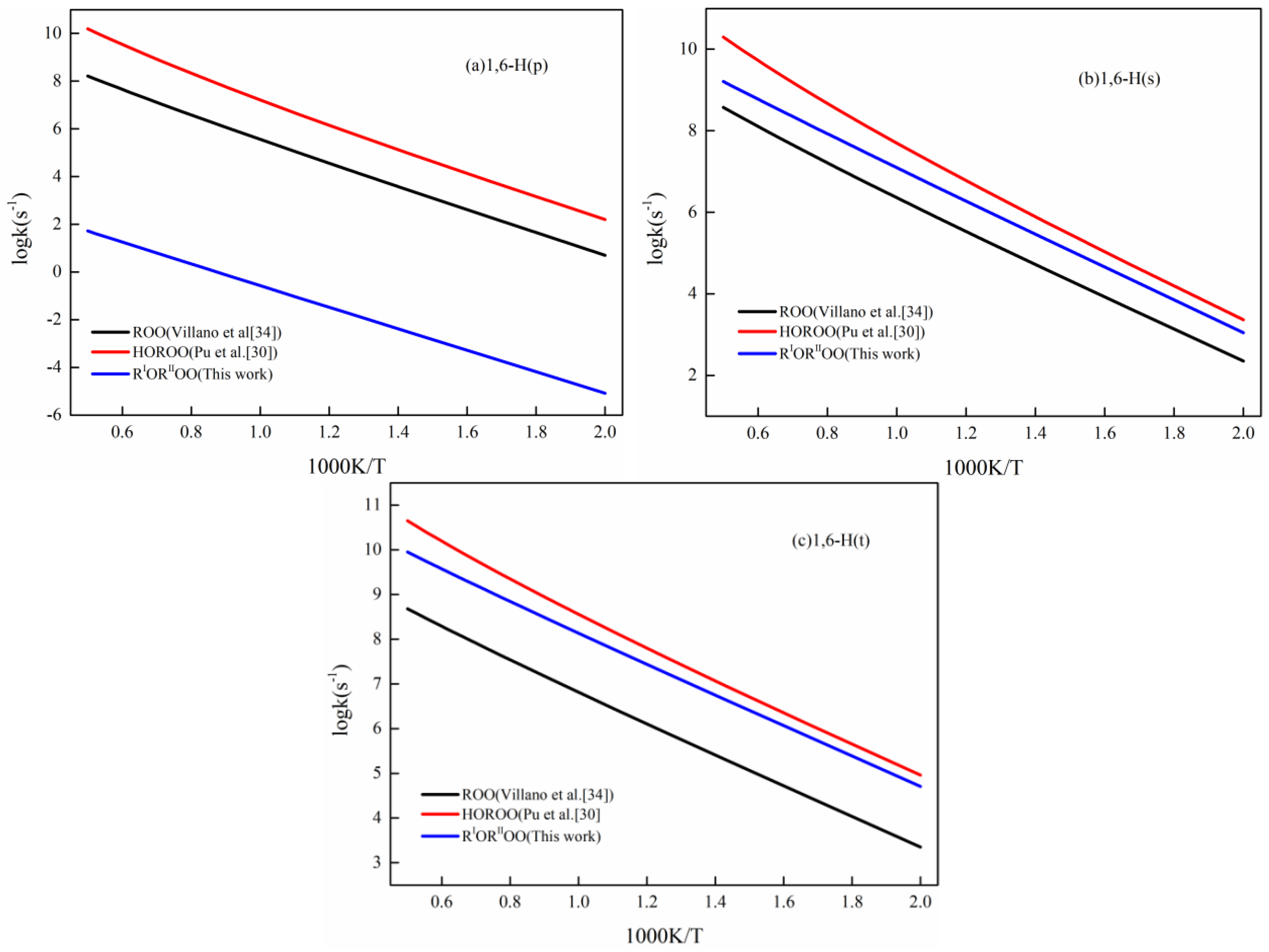
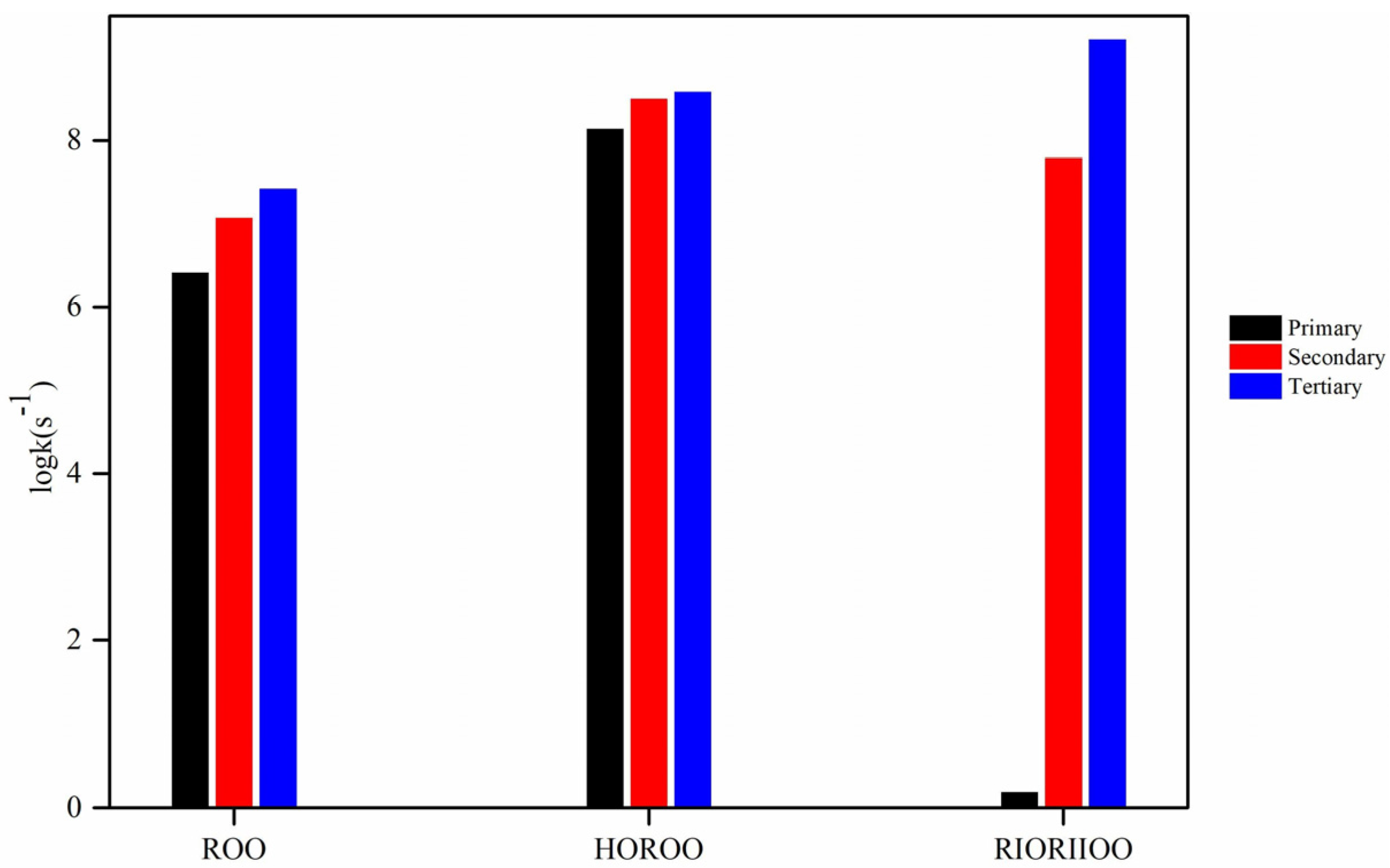

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction Class | Reaction Equation |
|---|---|
| 1,3-H(s) | |
| R1 | CH3OCH2OO·→ CH3OC·HOOH |
| R2 | CH3CH2OCH2OO· → CH3CH2OC·HOOH |
| R3 | CH3(CH2)2OCH2OO· → CH3(CH2)2OC·HOOH |
| R4 | CH3(CH2)3OCH2OO· → CH3(CH2)3OC·HOOH |
| 1,3-H(t) | |
| R5 | CH3OCH(CH3)OO· → CH3OC·(CH3)OOH |
| R6 | CH3OCH(C2H5)OO· → CH3OC·(C2H5)OOH |
| R7 | CH3CH2OCH(CH3)OO· → CH3CH2OC·(CH3)OOH |
| R8 | CH3(CH2)2OCH(CH3)OO· → CH3(CH2)2OC·(CH3)OOH |
| R9 | CH3(CH2)3OCH(CH3)OO· → CH3(CH2)3OC·(CH3)OOH |
| 1,5-H(p) | |
| R10 | CH3OCH2OO· → ·CH2OCH2OOH |
| R11 | CH3OCH(CH3)OO· → ·CH2OCHCH(CH3)OOH |
| R12 | CH3OCH(C2H5)OO· → ·CH2OCH(C2H5)OOH |
| 1,5-H(s) | |
| R13 | CH3CH2OCH2OO· → CH3C·HOCH2OOH |
| R14 | CH3(CH2)2OCH2OO·→ CH3CH2C·HOCH2OOH |
| R15 | CH3CH2OCH(CH3)OO· → CH3C·HOCH(CH3)OOH |
| R16 | CH3CH2OCH(C2H5)OO· → CH3C·HOCH(C2H5)OOH |
| R17 | CH3CH(CH3)CH2OCH(CH3)OO· → CH3CH(CH3)C·HOCH(CH3)OOH |
| 1,5-H(t) | |
| R18 | CH3CH(CH3)OCH2OO·→ CH3C·(CH3)OCH2OOH |
| R19 | CH3CH(CH3)OCH(CH3)OO·→ CH3C·(CH3)OCH(CH3)OOH |
| 1,6-H(p) | |
| R20 | CH3CH2OCH2OO· → ·CH2CH2OCH2OOH |
| R21 | CH3CH2OCH(CH3)OO· → ·CH2CH2OCH(CH3)OOH |
| R22 | CH3CH2OCH(C2H5)OO· → ·CH2CH2OCH(C2H5)OOH |
| 1,6-H(s) | |
| R23 | CH3(CH2)2OCH2OO· → CH3C·HCH2OCH2OOH |
| R24 | CH3(CH2)2OCH(CH3)OO·· → CH3C·HCH2OCH(CH3)OOH |
| R25 | CH3(CH2)3OCH2OO· → CH3CH2C·HCH2OCH2OOH |
| R26 | CH3CH(CH3)CH2CH(CH3)OCH(CH3)OO· → CH3CH(CH3)C·HCH(CH3)OCH(CH3)OOH |
| 1,6-H(t) | |
| R27 | CH3CH(CH3)CH2OCH2OO· → CH3C·(CH3)CH2OCH2OOH |
| R28 | CH3CH2CH(CH3)CH2OCH2OO· → CH3CH2C·(CH3)CH2OCH2OOH |
| R29 | CH3CH2CH(CH3)CH(CH3)OCH2OO· → CH3CH2C·(CH3)CH(CH3)OCH2OOH |
| R30 | CH3CH2CH(CH3)CH(CH3)OCH(CH3)OO· → CH3CH2C·(CH3)CH(CH3)OCH(CH3)OOH |
| 1,7-H(p) | |
| R31 | CH3CH2CH2OCH2OO· → ·CH2CH2CH2OCH2OOH |
| R32 | CH3CH(CH3)CH2OCH2OO· → ·CH2CH(CH3)CH2OCH2OOH |
| R33 | CH3CH(CH3)CH(CH3)OCH2OO· → ·CH2CH(CH3)CH(CH3)OCH2OOH |
| R34 | CH3CH(CH3)CH(CH3)OCH(CH3)OO· → ·CH2CH(CH3)CH(CH3)OCH(CH3)OOH |
| 1,7-H(s) | |
| R35 | CH3(CH2)3OCH2OO· → CH3C·H(CH2)2OCH2OOH |
| R36 | CH3CH2CH(CH3)CH2OCH2OO· → CH3C·HCH(CH3)CH2OCH2OOH |
| R37 | CH3CH2CH(CH3)CH(CH3)OCH2OO· → CH3C·HCH(CH3)CH(CH3)OCH2OOH |
| R38 | CH3CH2CH(CH3)CH(CH3)OCH(CH3)OO· → CH3C·HCH(CH3)CH(CH3)OCH(CH3)OOH |
| 1,7-H(t) | |
| R39 | CH3CH(CH3)CH2CH2OCH2OO· → CH3C·(CH3)(CH2)2OCH2OOH |
| R40 | CH3CH(CH3)CH(CH3)CH2OCH2OO· → CH3C·(CH3)CH(CH3)CH2OCH2OOH |
| R41 | CH3CH(CH3)CH(CH3)CH(CH3)OCH2OO· → CH3C·(CH3)CH(CH3)CH(CH3)OCH2OOH |
| Subclass | 1,3-H | 1,5-H | 1,6-H | 1,7-H |
|---|---|---|---|---|
| p | NA a | 0.33 | 0.54 | 1.13 |
| s | 0.23 | 1.34 | 0.80 | 1.25 |
| t | 0.64 | 2.69 | 0.94 | 0.90 |
| Avg b | 1.16 * | 7.19 * | 6.85 * | 6.70 * |
| Reaction Class | Reaction | CBS–QB3 | IRM | ΔV |
|---|---|---|---|---|
| 1,3-H(s) | R2 | 44.37 | 44.34 | 0.03 |
| 1,3-H(t) | R6 | 44.67 | 44.77 | −0.10 |
| 1,5-H(p) | R11 | 23.08 | 23.02 | 0.06 |
| 1,5-H(s) | R14 | 21.98 | 22.21 | −0.23 |
| 1,5-H(t) | R19 | 15.46 | 16.27 | −0.81 |
| 1,6-H(p) | R21 | 27.39 | 27.40 | −0.01 |
| 1,6-H(s) | R24 | 24.11 | 24.43 | −0.32 |
| 1,6-H(t) | R28 | 20.04 | 20.74 | −0.70 |
| 1,7-H(p) | R32 | 25.19 | 24.63 | 0.56 |
| 1,7-H(s) | R36 | 21.35 | 21.32 | 0.03 |
| 1,7-H(t) | R40 | 18.13 | 18.18 | −0.05 |
| Reaction Class | Reaction | CBS–QB3 | IRM | Curran et al. a [32] | ΔH b |
|---|---|---|---|---|---|
| 1,3-H(s) | R2 | −46.33 | −46.26 | NA | −0.07 |
| 1,3-H(t) | R6 | −46.33 | −46.60 | NA | 0.27 |
| 1,5-H(p) | R10 | NA | 9.77 | 9.30 | NA |
| 1,5-H(p) | R11 | 12.16 | 12.34 | NA | −0.18 |
| 1,5-H(t) | R19 | 10.53 | 10.21 | NA | 0.32 |
| 1,6-H(p) | R21 | 17.08 | 17.08 | NA | 0.00 |
| 1,6-H(s) | R24 | 14.29 | 14.08 | NA | 0.21 |
| 1,6-H(t) | R28 | 11.29 | 11.43 | NA | −0.14 |
| 1,7-H(p) | R32 | 15.38 | 15.42 | NA | −0.04 |
| 1,7-H(s) | R36 | 12.54 | 12.28 | NA | 0.26 |
| 1,7-H(t) | R40 | 9.76 | 9.46 | NA | 0.30 |
| Reaction | RIORIIOO· | ROO· | ||
|---|---|---|---|---|
| This Work | Villano et al. a [7] | Bugler et al. b [34] | ||
| R10 | COCOO· → ·COCOO | 7.22 | 7.67 (1.06) | 2.10 × 101 (2.91) |
| R13 | CCOCOO· → CC·OCOO | 3.67 × 103 | 2.84 × 102 (12.92) | 5.68 × 102 (6.46) |
| R18 | C2COCOO· → C2C·OCOO | 8.61 × 105 | 1.61 × 103 (5.35 × 102) | 8.22 × 103 (1.05 × 102) |
| R20 | CCOCOO· → ·CCOCOO | 6.76 × 104 | 3.21 (2.11 × 104) | 6.36 (1.06 × 104) |
| Reaction Class | Modified Arrhenius Parameters | 500 K | 1000 K | ||
|---|---|---|---|---|---|
| A (s−1) | n | E (kcal/mol) | f | f | |
| 1,3-H(s) | 1.36 × 109 | 1.26 | 37.74 | 1.27 | 1.38 |
| 1,3-H(t) | 9.58 × 109 | 0.86 | 39.05 | 1.98 | 2.34 |
| 1,5-H(p) | 2.03 × 1013 | 0.31 | 16.96 | 3.54 × 103 | 2.43 × 103 |
| 1,5-H(s) | 4.47 × 109 | 0.56 | 16.26 | 25.20 | 14.90 |
| 1,5-H(t) | 1.22 × 1010 | 0.58 | 11.06 | 14.20 | 4.17 |
| 1,6-H(p) | 1.19 × 1013 | 0.26 | 20.27 | 4.71 × 103 | 2.63 × 103 |
| 1,6-H(s) | 2.15 × 109 | 0.55 | 17.78 | 2.87 | 3.83 |
| 1,6-H(t) | 2.62 × 109 | 0.65 | 14.79 | 9.01 | 4.81 |
| 1,7-H(p) | 7.37 × 1012 | 0.16 | 17.84 | 2.46 × 103 | 1.59 × 103 |
| 1,7-H(s) | 2.32 × 109 | 0.56 | 14.87 | 20.00 | 18.20 |
| 1,7-H(t) | 2.19 × 1010 | 0.47 | 12.97 | 2.78 | 4.84 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, X.; Li, Z. Reaction Rate Rules of Intramolecular H-Migration Reaction Class for RIORIIOO·Radicals in Ether Combustion. Molecules 2024, 29, 4387. https://doi.org/10.3390/molecules29184387
Sun X, Li Z. Reaction Rate Rules of Intramolecular H-Migration Reaction Class for RIORIIOO·Radicals in Ether Combustion. Molecules. 2024; 29(18):4387. https://doi.org/10.3390/molecules29184387
Chicago/Turabian StyleSun, Xiaohui, and Zerong Li. 2024. "Reaction Rate Rules of Intramolecular H-Migration Reaction Class for RIORIIOO·Radicals in Ether Combustion" Molecules 29, no. 18: 4387. https://doi.org/10.3390/molecules29184387
APA StyleSun, X., & Li, Z. (2024). Reaction Rate Rules of Intramolecular H-Migration Reaction Class for RIORIIOO·Radicals in Ether Combustion. Molecules, 29(18), 4387. https://doi.org/10.3390/molecules29184387