
Driving Forces in the Formation of Paracetamol Cocrystals and Solvate with Naphthalene, Quinoline and Acridine


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Cocrystal Preparation and Thermal Analysis
2.2. IR Spectroscopy
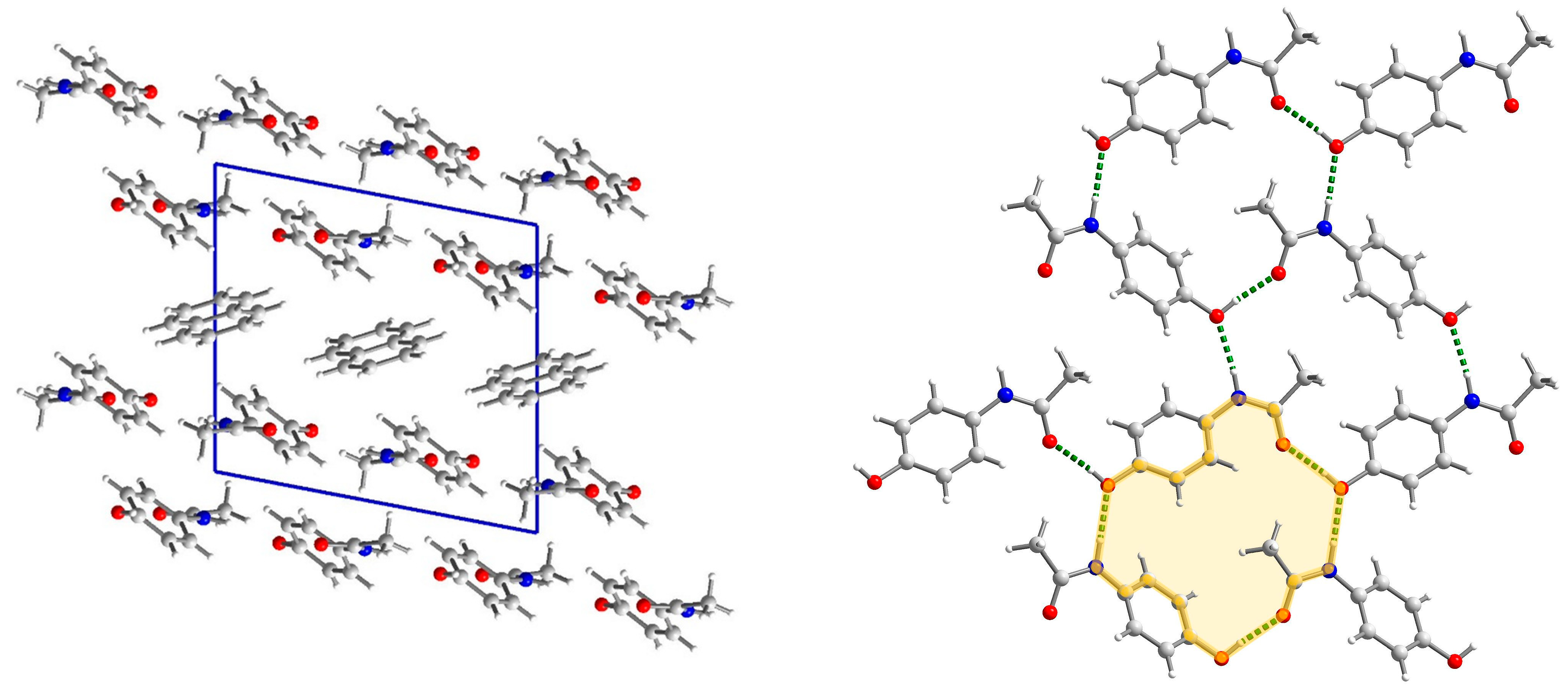
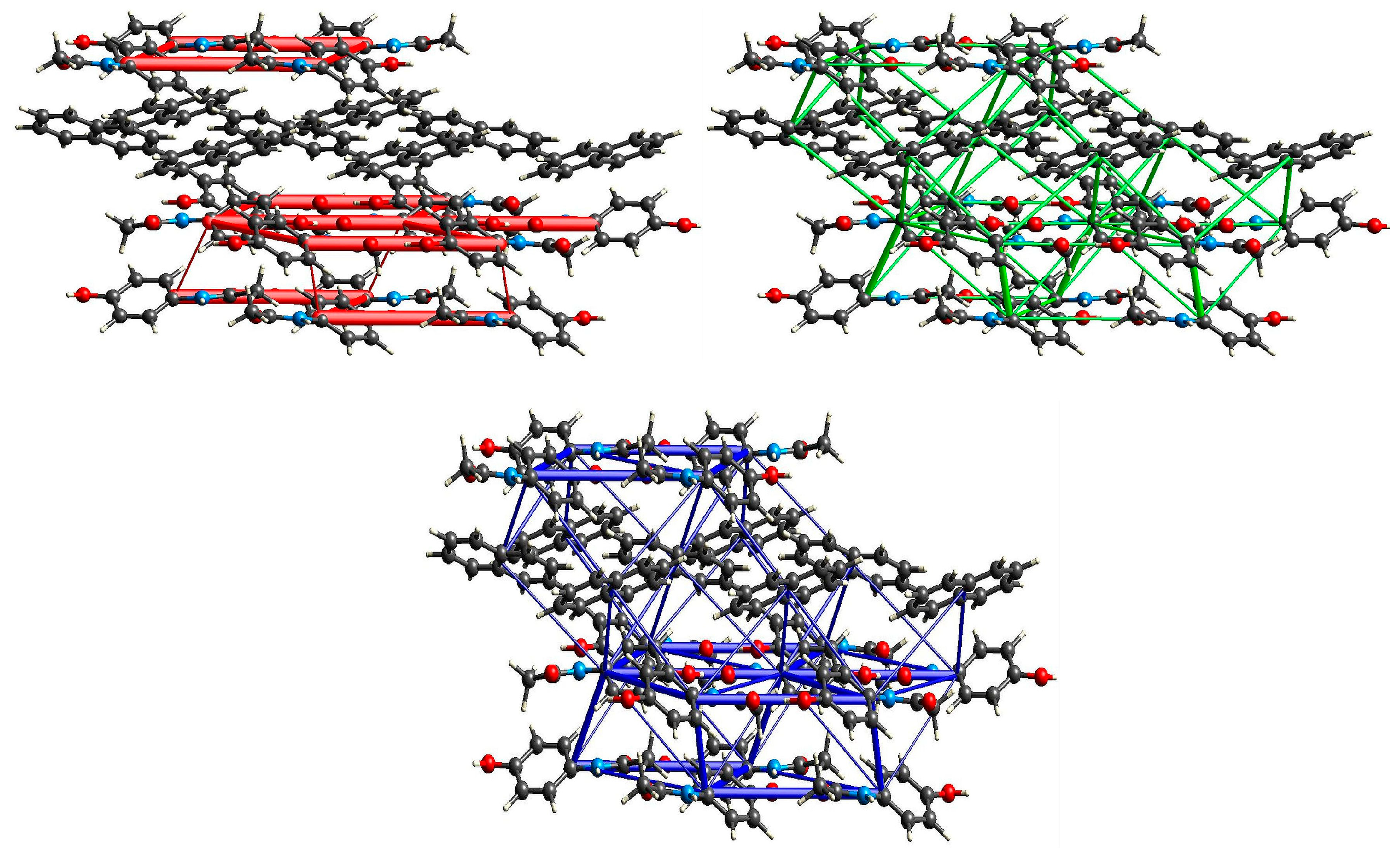
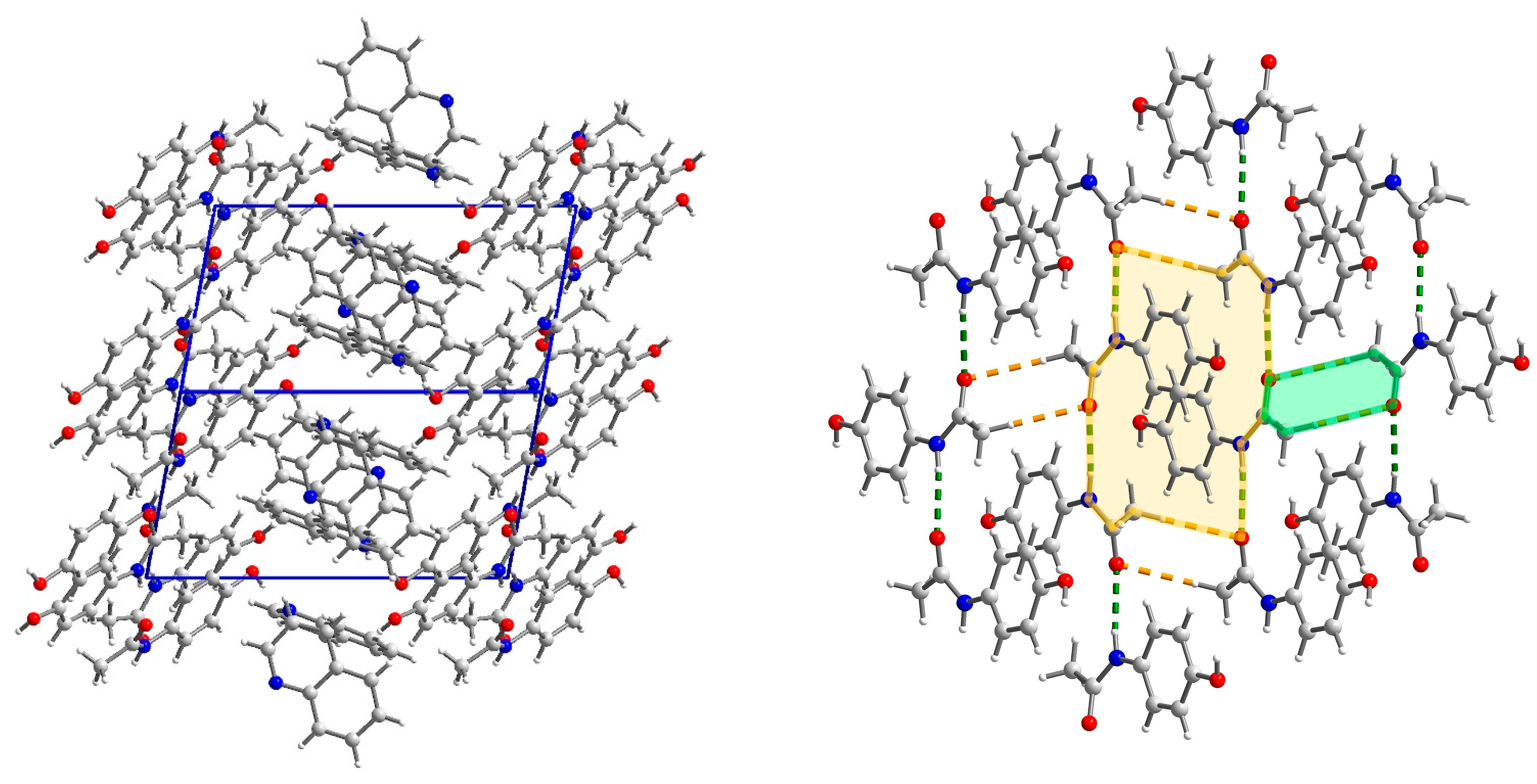
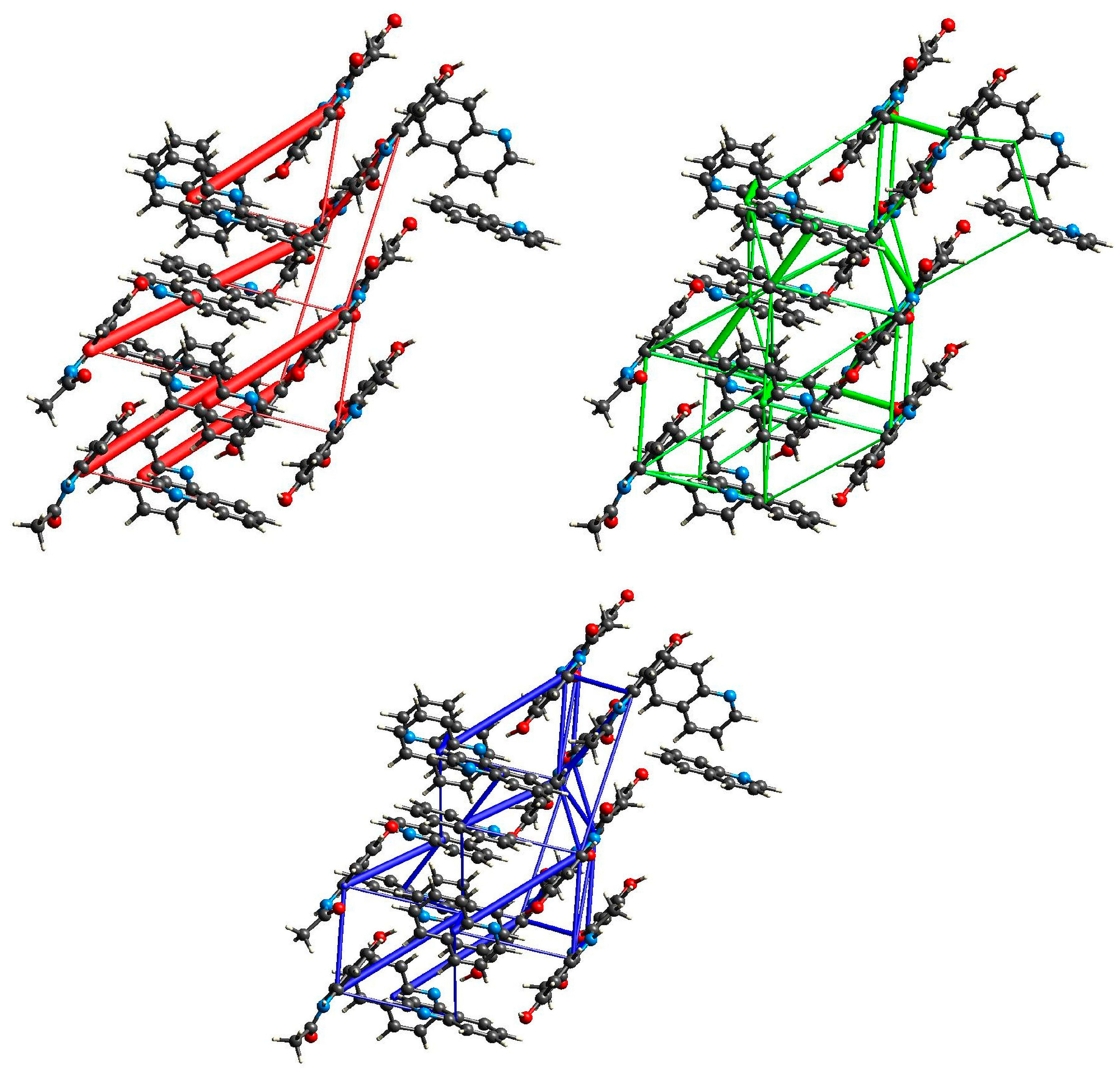
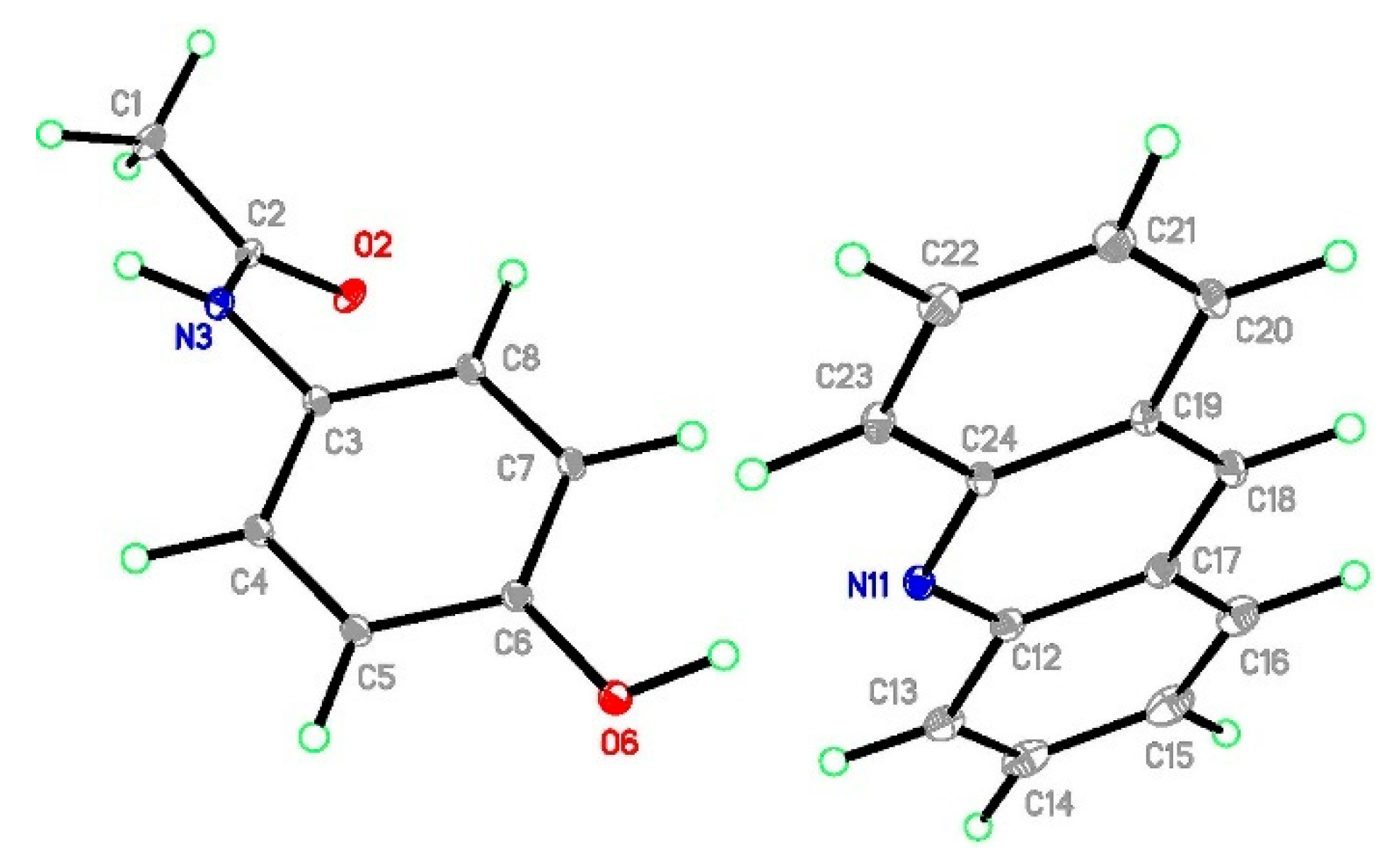
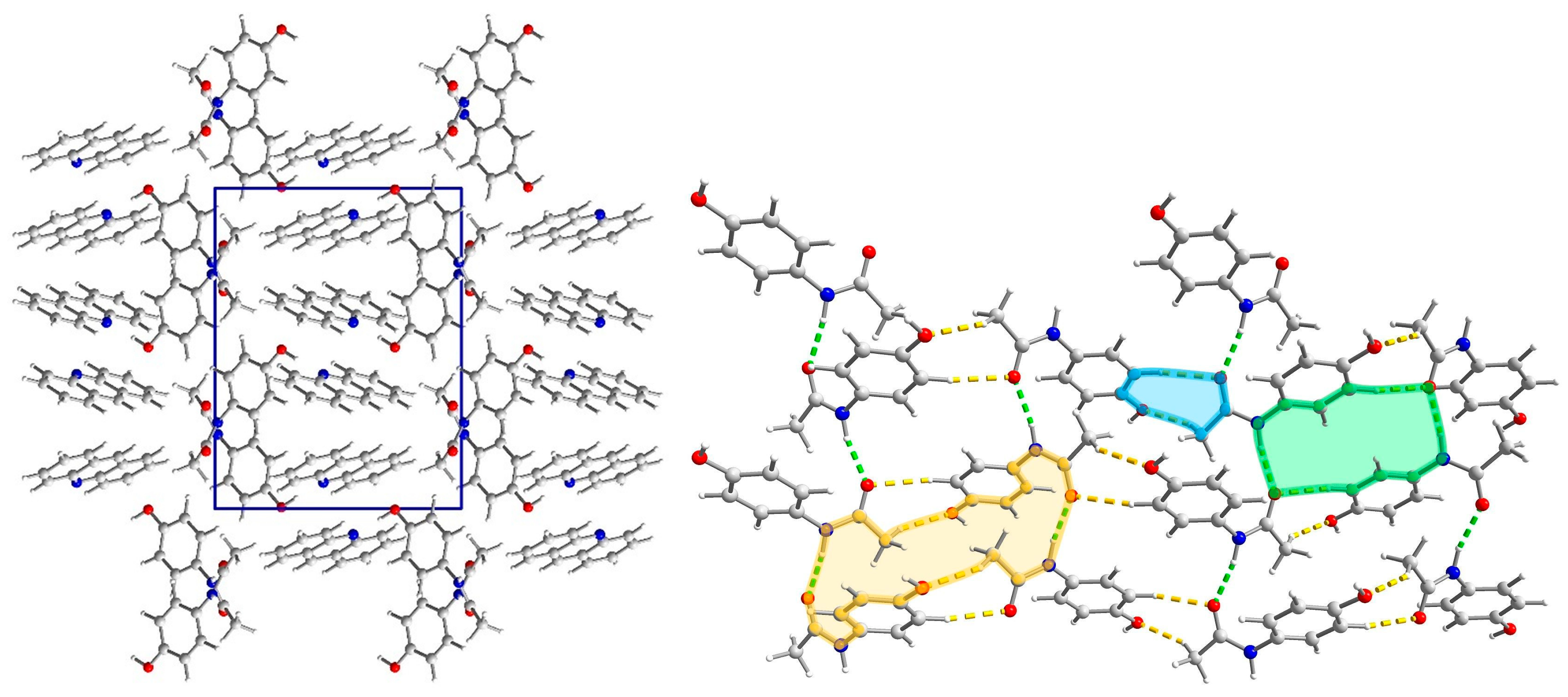
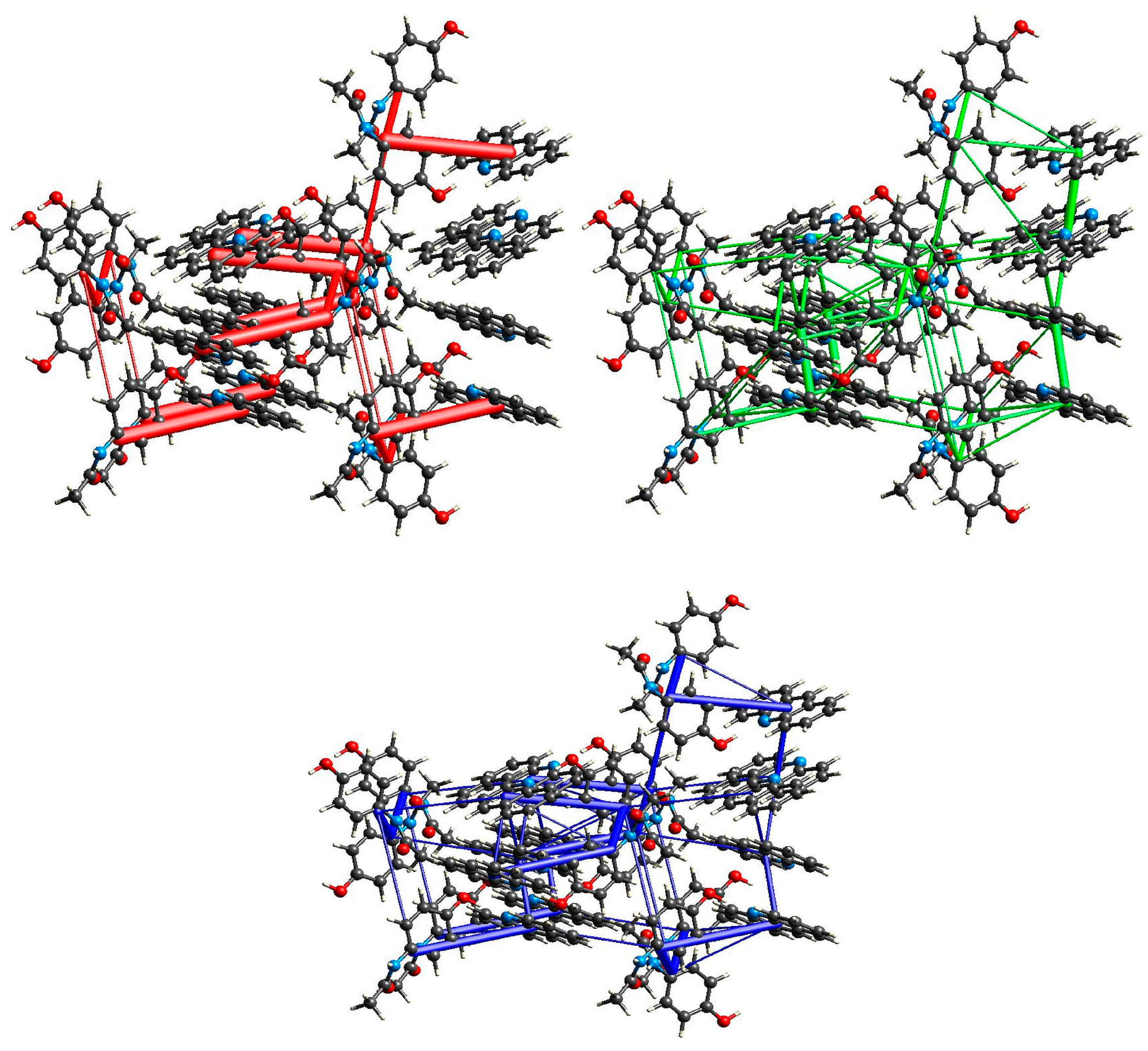
2.3. Structural Studies
2.3.1. Structure of (par)2∙(nap) (1)
2.3.2. Structure of (par)∙(quin) (2)
2.3.3. Structure of (par)∙(acr) (3)
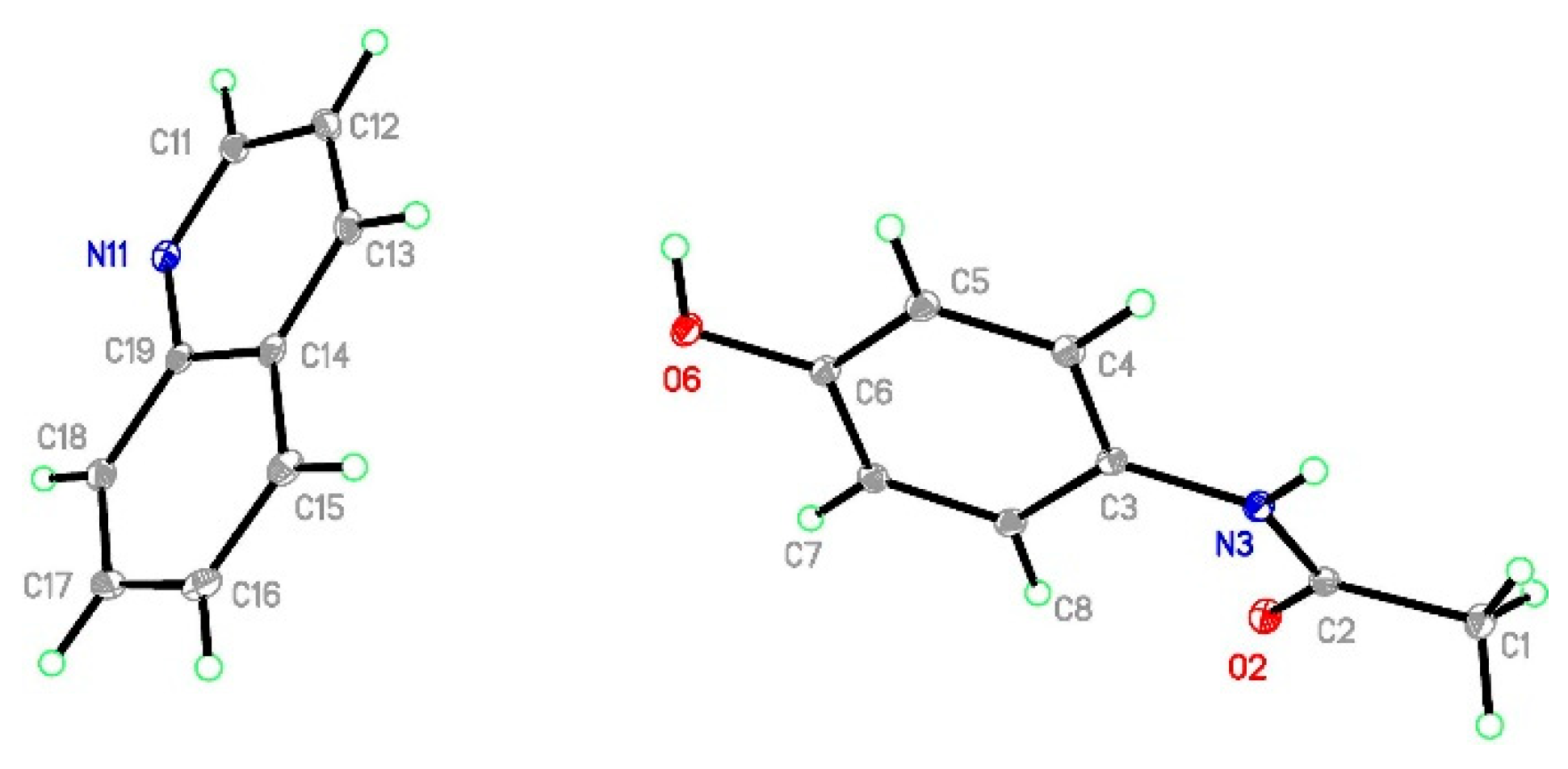
2.3.4. Structural Studies of Selected Coformers
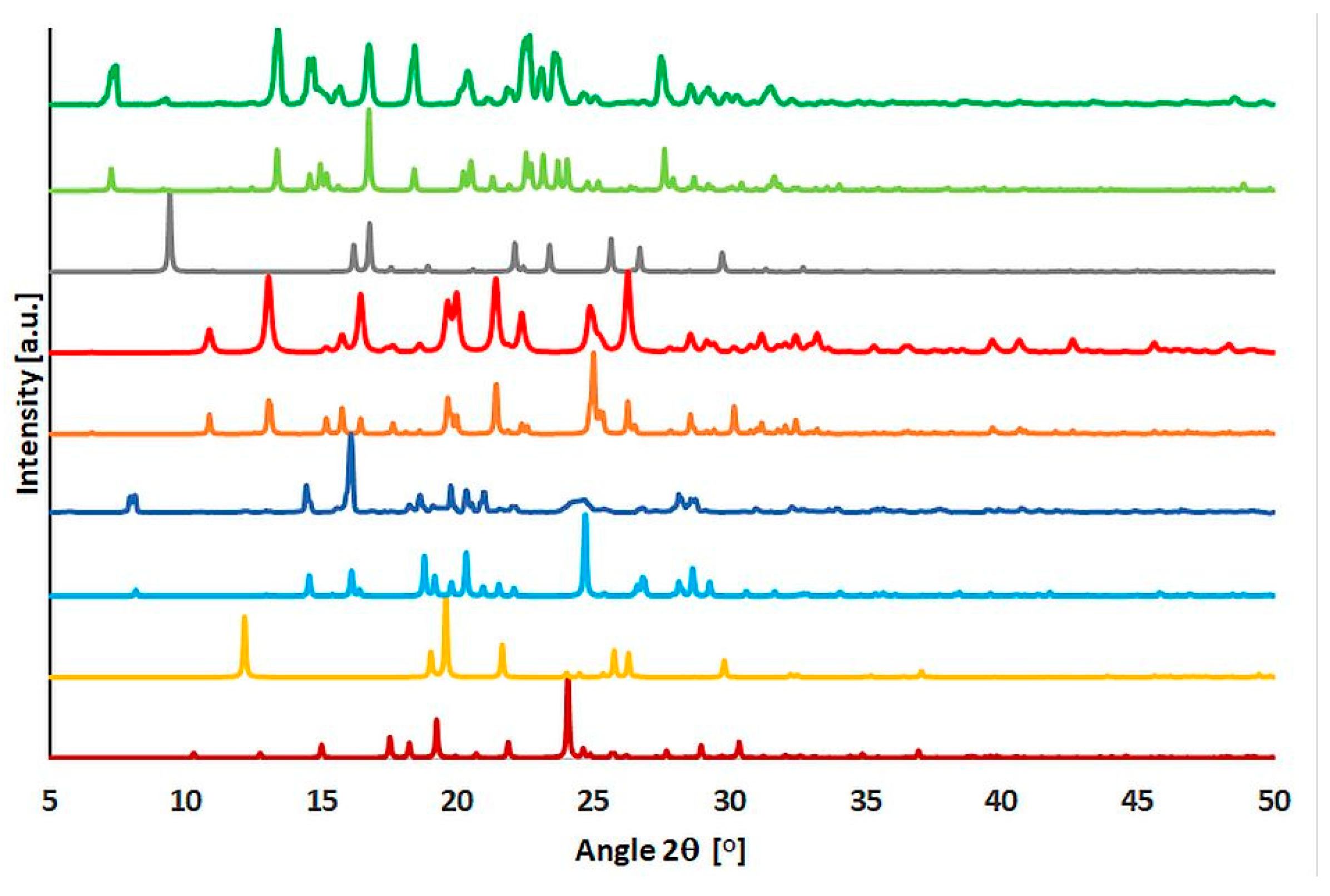
2.4. Powder Diffraction Experiments
2.5. XAS Results
3. Materials and Methods
3.1. Materials and General Procedure
3.2. Preparation of (par)2∙(nap)
3.3. Preparation of (par)∙(quin)
3.4. Preparation of (par)∙(acr)
3.5. Single Crystal X-ray Diffraction Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Srivastava, K.; Shimpi, M.R.; Srivastava, A.; Tandon, P.; Sinha, K.; Velaga, S.P. Vibrational Analysis and Chemical Activity of Paracetamol-Oxalic Acid Cocrystal Based on Monomer and Dimer Calculations: DFT and AIM Approach. RSC Adv. 2016, 6, 10024–10037. [Google Scholar] [CrossRef]
- Wichianphong, N.; Charoenchaitrakool, M. Application of Box-Behnken Design for Processing of Mefenamic Acid-Paracetamol Cocrystals Using Gas Anti-Solvent (GAS) Process. J. CO2 Util. 2018, 26, 212–220. [Google Scholar] [CrossRef]
- Srirambhatla, V.K.; Kraft, A.; Watt, S.; Powell, A.V. Crystal Design Approaches for the Synthesis of Paracetamol Co-Crystals. Cryst. Growth Des. 2012, 12, 4870–4879. [Google Scholar] [CrossRef]
- Bučar, D.K.; Elliott, J.A.; Eddleston, M.D.; Cockcroft, J.K.; Jones, W. Sonocrystallization Yields Monoclinic Paracetamol with Significantly Improved Compaction Behavior. Angew. Chem. Int. Ed. 2015, 54, 249–253. [Google Scholar] [CrossRef]
- Lee, W.M. Acetaminophen and the U.S. Acute Liver Failure Study Group: Lowering the Risks of Hepatic Failure. Hepatology 2004, 40, 6–9. [Google Scholar] [CrossRef]
- Maeno, Y.; Fukami, T.; Kawahata, M.; Yamaguchi, K.; Tagami, T.; Ozeki, T.; Suzuki, T.; Tomono, K. Novel Pharmaceutical Cocrystal Consisting of Paracetamol and Trimethylglycine, a New Promising Cocrystal Former. Int. J. Pharm. 2014, 473, 179–186. [Google Scholar] [CrossRef]
- Suzuki, N.; Kanazawa, T.; Takatori, K.; Suzuki, T.; Fukami, T. Crystal Structure Analysis and Pharmaceutical Properties of Amide Salts Consisting of Paracetamol/Sulfonic Acids as Solid Forms Prepared by Grinding. Cryst. Growth Des. 2020, 20, 590–599. [Google Scholar] [CrossRef]
- Trzybiński, D.; Domagała, S.; Kubsik, M.; Woźniak, K. Solid-State Analysis of Monohydrated Halide Salts of Paracetamol. Cryst. Growth Des. 2016, 16, 1156–1161. [Google Scholar] [CrossRef]
- Chadha, R.; Kuhad, A.; Arora, P.; Kishor, S. Characterisation and Evaluation of Pharmaceutical Solvates of Atorvastatin Calcium by Thermoanalytical and Spectroscopic Studies. Chem. Cent. J. 2012, 6, 114. [Google Scholar] [CrossRef]
- Elder, D.P.; Holm, R.; De Diego, H.L. Use of Pharmaceutical Salts and Cocrystals to Address the Issue of Poor Solubility. Int. J. Pharm. 2013, 453, 88–100. [Google Scholar] [CrossRef]
- Fael, H.; Demirel, A.L. Indomethacin Co-Amorphous Drug-Drug Systems with Improved Solubility, Supersaturation, Dissolution Rate and Physical Stability. Int. J. Pharm. 2021, 600, 120448. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Jin, Y.; Li, S.; Yang, Z.; Shi, B.; Chang, C.; Abramov, Y.A. Virtual Coformer Screening by Crystal Structure Predictions: Crucial Role of Crystallinity in Pharmaceutical Cocrystallization. J. Phys. Chem. Lett. 2020, 11, 8832–8838. [Google Scholar] [CrossRef] [PubMed]
- Elbagerma, M.A.; Edwards, H.G.M.; Munshi, T.; Scowen, I.J. Identification of a New Cocrystal of Citric Acid and Paracetamol of Pharmaceutical Relevance. CrystEngComm 2011, 13, 1877–1884. [Google Scholar] [CrossRef]
- Srivastava, K.; Shukla, A.; Karthick, T.; Velaga, S.P.; Tandon, P.; Sinha, K.; Shimpi, M.R. Molecular Structure, Spectroscopic Signature and Reactivity Analyses of Paracetamol Hydrochloride Monohydrate Salt Using Density Functional Theory Calculations. CrystEngComm 2019, 21, 857–865. [Google Scholar] [CrossRef]
- Islam, N.U.; Khan, E.; Umar, M.N.; Shah, A.; Zahoor, M.; Ullah, R.; Bari, A. Enhancing Dissolution Rate and Antibacterial Efficiency of Azithromycin through Drug-Drug Cocrystals with Paracetamol. Antibiotics 2021, 10, 939. [Google Scholar] [CrossRef]
- Suzuki, N.; Kawahata, M.; Yamaguchi, K.; Suzuki, T.; Tomono, K.; Fukami, T. Comparison of the Relative Stability of Pharmaceutical Cocrystals Consisting of Paracetamol and Dicarboxylic Acids. Drug Dev. Ind. Pharm. 2018, 44, 582–589. [Google Scholar] [CrossRef]
- Al-Ani, A.J.; Sugden, P.; Wilson, C.C.; Castro-Dominguez, B. Elusive Seed Formation via Electrical Confinement: Control of a Novel Cocrystal in Cooling Crystallization. Cryst. Growth Des. 2021, 21, 3310–3315. [Google Scholar] [CrossRef]
- Ahmed, H.; Shimpi, M.R.; Velaga, S.P. Relationship between Mechanical Properties and Crystal Structure in Cocrystals and Salt of Paracetamol. Drug Dev. Ind. Pharm. 2017, 43, 89–97. [Google Scholar] [CrossRef]
- Kennedy, A.R.; King, N.L.C.; Oswald, I.D.H.; Rollo, D.G.; Spiteri, R.; Walls, A. Structural Study of Salt Forms of Amides; Paracetamol, Benzamide and Piperine. J. Mol. Struct. 2018, 1154, 196–203. [Google Scholar] [CrossRef]
- Cruz-Cabeza, A.J. Acid-Base Crystalline Complexes and the PKa Rule. CrystEngComm 2012, 14, 6362–6365. [Google Scholar] [CrossRef]
- Karki, S.; Friščić, T.; Fabián, L.; Laity, P.R.; Day, G.M.; Jones, W. Improving Mechanical Properties of Crystalline Solids by Cocrystal Formation: New Compressible Forms of Paracetamol. Adv. Mater. 2009, 21, 3905–3909. [Google Scholar] [CrossRef]
- Zhang, C.; Xiong, Y.; Jiao, F.; Wang, M.; Li, H. Redefining the Term of “Cocrystal” and Broadening Its Intention. Cryst. Growth Des. 2019, 19, 1471–1478. [Google Scholar] [CrossRef]
- Aitipamula, S.; Banerjee, R.; Bansal, A.K.; Biradha, K.; Cheney, M.L.; Choudhury, A.R.; Desiraju, G.R.; Dikundwar, A.G.; Dubey, R.; Duggirala, N.; et al. Polymorphs, Salts, and Cocrystals: What’s in a Name? Cryst. Growth Des. 2012, 12, 2147–2152. [Google Scholar] [CrossRef]
- Zakharov, B.A.; Ogienko, A.G.; Yunoshev, A.S.; Ancharov, A.I.; Boldyreva, E.V. Bis(Paracetamol) Pyridine-a New Elusive Paracetamol Solvate: From Modeling the Phase Diagram to Successful Single-Crystal Growth and Structure-Property Relations. CrystEngComm 2015, 17, 7543–7550. [Google Scholar] [CrossRef]
- Latif, S.; Abbas, N.; Hussain, A.; Arshad, M.S.; Bukhari, N.I.; Afzal, H.; Riffat, S.; Ahmad, Z. Development of Paracetamol-Caffeine Co-Crystals to Improve Compressional, Formulation and in Vivo Performance. Drug Dev. Ind. Pharm. 2018, 44, 1099–1108. [Google Scholar] [CrossRef]
- Li, H.; Wang, L.; Ye, X.; Yao, C.; Song, S.; Qu, Y.; Jiang, J.; Wang, H.; Han, P.; Liu, Y.; et al. Efficient Screening of Pharmaceutical Cocrystals by Microspacing In-Air Sublimation. J. Am. Chem. Soc. 2024, 146, 11592–11598. [Google Scholar] [CrossRef]
- Sathisaran, I.; Dalvi, S.V. Cocrystallization of an Antiretroviral Drug Nevirapine: An Eutectic, a Cocrystal Solvate, and a Cocrystal Hydrate. Cryst. Growth Des. 2021, 21, 2076–2092. [Google Scholar] [CrossRef]
- Roy, S.; Gaur, R.; Paul, M.; Rajkumar, M.; Desiraju, G.R. Synthetic Strategies toward Higher Cocrystals of Some Resorcinols. Cryst. Growth Des. 2022, 22, 7578–7589. [Google Scholar] [CrossRef]
- Paul, M.; Desiraju, G.R. From a Binary to a Quaternary Cocrystal: An Unusual Supramolecular Synthon. Angew. Chem. 2019, 131, 12155–12159. [Google Scholar] [CrossRef]
- Mir, N.A.; Dubey, R.; Desiraju, G.R. Strategy and Methodology in the Synthesis of Multicomponent Molecular Solids: The Quest for Higher Cocrystals. Acc. Chem. Res. 2019, 52, 2210–2220. [Google Scholar] [CrossRef]
- Saha, S.; Desiraju, G.R. Acid⋯Amide Supramolecular Synthon in Cocrystals: From Spectroscopic Detection to Property Engineering. J. Am. Chem. Soc. 2018, 140, 6361–6373. [Google Scholar] [CrossRef] [PubMed]
- Jain, H.; Sutradhar, D.; Roy, S.; Desiraju, G.R. Synthetic Approaches to Halogen Bonded Ternary Cocrystals. Angew. Chem. Int. Ed. 2021, 60, 12841–12846. [Google Scholar] [CrossRef]
- Hiendrawan, S.; Veriansyah, B.; Widjojokusumo, E.; Soewandhi, S.N.; Wikarsa, S.; Tjandrawinata, R.R. Physicochemical and Mechanical Properties of Paracetamol Cocrystal with 5-Nitroisophthalic Acid. Int. J. Pharm. 2016, 497, 106–113. [Google Scholar] [CrossRef]
- Srivastava, K.; Khan, E.; Shimpi, M.R.; Tandon, P.; Sinha, K.; Velaga, S.P. Molecular Structure and Hydrogen Bond Interactions of a Paracetamol-4,4′-Bipyridine Cocrystal Studied Using a Vibrational Spectroscopic and Quantum Chemical Approach. CrystEngComm 2018, 20, 213–222. [Google Scholar] [CrossRef]
- Manonmani, M.; Balakrishnan, C.; Ahamed, S.R.; Vinitha, G.; Meenakshisundaram, S.P.; Sockalingam, R.M. Cocrystallization of Paracetamol-Picric Acid: Hirshfeld Surface Analysis, Supramolecular Architecture and Third-Order Nonlinear Optical Properties. J. Mol. Struct. 2019, 1190, 1–10. [Google Scholar] [CrossRef]
- Ayudhya, T.; Raymond, C.; Dingra, N. Synthesis and Structure of a 1:1 Co-Crystal of Hexamethylenetetramine Carboxyborane and Acetaminophen. Acta Crystallogr. E Crystallogr. Commun. 2020, 76, 1854–1858. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, M.K.; Patil, S.; Shettigar, H.; Jana, S. Effect of Biofield Treatment on Spectral Properties of Paracetamol and Piroxicam. Chem. Sci. J. 2015, 6, 100098. [Google Scholar] [CrossRef]
- Pauzat, F.; Talbi, D.; Miller, M.D.; DeFrees, D.J.; Ellinger, Y. Theoretical IR Spectra of Ionized Naphthalene. J. Phys. Chem. 1992, 96, 7882–7886. [Google Scholar] [CrossRef]
- Ricks, A.M.; Douberly, G.E.; Duncan, M.A. The Infrared Spectrum of Protonated Naphthalene and Its Relevance for the Unidentified Infrared Bands. Astrophys. J. 2009, 702, 301–306. [Google Scholar] [CrossRef]
- Lagutschenkov, A.; Dopfer, O. Infrared Spectrum of a Protonated Fluorescence Dye: Acridine Orange. J. Mol. Spectrosc. 2011, 268, 66–77. [Google Scholar] [CrossRef]
- Prochorow, J.; Deperasinska, I.; Morawskí, O. Fluorescence Excitation and Fluorescence Spectra of Jet-Cooled Acridine Molecules: Acridine Dimer Formation and Structure. Chem. Phys. Lett. 2000, 316, 24–30. [Google Scholar] [CrossRef]
- Özel, A.E.; Büyükmurat, Y.; Akyüz, S. Infrared-Spectra and Normal-Coordinate Analysis of Quinoline and Quinoline Complexes. J. Mol. Struct. 2001, 565–566, 455–462. [Google Scholar] [CrossRef]
- Etter, M.C. Encoding and Decoding Hydrogen-Bond Patterns of Organic Compounds. Acc. Chem. Res. 1990, 23, 120–126. [Google Scholar] [CrossRef]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld Surface Analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Spackman, M.A.; McKinnon, J.J. Fingerprinting Intermolecular Interactions in Molecular Crystals. CrystEngComm 2002, 4, 378–392. [Google Scholar] [CrossRef]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A Program for Hirshfeld Surface Analysis, Visualization and Quantitative Analysis of Molecular Crystals. J. Appl. Crystallogr. 2021, 54, 1006–1011. [Google Scholar] [CrossRef]
- Mackenzie, C.F.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer Model Energies and Energy Frameworks: Extension to Metal Coordination Compounds, Organic Salts, Solvates and Open-Shell Systems. IUCrJ 2017, 4, 575–587. [Google Scholar] [CrossRef]
- Stone, K.H.; Lapidus, S.H.; Stephens, P.W. Implementation and Use of Robust Refinement in Powder Diffraction in the Presence of Impurities. J. Appl. Crystallogr. 2009, 42, 385–391. [Google Scholar] [CrossRef]
- Fioretti, A.N.; Schwartz, C.P.; Vinson, J.; Nordlund, D.; Prendergast, D.; Tamboli, A.C.; Caskey, C.M.; Tuomisto, F.; Linez, F.; Christensen, S.T.; et al. Understanding and Control of Bipolar Self-Doping in Copper Nitride. J. Appl. Phys. 2016, 119, 181508. [Google Scholar] [CrossRef]
- Shimada, H.; Fukao, T.; Minami, H.; Ukai, M.; Fujii, K.; Yokoya, A.; Fukuda, Y.; Saitoh, Y. Nitrogen K-Edge X-ray Absorption near Edge Structure (XANES) Spectra of Purine-Containing Nucleotides in Aqueous Solution. J. Chem. Phys. 2014, 141, 055102. [Google Scholar] [CrossRef]
- Shimada, H.; Minami, H.; Okuizumi, N.; Sakuma, I.; Ukai, M.; Fujii, K.; Yokoya, A.; Fukuda, Y.; Saitoh, Y. Nitrogen K-Edge x-Ray Absorption near Edge Structure of Pyrimidine-Containing Nucleotides in Aqueous Solution. J. Chem. Phys. 2015, 142, 175102. [Google Scholar] [CrossRef] [PubMed]
- Kruse, J.; Schlichting, A.; Siemens, J.; Regier, T.; Leinweber, P. Pyrolysis-Field Ionization Mass Spectrometry and Nitrogen K-Edge XANES Spectroscopy Applied to Bulk Soil Leachates-a Case Study. Sci. Total Environ. 2010, 408, 4910–4915. [Google Scholar] [CrossRef] [PubMed]
- Leinweber, P.; Kruse, J.; Walley, F.L.; Gillespie, A.; Eckhardt, K.U.; Blyth, R.I.R.; Regier, T. Nitrogen K-Edge XANES—An Overview of Reference Compounds Used to Identify “unknown” Organic Nitrogen in Environmental Samples. J. Synchrotron Radiat. 2007, 14, 500–511. [Google Scholar] [CrossRef]
- Turner, J.A.; Thomas, K.M.; Russell, A.E. The Identification of Oxygen Functional Groups in Carbonaceous Materials by Oxygen K-edge XANES. Carbon 1997, 35, 983–992. [Google Scholar] [CrossRef]
- MacRae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From Visualization to Analysis, Design and Prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Ravel, B.; Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: Data Analysis for X-ray Absorption Spectroscopy Using IFEFFIT. J. Synchrotron Radiat. 2005, 12, 537–541. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Fnnctional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colic-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Oxford Diffraction Ltd. CrysAlis Red and CrysAlis CCD; Oxford Diffraction Ltd.: Abingdon, UK, 2000. [Google Scholar]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An Update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Diamond—Crystal and Molecular Structure Visualization, Crystal Impact—Dr. H. Putz & Dr. K. Brandenburg GbR, Kreuzherrenstr. 102, 53227 Bonn, Germany. Available online: https://www.crystalimpact.de/diamond (accessed on 15 September 2024).
- Lanzilotto, V.; Silva, J.L.; Zhang, T.; Stredansky, M.; Grazioli, C.; Simonov, K.; Giangrisostomi, E.; Ovsyannikov, R.; De Simone, M.; Coreno, M.; et al. Spectroscopic Fingerprints of Intermolecular H-Bonding Interactions in Carbon Nitride Model Compounds. Chem. A Eur. J. 2018, 24, 14198–14206. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muzioł, T.M.; Bronikowska, E. Driving Forces in the Formation of Paracetamol Cocrystals and Solvate with Naphthalene, Quinoline and Acridine. Molecules 2024, 29, 4437. https://doi.org/10.3390/molecules29184437
Muzioł TM, Bronikowska E. Driving Forces in the Formation of Paracetamol Cocrystals and Solvate with Naphthalene, Quinoline and Acridine. Molecules. 2024; 29(18):4437. https://doi.org/10.3390/molecules29184437
Chicago/Turabian StyleMuzioł, Tadeusz M., and Emilia Bronikowska. 2024. "Driving Forces in the Formation of Paracetamol Cocrystals and Solvate with Naphthalene, Quinoline and Acridine" Molecules 29, no. 18: 4437. https://doi.org/10.3390/molecules29184437
APA StyleMuzioł, T. M., & Bronikowska, E. (2024). Driving Forces in the Formation of Paracetamol Cocrystals and Solvate with Naphthalene, Quinoline and Acridine. Molecules, 29(18), 4437. https://doi.org/10.3390/molecules29184437