
Synthesis of Novel Planar-Chiral Charge-Compensated nido-Carborane-Based Amino Acid

 , , , , and
, , , , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
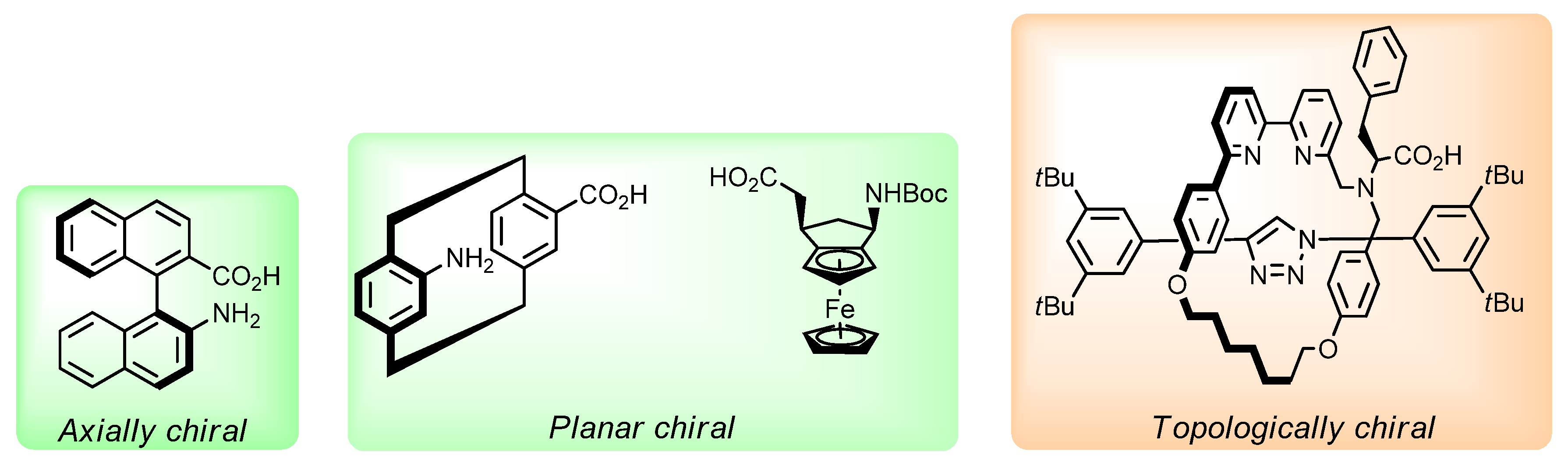
1. Introduction
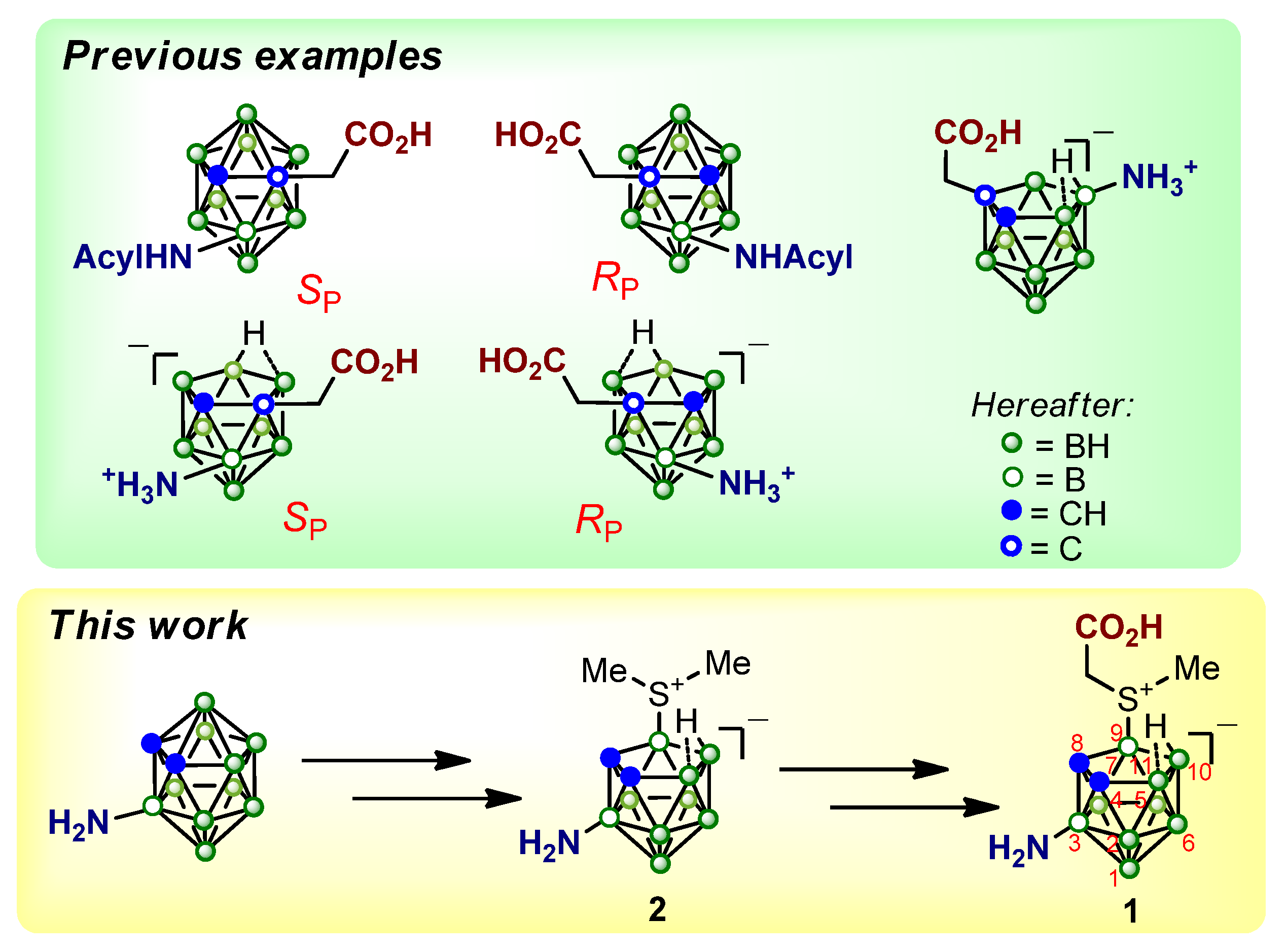
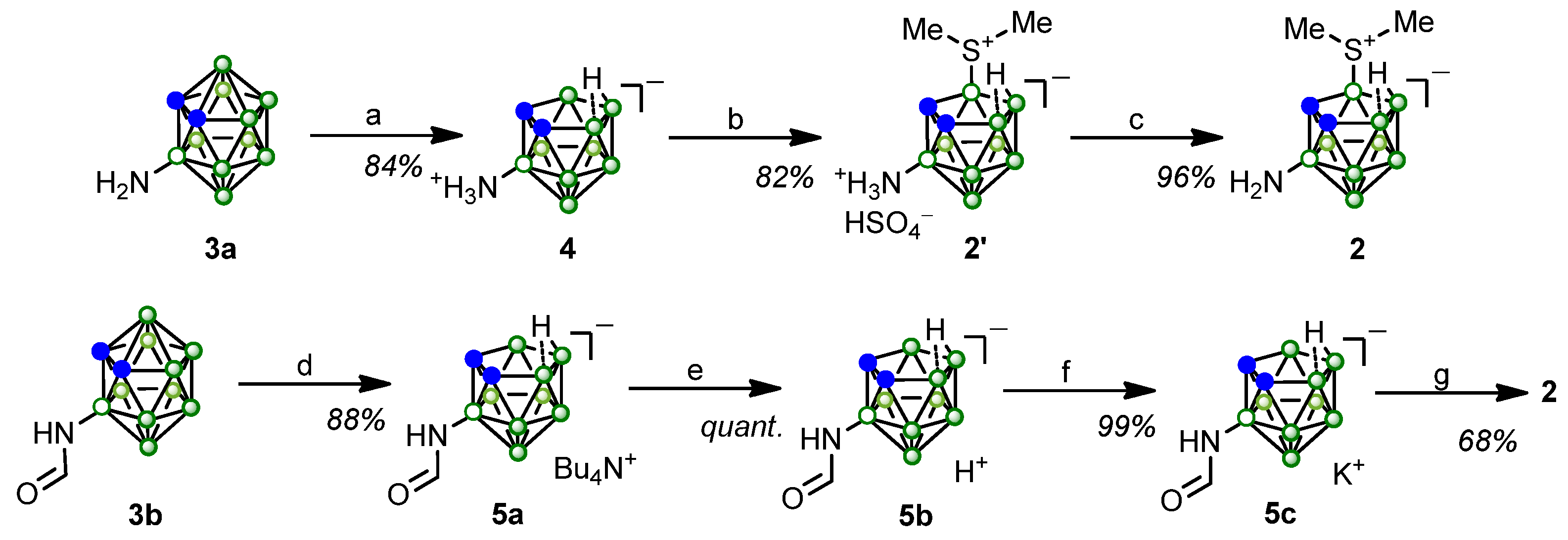
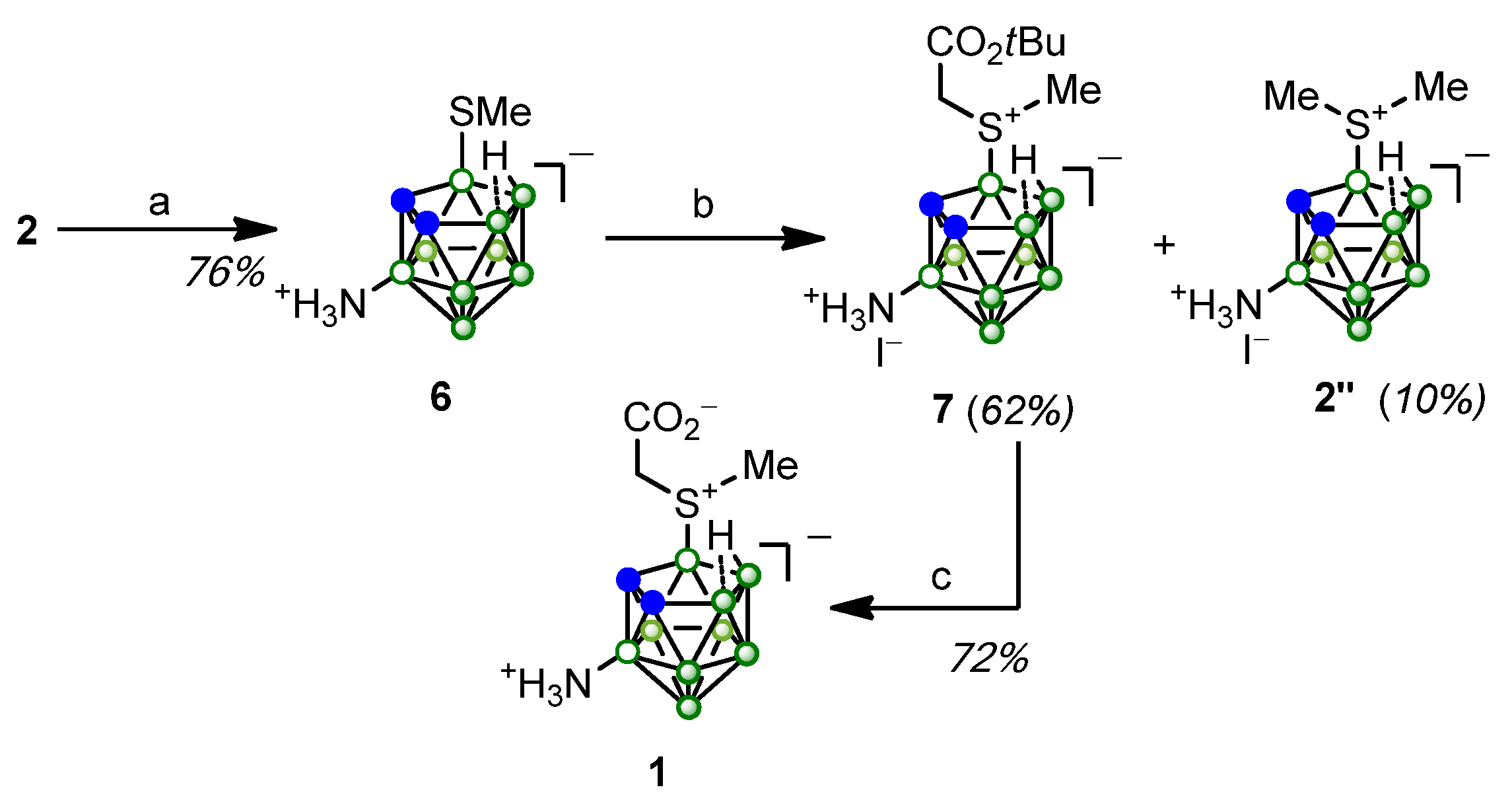
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Eliel, E.L.; Wilen, S.H.; Mander, L.N. Stereochemistry of Organic Compounds; John Wiley & Sons, Inc.: New York, NY, USA, 1994; 1267p. [Google Scholar]
- Wang, Y.; Xu, J.; Wang, Y.; Chen, H. Emerging chirality in nanoscience. Chem. Soc. Rev. 2013, 42, 2930–2962. [Google Scholar] [CrossRef] [PubMed]
- Tverdislov, V.A.; Malyshko, E.V. Chiral Dualism as an Instrument of Hierarchical Structure Formation in Molecular Biology. Symmetry 2020, 12, 587. [Google Scholar] [CrossRef]
- Peluso, P.; Chankvetadze, B. Recognition in the Domain of Molecular Chirality: From Noncovalent Interactions to Separation of Enantiomers. Chem. Rev. 2022, 122, 13235–13400. [Google Scholar] [CrossRef] [PubMed]
- Hassan, Z.; Spuling, E.; Knoll, D.M.; Lahann, J.; Bräse, S. Planar chiral [2.2]paracyclophanes: From synthetic curiosity to applications in asymmetric synthesis and materials. Chem. Soc. Rev. 2018, 47, 6947–6963. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Peng, C.; Wang, Y.-T.; Zhan, G.; Han, B. Recent progress on the construction of axial chirality through transition-metal-catalyzed benzannulation. Org. Chem. Front. 2021, 8, 2772–2785. [Google Scholar] [CrossRef]
- López, R.; Palomo, C. Planar Chirality: A Mine for Catalysis and Structure Discovery. Angew. Chem. Int. Ed. 2022, 61, e202113504. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, Z.-G.; Shi, F. Advances in catalytic enantioselective synthesis of chiral helicenes and helicenoids. Chem Catal. 2022, 2, 3077–3111. [Google Scholar] [CrossRef]
- Wang, Z.; Meng, L.; Liu, X.; Zhang, L.; Yu, Z.; Wu, G. Recent progress toward developing axial chirality bioactive compounds. Eur. J. Med. Chem. 2022, 243, 114700. [Google Scholar] [CrossRef]
- Laws, D., III; Poff, C.D.; Heyboer, E.M.; Blakey, S.B. Synthesis, stereochemical assignment, and enantioselective catalytic activity of late transition metal planar chiral complexes. Chem. Soc. Rev. 2023, 52, 6003–6030. [Google Scholar] [CrossRef]
- Blaskovich, M.A.T. Unusual Amino Acids in Medicinal Chemistry. J. Med. Chem. 2016, 59, 10807–10836. [Google Scholar] [CrossRef]
- Agirre, M.; Arrieta, A.; Arrastia, I.; Cossío, F.P. Organocatalysts Derived from Unnatural α-Amino Acids: Scope and Applications. Chem.—Asian J. 2019, 14, 44–66. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Deng, H.; Li, X.; Quan, Z.-S. Application of Amino Acids in the Structural Modification of Natural Products: A Review. Front. Chem. 2021, 9, 650569. [Google Scholar] [CrossRef] [PubMed]
- Zahradníčková, H.; Opekar, S.; Řimnáčová, L.; Šimek, P.; Moos, M. Chiral secondary amino acids, their importance, and methods of analysis. Amino Acids 2022, 54, 687–719. [Google Scholar] [CrossRef] [PubMed]
- Bongioanni, A.; Bueno, M.S.; Mezzano, B.A.; Longhi, M.R.; Garnero, C. Amino acids and its pharmaceutical applications: A mini-review. Int. J. Pharm. 2022, 613, 121375. [Google Scholar] [CrossRef] [PubMed]
- Kamanna, K. Organocatalysts based on natural and modified amino acids for asymmetric reactions. Phys. Sci. Rev. 2022, 7, 429–467. [Google Scholar] [CrossRef]
- Tichý, M.; Holanová, J.; Závada, J. ω-Amino acids and lactams with chiral biaryl axis. Tetrahedron Asymmetry 1998, 9, 3497–3504. [Google Scholar] [CrossRef]
- Ridvan, L.; Buděšínský, M.; Tichý, M.; Maloň, P.; Závada, J.; Podlaha, J.; Císařová, I. Axially Chiral Bis(α-Amino Acid)s and Dioxopiperazines. Synthesis and Configurational Assignment. Tetrahedron 1999, 55, 12331–12348. [Google Scholar] [CrossRef]
- Kano, T.; Takai, J.; Tokuda, O.; Maruoka, K. Design of an Axially Chiral Amino Acid with a Binaphthyl Backbone as an Organocatalyst for a Direct Asymmetric Aldol Reaction. Angew. Chem. Int. Ed. 2005, 44, 3055–3057. [Google Scholar] [CrossRef]
- Kano, T.; Tokuda, O.; Takai, J.; Maruoka, K. Design of a Binaphthyl-Based Axially Chiral Amino Acid as an Organocatalyst for Direct Asymmetric Aldol Reactions. Chem.—Asian J. 2006, 1–2, 210–215. [Google Scholar] [CrossRef]
- Furuta, T.; Yamamoto, J.; Kitamura, Y.; Hashimoto, A.; Masu, H.; Azumaya, I.; Kan, T.; Kawabata, T. Synthesis of Axially Chiral Amino Acid and Amino Alcohols via Additive–Ligand-free Pd-Catalyzed Domino Coupling Reaction and Subsequent Transformations of the Product Amidoaza [5]helicene. J. Org. Chem. 2010, 75, 7010–7013. [Google Scholar] [CrossRef]
- Furuta, T. Biaryl Amino Acids and Their Surrogates: A Unique Class of Unnatural Amino Acid. In Designed Molecular Space in Material Science and Catalysis; Shirakawa, S., Ed.; Springer Nature: Singapore, 2018; pp. 123–145. [Google Scholar] [CrossRef]
- Bulman Page, P.C.; Kinsey, F.S.; Chan, Y.; Strutt, I.R.; Slawin, A.M.Z.; Jones, G.A. Novel binaphthyl and biphenyl α- and β-amino acids and esters: Organocatalysis of asymmetric Diels–Alder reactions. A combined synthetic and computational study. Org. Biomol. Chem. 2018, 16, 7400–7416. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Shi, Q.; Hu, W.; Chen, T.; Guo, Y.; Hu, Z.; Gong, M.; Guo, J.; Wei, D.; Fu, Z.; et al. Organocatalytic asymmetric N-sulfonyl amide C–N bond activation to access axially chiral biaryl amino acids. Nat. Commun. 2020, 11, 946. [Google Scholar] [CrossRef] [PubMed]
- Cen, S.; Li, S.-S.; Zhao, Y.; Zhao, M.-X.; Zhang, Z. Catalytic Asymmetric Synthesis of Unnatural Axially Chiral Biaryl δ-Amino Acid Derivatives via a Chiral Phenanthroline-Potassium Catalyst-Enabled Dynamic Kinetic Resolution. Angew. Chem. Int. Ed. 2024, 63, e202407920. [Google Scholar] [CrossRef] [PubMed]
- Ansari, S.; Knipe, P.C. Atropisomeric Foldamers. ChemPlusChem 2024, 89, e202400218. [Google Scholar] [CrossRef] [PubMed]
- Salter, R.; Pickett, T.E.; Richards, C.J. 2-Nitroferrocenyloxazolines: Precursors of nitrofulvalenes and derivatives of (pS)- and (pR)-2-aminoferrocenecarboxylic acids. Tetrahedron Asymmetry 1998, 9, 4239–4247. [Google Scholar] [CrossRef]
- Hunold, A.; Neundorf, I.; James, P.; Neudörfl, J.; Schmalz, H.-G. Stereoselective Synthesis of New Ferrocene-Derived Amino Acid Building Blocks. Eur. J. Org. Chem. 2009, 2009, 4429–4440. [Google Scholar] [CrossRef]
- Werner, G.; Butenschön, H. Improved Syntheses of 1,2-Disubstituted Ferrocenes. Eur. J. Inorg. Chem. 2017, 2017, 378–387. [Google Scholar] [CrossRef]
- Kadari, L.; Erb, W.; Halauko, Y.S.; Ivashkevich, O.A.; Matulis, V.E.; Lyakhov, D.; Roisnel, T.; Krishna, P.R.; Mongin, F. On the N-Arylation of Acetamide using 2-, 3- and 1′-Substituted Iodoferrocenes. Eur. J. Inorg. Chem. 2021, 2021, 377–391. [Google Scholar] [CrossRef]
- Pelter, A.; Crump, R.A.N.C.; Kidwell, H. Chiral [2:2]Paracyclophanes. 1. Synthesis and Characterization of Unique Homochiral Amino-Acids derived from [2:2]Paracyclophane. Tetrahedron Lett. 1996, 37, 1273–1276. [Google Scholar] [CrossRef]
- Pelter, A.; Crump, R.A.N.C.; Kidwell, H. Chiral [2.2]paracyclophanes—III. The preparation of unique homochiral amino-acids derived from [2.2]paracyclophane. Tetrahedron Asymmetry 1997, 8, 3873–3880. [Google Scholar] [CrossRef]
- Marchand, A.; Maxwell, A.; Mootoo, B.; Pelter, A.; Reid, A. Oxazoline Mediated Routes to a Unique Amino-Acid, 4-Amino-13-carboxy[2.2]paracyclophane, of Planar Chirality. Tetrahedron 2000, 56, 7331–7338. [Google Scholar] [CrossRef]
- Jayasundera, K.P.; Kusmus, D.N.M.; Deuilhé, L.; Etheridge, L.; Farrow, Z.; Lun, D.J.; Kaur, G.; Rowlands, G.J. The synthesis of substituted amino[2.2]paracyclophanes. Org. Biomol. Chem. 2016, 14, 10848–10860. [Google Scholar] [CrossRef] [PubMed]
- Felder, S.; Micouin, L.; Benedetti, E. Para-Functionalization of N-Substituted 4-Amino[2.2]paracyclophanes by Regioselective Formylation. Eur. J. Org. Chem. 2021, 2021, 4015–4018. [Google Scholar] [CrossRef]
- Yu, S.; Bao, H.; Zhang, D.; Yang, X. Kinetic resolution of substituted amido[2.2]paracyclophanes via asymmetric electrophilic amination. Nat. Commun. 2023, 14, 5239. [Google Scholar] [CrossRef] [PubMed]
- Gibson, S.E.; Jones, J.O.; Kalindjian, S.B.; Knight, J.D.; Mainolfi, N.; Rudd, M.; Steed, J.W.; Tozer, M.J.; Wright, P.T. Synthesis of meta- and paracyclophanes containing unsaturated amino acid residues. Tetrahedron 2004, 60, 6945–6958. [Google Scholar] [CrossRef]
- Kotha, S.; Halder, S.; Damodharan, L.; Pattabhi, V. First and unexpected synthesis of macrocyclic cyclophane-based unusual α-amino acid derivatives by phosphazene base without high dilution conditions. Bioorg. Med. Chem. Lett. 2002, 12, 1113–1115. [Google Scholar] [CrossRef]
- Kotha, S.; Halder, S. Synthesis of macrocyclic cyclophane-based unusual α-amino acid derivatives. Arkivoc 2005, iii, 56–66. [Google Scholar] [CrossRef]
- Tanaka, M.; Anan, K.; Demizu, Y.; Kurihara, M.; Doi, M.; Suemune, H. Side-Chain Chiral Centers of Amino Acid and Helical-Screw Handedness of Its Peptides. J. Am. Chem. Soc. 2005, 127, 11570–11571. [Google Scholar] [CrossRef]
- Barišić, L.; Čakić, M.; Mahmoud, K.A.; Liu, Y.; Kraatz, H.-B.; Pritzkow, H.; Kirin, S.I.; Metzler-Nolte, N.; Rapić, V. Helically Chiral Ferrocene Peptides Containing 1′-Aminoferrocene-1-Carboxylic Acid Subunits as Turn Inducers. Chem.—Eur. J. 2006, 12, 4965–4980. [Google Scholar] [CrossRef]
- Liu, Y.; Gao, X.; Lu, F.; Hu, M.; Shi, L.; Zheng, L. Reversible helical chirality of perylene bisimide aggregates: Amino acid-directed chiral transfer and chiral inversion. Soft Matter 2017, 13, 3072–3075. [Google Scholar] [CrossRef]
- Ueda, A.; Makura, Y.; Kakazu, S.; Kato, T.; Umeno, T.; Hirayama, K.; Doi, M.; Oba, M.; Tanaka, M. E-Selective Ring-Closing Metathesis in α-Helical Stapled Peptides Using Carbocyclic α,α-Disubstituted α-Amino Acids. Org. Lett. 2022, 24, 1049–1054. [Google Scholar] [CrossRef] [PubMed]
- Ousaka, N.; MacLachlan, M.J.; Akine, S. Stapling strategy for slowing helicity interconversion of α-helical peptides and isolating chiral auxiliary-free one-handed forms. Nat. Commun. 2023, 14, 6834. [Google Scholar] [CrossRef] [PubMed]
- Wanka, L.; Cabrele, C.; Vanejews, M.; Schreiner, P.R. γ-Aminoadamantanecarboxylic Acids through Direct C–H Bond Amidations. Eur. J. Org. Chem. 2007, 2007, 1474–1490. [Google Scholar] [CrossRef]
- Aoyama, M.; Hara, S. Synthesis of optically active fluoroadamantane derivative having different substituents on its tert-carbons and its use as non-racemizable source for new optically active adamantine derivatives. Tetrahedron 2013, 69, 10357–10360. [Google Scholar] [CrossRef]
- Yasue, R.; Yoshida, K. Enantioselective Desymmetrization of 1,3-Disubstituted Adamantane Derivatives via Rhodium-Catalyzed C–H Bond Amination: Access to Optically Active Amino Acids Containing Adamantane Core. Adv. Synth. Catal. 2021, 363, 1662–1671. [Google Scholar] [CrossRef]
- Shirakawa, S.; Tanaka, Y.; Kobari, T.; Shimizu, S. Synthesis and optical resolution of an inherently chiral calix[4]arene amino acid. New J. Chem. 2008, 32, 1835–1838. [Google Scholar] [CrossRef]
- Shirakawa, S.; Shimizu, S. Synthesis of an Inherently Chiral Calix[4]arene Amino Acid and Its Derivatives: Their Application to Asymmetric Reactions as Organocatalysts. Eur. J. Org. Chem. 2009, 2009, 1916–1924. [Google Scholar] [CrossRef]
- Bianco, A.; Maggini, M.; Scorrano, G.; Toniolo, C.; Marconi, G.; Villani, C.; Prato, M. Synthesis, Chiroptical Properties, and Configurational Assignment of Fulleroproline Derivatives and Peptides. J. Am. Chem. Soc. 1996, 118, 4072–4080. [Google Scholar] [CrossRef]
- Ioutsi, V.A.; Zadorin, A.A.; Khavrel, P.A.; Belov, N.M.; Ovchinnikova, N.S.; Goryunkov, A.A.; Kharybin, O.N.; Nikolaev, E.N.; Yurovskaya, M.A.; Sidorov, L.N. Diastereoselective lithium salt-assisted 1,3-dipolar cycloaddition of azomethine ylides to the fullerene C60. Tetrahedron 2010, 66, 3037–3041. [Google Scholar] [CrossRef]
- Levitskiy, O.A.; Grishin, Y.K.; Semivrazhskaya, O.O.; Ambartsumyan, A.A.; Kochetkov, K.A.; Magdesieva, T.V. Individual (f,tA)- and (f,tC)-Fullerene-Based Nickel(II) Glycinates: Protected Chiral Amino Acids Directly Linked to a Chiral π-Electron System. Angew. Chem. Int. Ed. 2017, 56, 2704–2708. [Google Scholar] [CrossRef]
- Rodríguez-Rubio, A.; Savoini, A.; Modicom, F.; Butler, P.; Goldup, S.M. A Co-conformationally “Topologically” Chiral Catenane. J. Am. Chem. Soc. 2022, 144, 11927–11932. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Rodríguez-Rubio, A.; Saady, A.; Tizzard, G.J.; Goldup, S.M. A chiral macrocycle for the stereoselective synthesis of mechanically planar chiral rotaxanes and catenanes. Chem 2023, 9, 1195–1207. [Google Scholar] [CrossRef]
- Bao, S.-J.; Zhang, H.-N.; Jin, G.-X. Stereoselective Self-Assembly of Chiral Metalla[2]Catenanes and Lemniscular Macrocycles. CCS Chem. 2024, 6, 2000–2010. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, Z.; Lv, S.; Zeng, X.; Sun, Y.; Li, H.; Zhang, R. The chiral interfaces fabricated by D/L-alanine-pillar[5]arenes for selectively absorbing ctDNA. Chem. Commun. 2019, 55, 778–781. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Fu, L.; Sun, B.; Qian, C.; Wang, R.; Jiang, J.; Lin, C.; Ma, J.; Wang, L. Competitve Selection of Conformation Chirality of Water-Soluble Pillar[5]arene Induced by Amino Acid Derivatives. Org. Lett. 2020, 22, 2266–2270. [Google Scholar] [CrossRef] [PubMed]
- Nazarova, A.; Padnya, P.; Kharlamova, A.; Petrov, K.; Yusupov, G.; Zelenikhin, P.; Bukharov, M.; Hua, B.; Huang, F.; Stoikov, I. Peptidomimetics based on ammonium decasubstituted pillar[5]arenes: Influence of the alpha-amino acid residue nature on cholinesterase inhibition. Bioorg. Chem. 2023, 141, 106927. [Google Scholar] [CrossRef]
- Sultanaev, V.; Yakimova, L.; Nazarova, A.; Mostovaya, O.; Sedov, I.; Davletshin, D.; Gilyazova, E.; Bulatov, E.; Li, Z.-T.; Zhang, D.-W.; et al. Decasubstituted Pillar[5]arene Derivatives Containing L-Tryptophan and L-Phenylalanine Residues: Non-Covalent Binding and Release of Fluorescein from Nanoparticles. Int. J. Mol. Sci. 2023, 24, 7700. [Google Scholar] [CrossRef]
- Shibatomi, K.; Kitahara, K.; Okimi, T.; Abe, Y.; Iwasa, S. Enantioselective fluorination of α-branched aldehydes and subsequent conversion to α-hydroxyacetals via stereospecific C–F bond cleavage. Chem. Sci. 2016, 7, 1388–1392. [Google Scholar] [CrossRef]
- Shibatomi, K.; Kitahara, K.; Sasaki, N.; Kawasaki, Y.; Fujisawa, I.; Iwasa, S. Enantioselective decarboxylative chlorination of β-ketocarboxylic acids. Nat. Commun. 2017, 8, 15600. [Google Scholar] [CrossRef]
- Lu, W.; Pei, X.; Murai, T.; Sasamori, T.; Tokitoh, N.; Kawabata, T.; Furuta, T. Asymmetric Intramolecular C–H Insertion Promoted by Dirhodium(II) Carboxylate Catalyst Bearing Axially Chiral Amino Acid Derivatives. Synlett 2017, 28, 679–683. [Google Scholar] [CrossRef]
- Mizutani, H.; Kawanishi, R.; Shibatomi, K. Enantioselective decarboxylative protonation and deuteration of β-ketocarboxylic acids. Chem. Commun. 2021, 57, 6676–6679. [Google Scholar] [CrossRef] [PubMed]
- Thoß, M.; Seidel, R.W.; Feigel, M. The artificial binaphthyl amino acid 6-amino-6′-carboxyethyl-2-methoxy-2′-hydroxy-1,1′-binaphthyl (Bna): Synthesis and assembly of Bna peptides. Tetrahedron 2010, 66, 8503–8511. [Google Scholar] [CrossRef]
- Grimes, R.N. Carboranes, 3rd ed.; Academic Press: Cambridge, MA, USA, 2016; 1041p. [Google Scholar]
- Sivaev, I.B. Polyhedral Boranes and Carboranes. In Comprehensive Organometallic Chemistry IV; Aldridge, S., Ed.; Elsevier: Amsterdam, The Netherlands, 2022; Volume 9, pp. 196–262. [Google Scholar] [CrossRef]
- Takagaki, M.; Kazuko, U.; Hosmane, N.S. Chapter 5: An Overview of Clinical and Biological Aspects of Current Boron Neutron Capture Therapy (BNCT) for Cancer Treatment. In Handbook of Boron Science with Applications in Organometallics, Catalysis, Materials and Medicine; Hosmane, N.S., Egaling, R., Eds.; World Scientific: Hackensack, NJ, USA, 2018; Volume 4, pp. 101–143. [Google Scholar] [CrossRef]
- Marfavi, A.; Kavianpoor, P.; Rendina, L.M. Carboranes in drug discovery, chemical biology and molecular imaging. Nat. Rev. Chem. 2022, 6, 486–504. [Google Scholar] [CrossRef] [PubMed]
- Marforio, T.D.; Carboni, A.; Calvaresi, M. In Vivo Application of Carboranes for Boron Neutron Capture Therapy (BNCT): Structure, Formulation and Analytical Methods for Detection. Cancers 2023, 15, 4944. [Google Scholar] [CrossRef]
- Xu, H.; Liu, J.; Li, R.; Lin, J.; Gui, L.; Wang, Y.; Jin, Z.; Xia, W.; Liu, Y.; Cheng, S.; et al. Novel promising boron agents for boron neutron capture therapy: Current status and outlook on the future. Coord. Chem. Rev. 2024, 511, 215795. [Google Scholar] [CrossRef]
- Couto, M.; Cerecetto, H. Advancements in the Synthesis and Biological Properties of Carboranes and High-Boron Related Compounds: A Comprehensive Exploration with Emphasis on BNCT Applications. J. Braz. Chem. Soc. 2024, 35, e-20240109. [Google Scholar] [CrossRef]
- Lanfranco, A.; Alberti, D.; Parisotto, S.; Renzi, P.; Lecomte, V.; Geninatti Crich, S.; Deagostino, A. Biotinylation of a MRI/Gd BNCT theranostic agent to access a novel tumour-targeted delivery system. Org. Biomol. Chem. 2022, 20, 5342–5354. [Google Scholar] [CrossRef]
- Lee, W.; Kim, K.W.; Lim, J.E.; Sarkar, S.; Kim, J.Y.; Chang, Y.; Yoo, J. In vivo evaluation of the effects of combined boron and gadolinium neutron capture therapy in mouse models. Sci. Rep. 2022, 12, 13360. [Google Scholar] [CrossRef]
- Shanmugam, M.; Kuthala, N.; Kong, X.; Chiang, C.-S.; Hwang, K.C. Combined Gadolinium and Boron Neutron Capture Therapies for Eradication of Head-and-Neck Tumor Using Gd10B6 Nanoparticles under MRI/CT Image Guidance. JACS Au 2023, 3, 2192–2205. [Google Scholar] [CrossRef]
- Gruzdev, D.A.; Levit, G.L.; Krasnov, V.P.; Charushin, V.N. Carborane-containing amino acids and peptides: Synthesis, properties, and applications. Coord. Chem. Rev. 2021, 433, 213753. [Google Scholar] [CrossRef]
- Levit, G.L.; Krasnov, V.P.; Gruzdev, D.A.; Demin, A.M.; Bazhov, I.V.; Sadretdinova, L.S.; Olshevskaya, V.A.; Kalinin, V.N.; Cheong, C.S.; Chupakhin, O.N.; et al. Synthesis of N-[(3-amino-1,2-dicarba-closo-dodecaboran-1-yl)acetyl] derivatives of α-amino acids. Collect. Czech. Chem. Commun. 2007, 72, 1697–1706. [Google Scholar] [CrossRef]
- Gruzdev, D.A.; Nuraeva, A.S.; Slepukhin, P.A.; Levit, G.L.; Zelenovskiy, P.S.; Shur, V.Y.; Krasnov, V.P. Piezoactive amino acid derivatives containing fragments of planar-chiral ortho-carboranes. J. Mater. Chem. C 2018, 6, 8638–8645. [Google Scholar] [CrossRef]
- Gruzdev, D.A.; Ustinova, V.O.; Chulakov, E.N.; Ol’shevskaya, V.A.; Slepukhin, P.A.; Levit, G.L.; Krasnov, V.P.; Charushin, V.N. Preparation of enantiomerically pure derivatives of (3-amino-1,2-dicarba-closo-dodecaboran-1-yl)acetic acid. J. Organomet. Chem. 2018, 876, 50–55. [Google Scholar] [CrossRef]
- Telegina, A.A.; Gruzdev, D.A.; Levit, G.L.; Krasnov, V.P. Synthesis of a novel planar-chiral nido-carborane amino acid. Russ. Chem. Bull. 2021, 70, 539–544. [Google Scholar] [CrossRef]
- Gruzdev, D.A.; Telegina, A.A.; Chulakov, E.N.; Levit, G.L.; Krasnov, V.P. (7,8-Dicarba-nido-undecaboran-7-yl)acetic acid: Synthesis of individual enantiomers and the first example of the determination of the absolute configuration of chiral monosubstituted nido-carborane. New J. Chem. 2022, 46, 17338–17347. [Google Scholar] [CrossRef]
- Nuraeva, A.S.; Vasileva, D.S.; Vasilev, S.G.; Zelenovskiy, P.S.; Gruzdev, D.A.; Krasnov, V.P.; Ol’shevskaya, V.A.; Kalinin, V.N.; Shur, V.Y. Piezoelectric and ferroelectric properties of organic single crystals and films derived from chiral 2-methoxy and 2-amino acids. Ferroelectrics 2016, 496, 1–9. [Google Scholar] [CrossRef]
- Gruzdev, D.A.; Telegina, A.A.; Levit, G.L.; Krasnov, V.P. N-Aminoacyl-3-amino-nido-carboranes as a Group of Boron-Containing Derivatives of Natural Amino Acids. J. Org. Chem. 2022, 87, 5437–5441. [Google Scholar] [CrossRef]
- Gruzdev, D.A.; Telegina, A.A.; Levit, G.L.; Ezhikova, M.A.; Kodess, M.I.; Krasnov, V.P. Synthesis of Charge-Compensated nido-Carboranyl Derivatives of Sulfur-Containing Amino Acids and Biotin. J. Org. Chem. 2023, 88, 14022–14032. [Google Scholar] [CrossRef]
- Telegina, A.A.; Gruzdev, D.A.; Ezhikova, M.A.; Kodess, M.I.; Ol’shevskaya, V.A.; Levit, G.L.; Krasnov, V.P. Synthesis of novel zwitterionic nido-carborane-containing derivatives of cysteine and methionine. Russ. Chem. Bull. 2024, 73, 1716–1724. [Google Scholar] [CrossRef]
- Anufriev, S.A.; Zakharova, M.V.; Sivaev, I.B.; Bregadze, V.I. New carborane-containing acids and amines. Russ. Chem. Bull. 2017, 66, 1643–1649. [Google Scholar] [CrossRef]
- Zakharkin, L.I.; Ol’shevskaya, V.A.; Sulaimankulova, D.D.; Antonovich, V.A. Cleavage of 3-amino-o-carborane and its N-derivatives by bases into 3-amino-7,8-dicarbaundecaborate anion and its N-derivatives. Russ. Chem. Bull. 1991, 40, 1026–1032. [Google Scholar] [CrossRef]
- Plešek, J.; Janoušek, Z.; Heřmánek, S. Four new (CH3)2S.C2B9H11 isomers. Collect. Czech. Chem. Commun. 1978, 43, 2862–2868. [Google Scholar] [CrossRef]
- Plešek, J.; Janoušek, Z.; Heřmánek, S. (9-Dimethyl sulfide)-7,8-dicarba-nido-undecaborane(11), 9-[(CH3)2S]-7,8-C2B9H11. Inorg. Synth. 1983, 22, 239–241. [Google Scholar] [CrossRef]
- Yan, Y.-K.; Mingos, D.M.P.; Müller, T.E.; Williams, D.J.; Kurmoo, M. Synthesis and Structure of a Charge-compensated Ferracarborane, commo-[3,3′-Fe{4-(Me2S)-l,2-C2B9H10}2], and Its Charge-transfer Salt with 2,3-Dichloro-5,6-dicyano-p-benzoquinone. J. Chem. Soc. Dalton Trans. 1994, 1735–1741. [Google Scholar] [CrossRef]
- Rosair, G.M.; Welch, A.J.; Weller, A.S.; Zahn, S.K. Sterically encumbered charge-compensated carbaboranes: Synthesis and reactivity. Molecular structure of 7-Ph-11-SMe2-7,8-nido-C2B9H10 and 1-Ph-3,3-(CO)2-7-SMe2-3,1,2-closo-RhC2B9H8. J. Organomet. Chem. 1997, 536–537, 299–308. [Google Scholar] [CrossRef]
- Meshcheryakov, V.I.; Kitaev, P.S.; Lyssenko, K.A.; Starikova, Z.A.; Petrovskii, P.V.; Janoušek, Z.; Corsini, M.; Laschi, F.; Zanello, P.; Kudinov, A.R. (Tetramethylcyclobutadiene)cobalt complexes with monoanionic carborane ligands [9-L-7,8-C2B9H10]− (L = SMe2, NMe3 and py). J. Organomet. Chem. 2005, 690, 4745–4754. [Google Scholar] [CrossRef]
- Vinogradov, M.M.; Nelyubina, Y.V.; Aliyeu, T.M. New aspects of acid-assisted nucleophilic substitution reactions of 11-vertex nido-carboranes. Polyhedron 2022, 214, 115654. [Google Scholar] [CrossRef]
- Telegina, A.A.; Gruzdev, D.A.; Levit, G.L.; Krasnov, V.P. Synthesis of new nido-carborane-containing 6-thiopurine derivatives. Russ. Chem. Bull. 2023, 72, 2860–2866. [Google Scholar] [CrossRef]
- Stewart, W.E.; Siddall, T.H., III. Nuclear Magnetic Resonance Studies of Amides. Chem. Rev. 1970, 70, 517–551. [Google Scholar] [CrossRef]
- Li, Y.-S.; Escobar, L.; Zhu, Y.-J.; Cohen, Y.; Ballester, P.; Rebek, J., Jr.; Yu, Y. Relative hydrophilicities of cis and trans formamides. Proc. Natl. Acad. Sci. USA 2019, 116, 19815–19820. [Google Scholar] [CrossRef]
- Chouhan, K.K.; Chowdhury, D.; Mukherjee, A. Transamidation of Aromatic Amines with Formamides Using Cyclic Dihydrogen Tetrametaphosphate. Org. Biomol. Chem. 2022, 20, 7929–7935. [Google Scholar] [CrossRef] [PubMed]
- Plešek, J.; Grüner, B.; Maloń, P. Synthesis and properties of (±)- and (+)-4-MeS-3-C2H5-1,2,3-C2CoB9H10. Collect. Czech. Chem. Commun. 1993, 58, 1087–1092. [Google Scholar] [CrossRef]
- Tian, Z.-Y.; Hu, Y.-T.; Teng, H.-B.; Zhang, C.-P. Application of arylsulfonium salts as arylation reagents. Tetrahedron Lett. 2018, 59, 299–309. [Google Scholar] [CrossRef]
- Tian, Z.-Y.; Ma, Y.; Zhang, C.-P. Alkylation Reactions with Alkylsulfonium Salts. Synthesis 2022, 54, 1478–1502. [Google Scholar] [CrossRef]
- Chen, J.; Liu, S.; Su, S.; Fan, R.; Zhang, R.; Meng, W.; Tan, J. Sulfonium-based precise alkyl transposition reactions. Sci. Adv. 2023, 9, eadi1370. [Google Scholar] [CrossRef]
- Anufriev, S.A.; Sivaev, I.B.; Suponitsky, K.Y.; Bregadze, V.I. Practical synthesis of 9-methylthio-7,8-nido-carborane [9-MeS-nido-7,8-C2B9H11]−. Some evidences of BH···X hydride-halogen bonds in 9-XCH2(Me)S-7,8-C2B9H11 (X = Cl, Br, I). J. Organomet. Chem. 2017, 849–850, 315–323. [Google Scholar] [CrossRef]
- Kultyshev, R.G.; Liu, J.; Meyers, E.A.; Shore, S.G. Synthesis and Characterization of Sulfide, Sulfide–Sulfonium, and Bissulfide Derivatives of [B12H12]2−. Additivity of Me2S and MeS− Substituent Effects in 11B NMR Spectra of Disubstituted Icosahedral Boron Clusters. Inorg. Chem. 2000, 39, 3333–3341. [Google Scholar] [CrossRef]
- Kultyshev, R.G.; Liu, J.; Liu, S.; Tjarks, W.; Soloway, A.H.; Shore, S.G. S-Alkylation and S-Amination of Methyl Thioethers–Derivatives of closo-[B12H12]2−. Synthesis of a Boronated Phosphonate, gem-Bisphosphonates, and Dodecaborane-ortho-carborane Oligomers. J. Am. Chem. Soc. 2002, 124, 2614–2624. [Google Scholar] [CrossRef]
- Kabytaev, K.Z.; Safronov, A.V.; Sevryugina, Y.V.; Barnes, C.L.; Jalisatgi, S.S.; Hawthorne, M.F. Novel Synthetic Approach to Charge-Compensated Phosphonio-nido-Carboranes. Synthesis and Structural Characterization of Neutral Mono and Bis(Phosphonio) nido-ortho-Carboranes. Inorg. Chem. 2015, 54, 4143–4150. [Google Scholar] [CrossRef]
- Zakharova, M.V.; Sivaev, I.B.; Anufriev, S.A.; Timofeev, S.V.; Suponitsky, K.Y.; Godovikov, I.A.; Bregadze, V.I. A new approach to the synthesis of functional derivatives of nido-carborane: Alkylation of [9-MeS-nido-7,8-C2B9H11]−. Dalton Trans. 2014, 43, 5044–5053. [Google Scholar] [CrossRef]
- Poater, J.; Viñas, C.; Bennour, I.; Escayola, S.; Solà, M.; Teixidor, F. Too Persistent to Give Up: Aromaticity in Boron Clusters Survives Radical Structural Changes. J. Am. Chem. Soc. 2020, 142, 9396–9407. [Google Scholar] [CrossRef] [PubMed]
- Zakharkin, L.I.; Kalinin, V.N.; Gedymin, V.V. Synthesis of 1,2-Dicarba-closo-dodecaboran(12)-3-yl-carboxylic Acids (3-O-Carboranecarboxylic Acids). Synth. Inorg. Met.-Org. Chem. 1971, 1, 45–50. [Google Scholar] [CrossRef]
- Meguellati, K.; Koripelly, G.; Ladame, S. DNA-Templated Synthesis of Trimethine Cyanine Dyes: A Versatile Fluorogenic Reaction for Sensing G-Quadruplex Formation. Angew. Chem. Int. Ed. 2010, 49, 2738–2742. [Google Scholar] [CrossRef] [PubMed]
- Armarego, W.L.F.; Chai, C.L.L. Purification of Laboratory Chemicals, 6th ed.; Butterworth Heinemann: Burlington, MA, USA, 2009; 743p. [Google Scholar]
- Janoušek, Z.; Heřmánek, S.; Plešek, J.; Štíbr, B. Tetracarba-dinido-docosaborane (C4B18H22), a new type of carborane, its chemistry and structure. Collect. Czech. Chem. Commun. 1974, 39, 2363–2373. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gruzdev, D.A.; Telegina, A.A.; Ezhikova, M.A.; Kodess, M.I.; Levit, G.L.; Krasnov, V.P. Synthesis of Novel Planar-Chiral Charge-Compensated nido-Carborane-Based Amino Acid. Molecules 2024, 29, 4487. https://doi.org/10.3390/molecules29184487
Gruzdev DA, Telegina AA, Ezhikova MA, Kodess MI, Levit GL, Krasnov VP. Synthesis of Novel Planar-Chiral Charge-Compensated nido-Carborane-Based Amino Acid. Molecules. 2024; 29(18):4487. https://doi.org/10.3390/molecules29184487
Chicago/Turabian StyleGruzdev, Dmitry A., Angelina A. Telegina, Marina A. Ezhikova, Mikhail I. Kodess, Galina L. Levit, and Victor P. Krasnov. 2024. "Synthesis of Novel Planar-Chiral Charge-Compensated nido-Carborane-Based Amino Acid" Molecules 29, no. 18: 4487. https://doi.org/10.3390/molecules29184487