
Molecular Simulation of the Water Diffusion Behavior and Electronic Properties of Boron-Nitride-Composited Mineral Oil


Abstract
1. Introduction
2. Results and Discussion
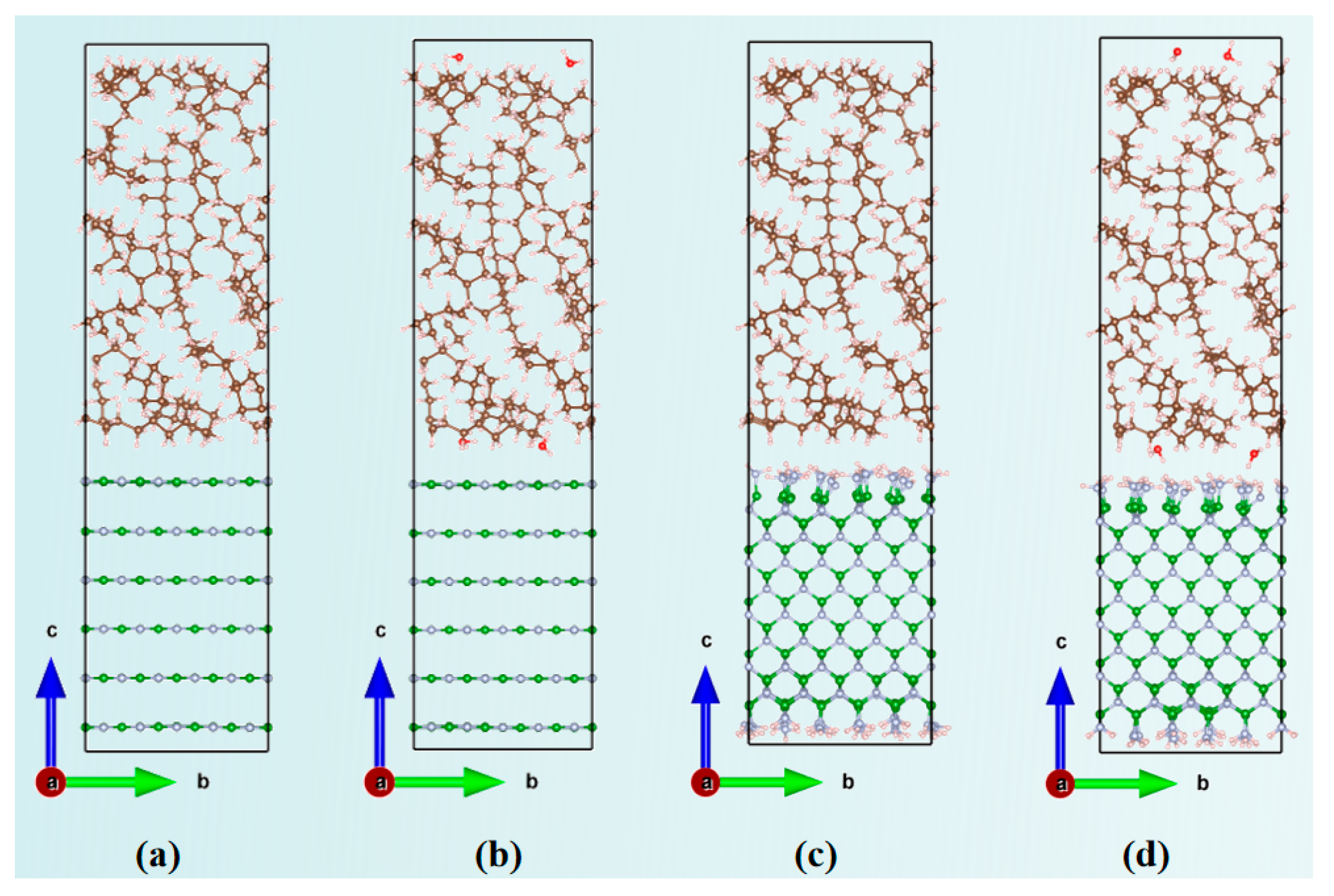
2.1. Diffusion Behavior of Water Molecules in MO/BN Composite Models
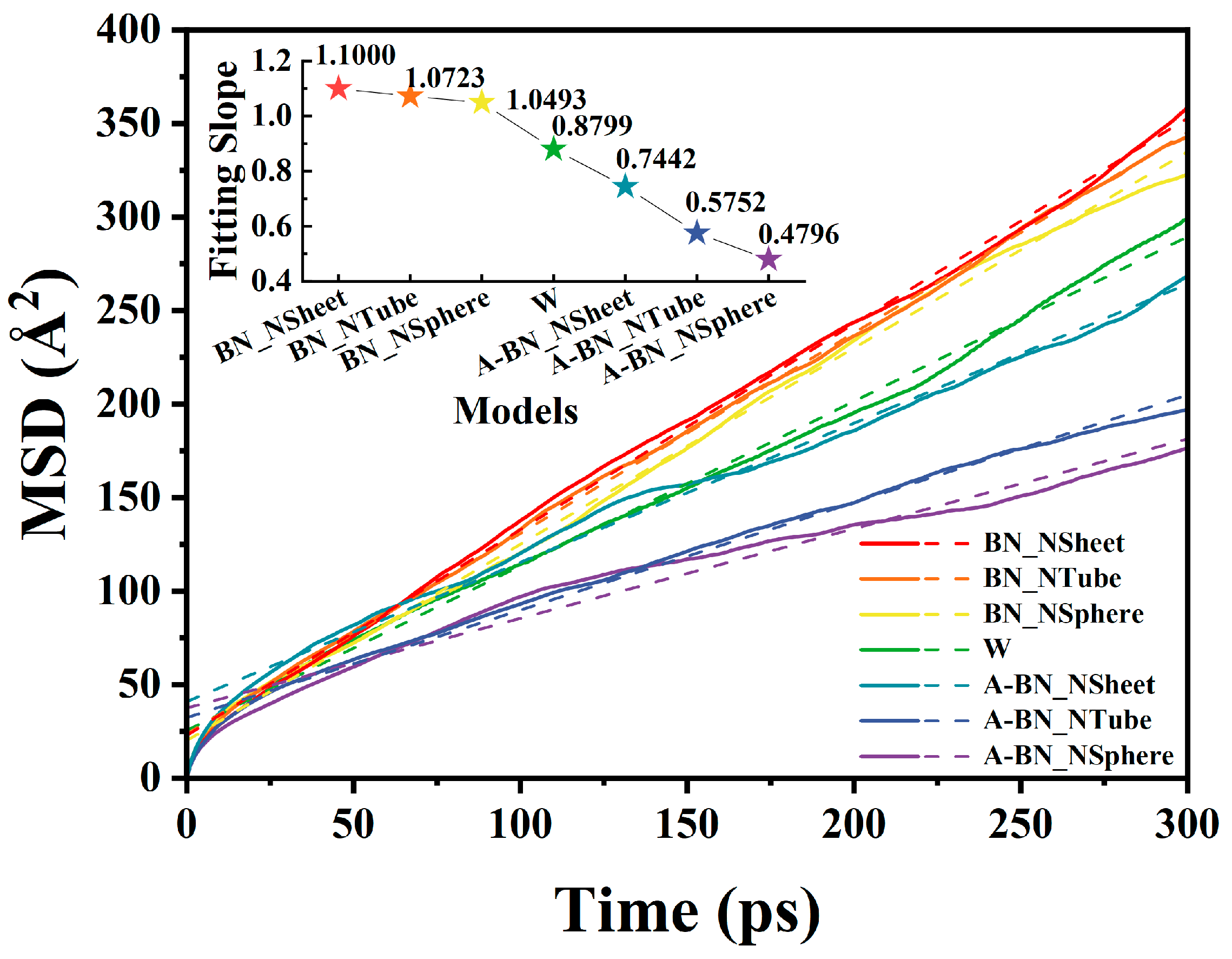
2.1.1. Diffusion Coefficients in Different BN-Doped MO Models
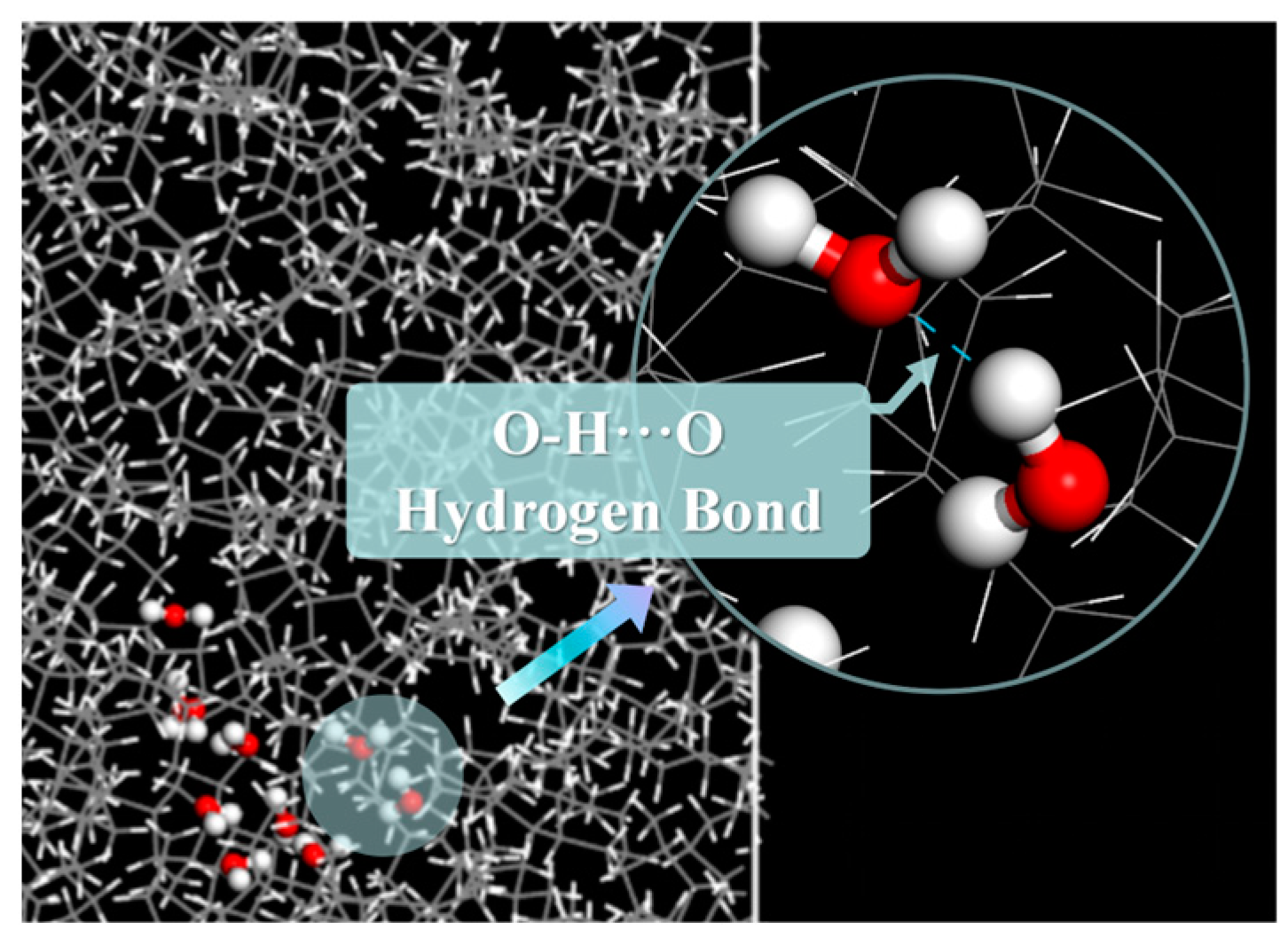
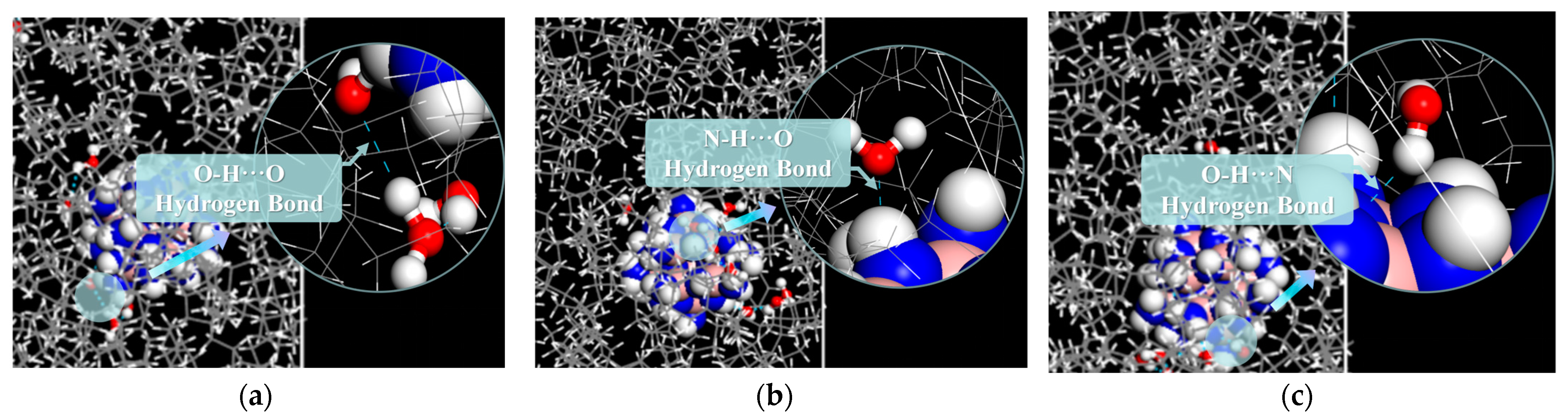
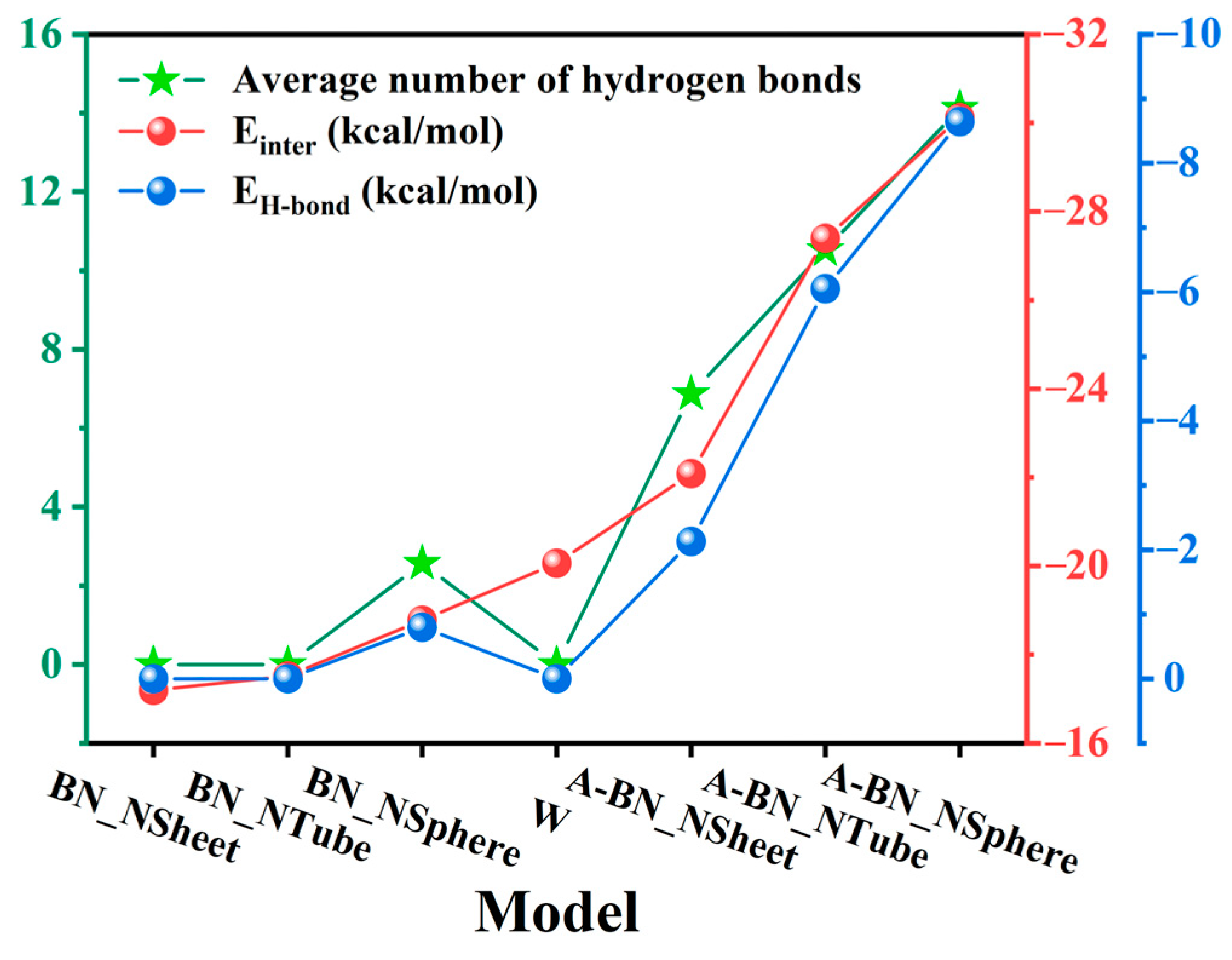
2.1.2. Average Number and Types of Hydrogen Bonds
2.1.3. Interaction Energies
2.1.4. Fractional Free Volumes
2.2. Effect of Different Temperatures on the Diffusion of Water Molecules in the A-BN_NSphere Model
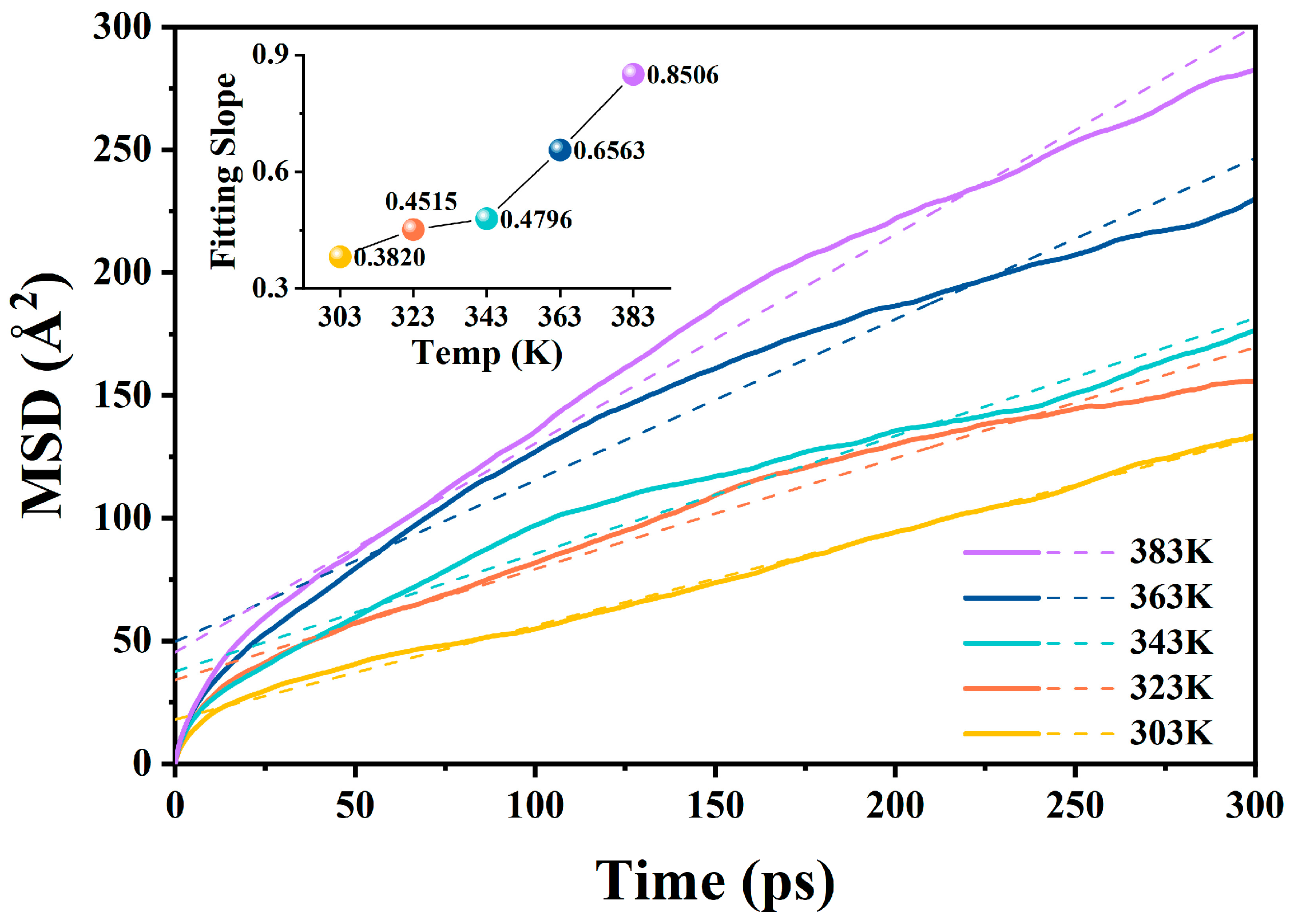
2.2.1. Diffusion Coefficients at Different Temperatures in the A-BN_NSphere Model
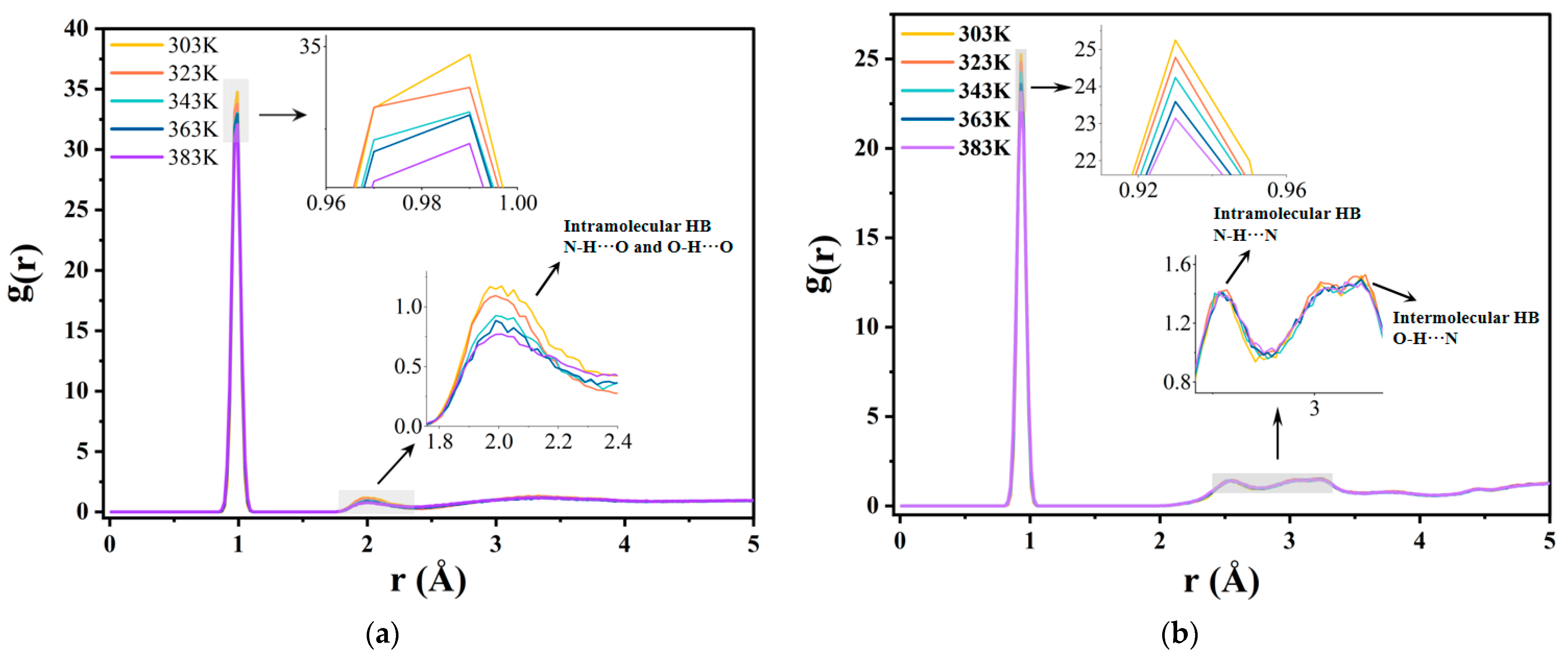
2.2.2. Hydrogen Bonds and Radial Distribution Functions
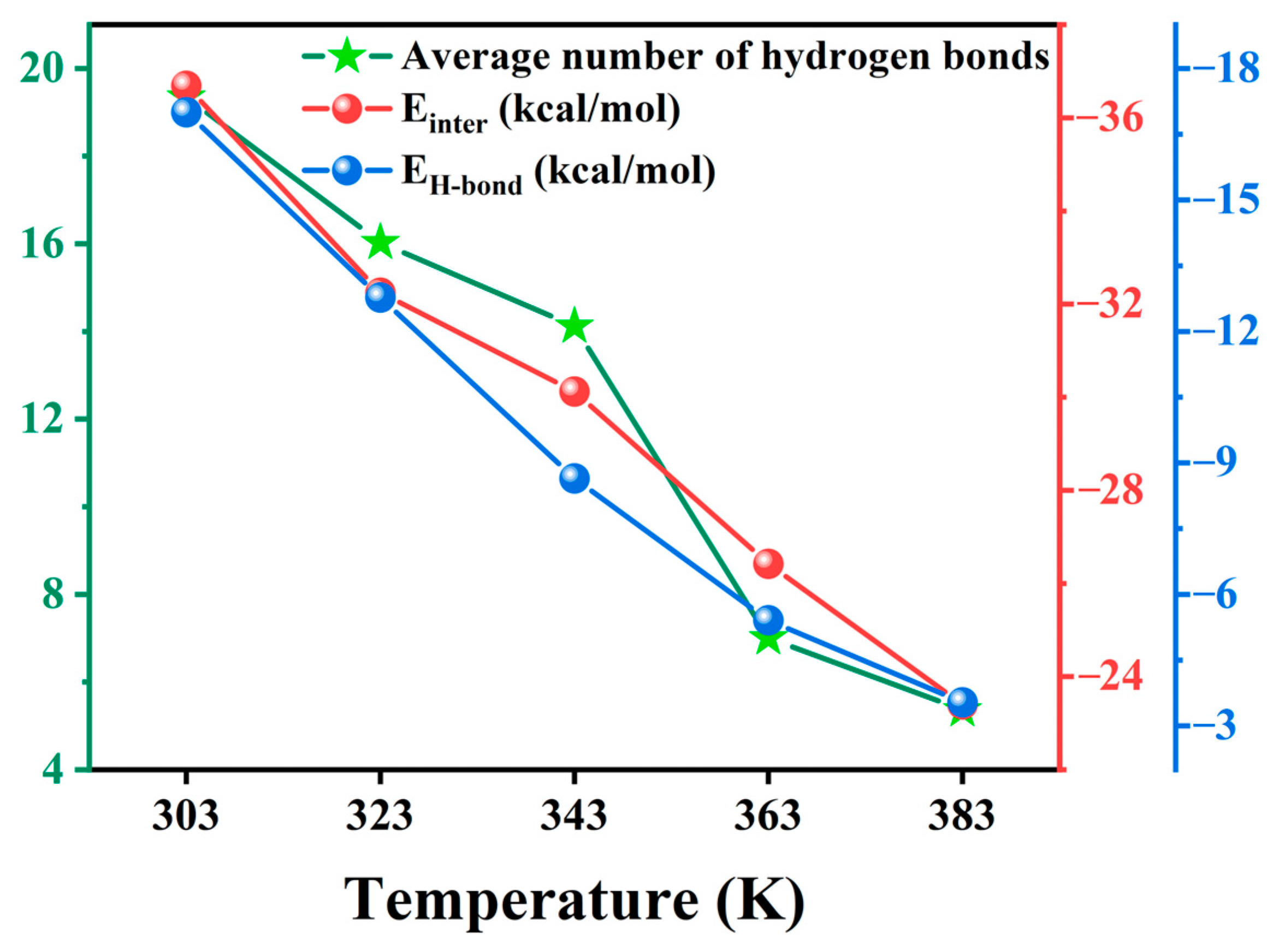
2.2.3. Temperature Dependence of Interaction Energy
2.3. Electrical Properties of BN/MO Interface Models
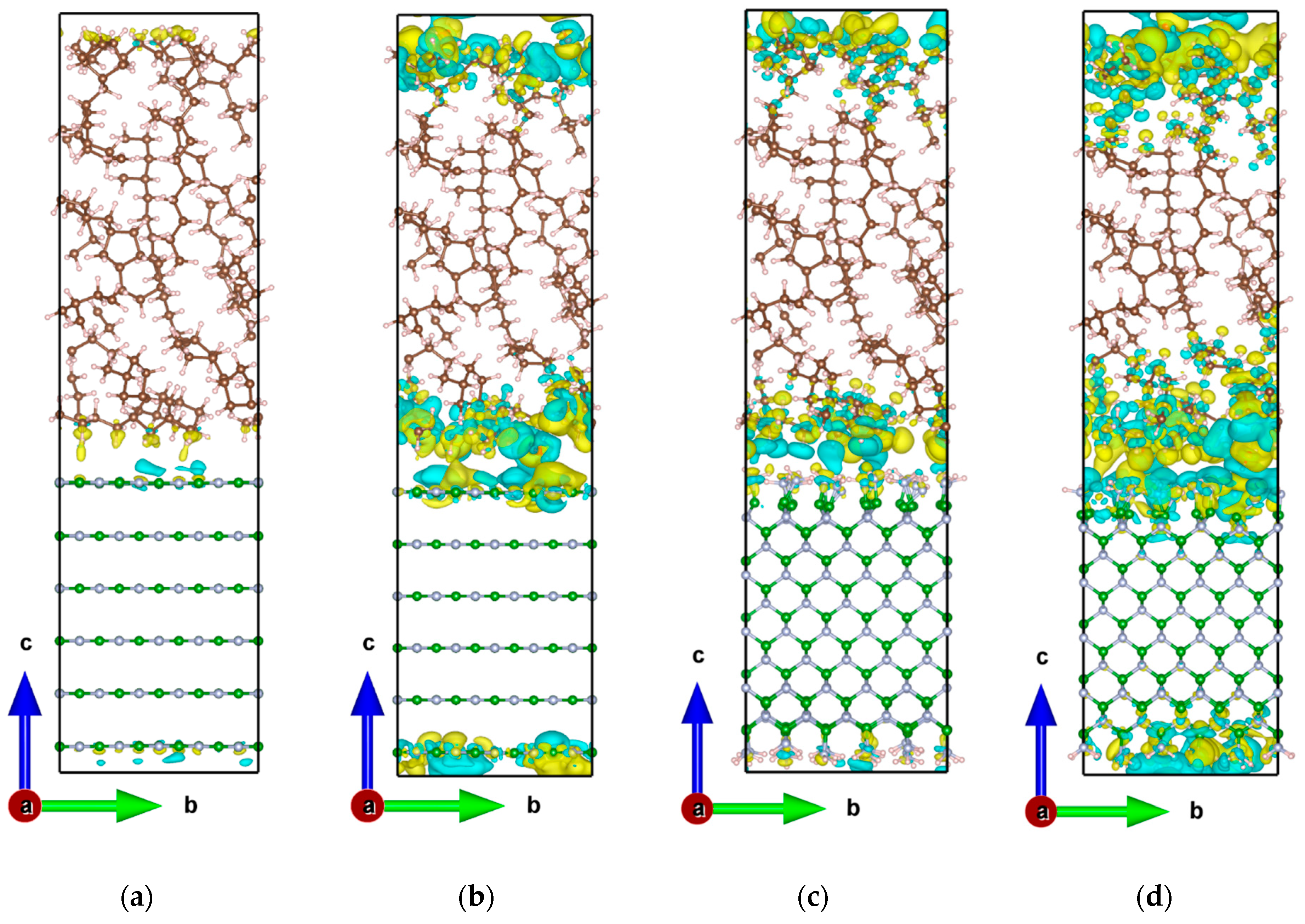
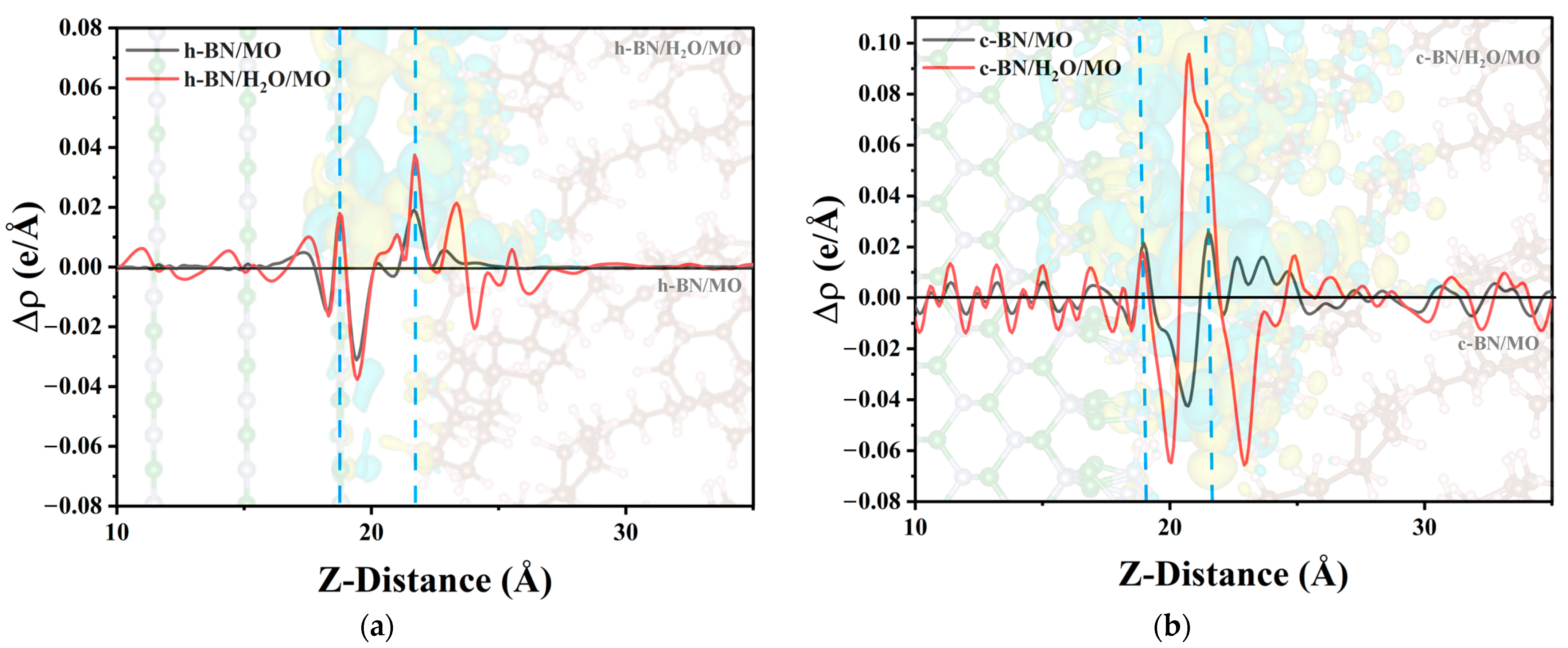
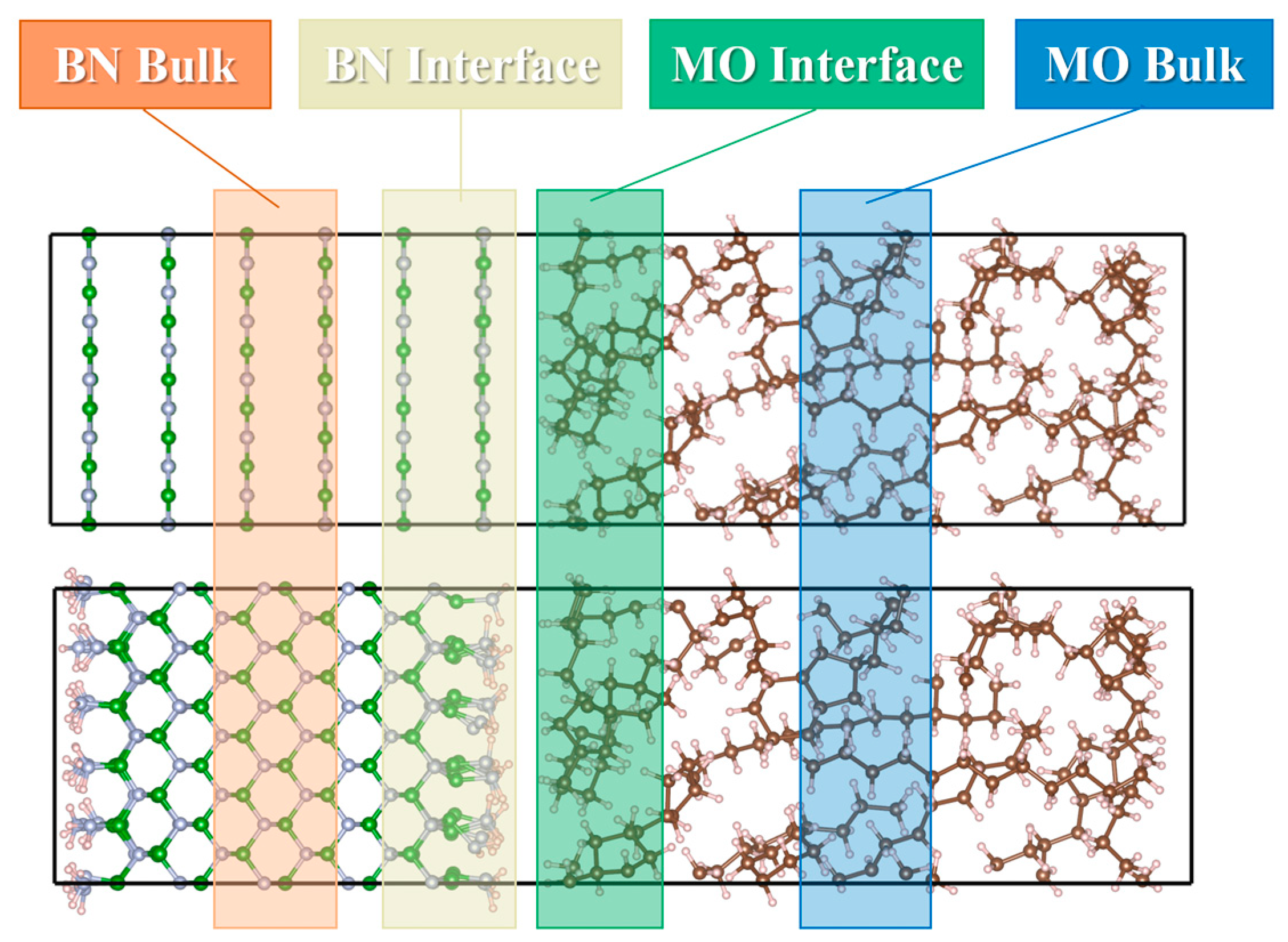
2.3.1. Charge Density Differences
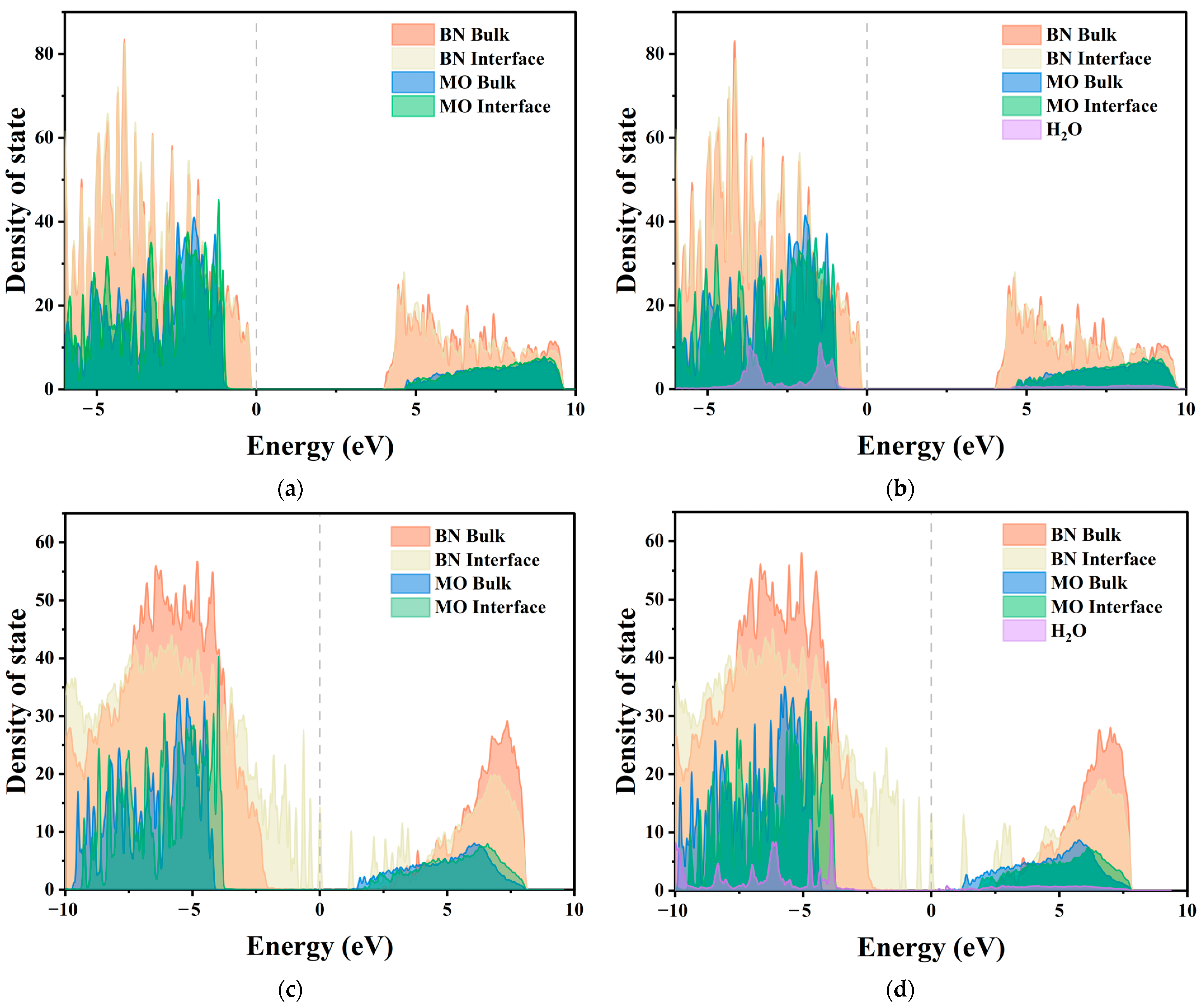
2.3.2. Local Density of States
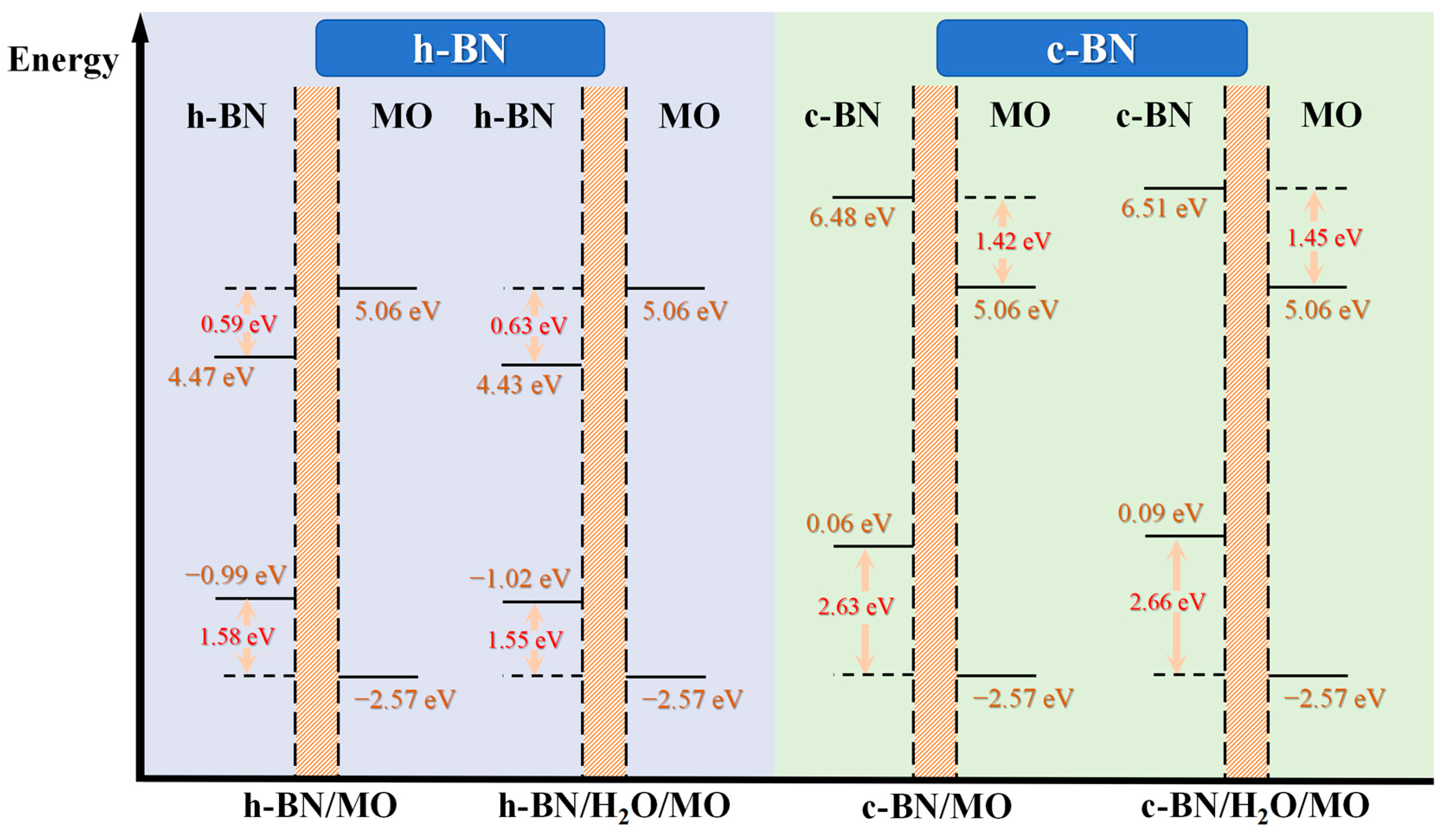
2.3.3. Potential Distributions and Barriers across the BN/MO Interface
3. Molecular Modeling and Simulation Details
3.1. Molecular Dynamics Simulations of Diffusion Behavior of Water Molecules in MO/BN Composite Models
3.2. DFT Calculations of Electrical Properties of MO/BN Interface Models
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lyutikova, M.; Korobeynikov, S.; Mohan Rao, U.; Fofana, I. Mixed Insulating Liquids with Mineral Oil for High Voltage Transformer Applications: A Review. IEEE Trans. Dielectr. Electr. Insul. 2022, 29, 454–461. [Google Scholar] [CrossRef]
- Rafiq, M.; Shafique, M.; Azam, A.; Ateeq, M.; Khan, I.A.; Hussain, A. Sustainable, Renewable and Environmental-Friendly Insulation Systems for High Voltages Applications. Molecules 2020, 25, 3901. [Google Scholar] [CrossRef] [PubMed]
- Gerber, D.L.; Ghatpande, O.A.; Nazir, M.; Heredia, W.G.B.; Feng, W.; Brown, R.E. Energy and Power Quality Measurement for Electrical Distribution in AC and DC Microgrid Buildings. Appl. Energy 2022, 308, 118308. [Google Scholar] [CrossRef]
- Zhu, M.; Gu, C.; Zhu, W. Investigation on the Interaction between Cellulosic Paper and Organic Acids Based on Molecular Dynamics. Molecules 2020, 25, 3938. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Wang, Y.; Zhang, Q.; Xiong, J.; Cao, W.; Lv, Z.; Wu, K. Dynamics of an Impurity Ion Transport in Oil-paper Insulation under Various Electric Fields. High Volt. 2024, 9, 66–80. [Google Scholar] [CrossRef]
- Wang, L.; Yin, F.; Shen, Y.; Tang, C. Reactive Molecular Dynamics Research on Influences of Water on Aging Characteristics of PMIA Insulation Paper. J. Appl. Phys. 2020, 127, 105107. [Google Scholar] [CrossRef]
- Zhang, J.; Tang, C.; Qiu, Q.; Yang, L. Effect of Water on the Diffusion of Small Molecular Weight Acids in Nano-SiO2 Modified Insulating Oil. J. Mol. Liq. 2020, 314, 113670. [Google Scholar] [CrossRef]
- Atalar, F.; Ersoy, A.; Rozga, P. Experimental Investigation of the Effect of Nano Particles on the Breakdown Strength of Transformer Liquids by Harmonic Current Analysis. Electr. Power Syst. Res. 2023, 221, 109390. [Google Scholar] [CrossRef]
- Madavan, R.; Saroja, S.; Karthick, A.; Murugesan, S.; Mohanavel, V.; Velmurugan, P.; Al Obaid, S.; Alfarraj, S.; Sivakumar, S. Performance Analysis of Mixed Vegetable Oil as an Alternative for Transformer Insulation Oil. Biomass Convers. Biorefinery 2022. [Google Scholar] [CrossRef]
- Gao, B.; Fang, Y.; Liu, K.; Yin, H.; Zeng, Y.; Wu, G. Molecular Dynamics Study on Thermal Decomposition Characteristics of Synthetic Ester Oil. Chem. Phys. Lett. 2023, 813, 140302. [Google Scholar] [CrossRef]
- Hao, Y.; Yu, N.; Qi, G.; Qin, J.; Zhang, R. Study on the Influence of Antioxidants on Transformer Oil Related Indicators. IOP Conf. Ser. Earth Environ. Sci. 2019, 223, 012032. [Google Scholar] [CrossRef]
- Zhou, P.; Liu, J.; Zhu, Z.; Liu, Z. Voltammetric Determination of the Antioxidant 2,6-Di-Tert-Butyl-p-Cresol (T501) in Transformer Oil on a Graphite Electrode in a Solution of Potassium Hydroxide and Ethanol. Anal. Lett. 2021, 54, 1112–1124. [Google Scholar] [CrossRef]
- Zhao, M.; Zhang, B.; Li, J.; Zhang, Q.; Li, H. Molecular Dynamics Study on the Diffusion Behavior of Water Molecules and the Dielectric Constant of Vegetable/Mineral Oil Blends. Molecules 2023, 28, 1067. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Chen, P.; Yu, S.; Zhang, P.; Li, J.; Li, S. Nanoparticle Dispersion and Distribution in XLPE and the Related DC Insulation Performance. IEEE Trans. Dielectr. Electr. Insul. 2018, 25, 2349–2357. [Google Scholar] [CrossRef]
- Yang, L.; Dai, J.; Dong, M.; Wang, L. Molecular Dynamics Simulation of Temperature Impact on the Viscosity of Transformer Oil-Based Nanofluids. In Proceedings of the 2016 International Conference on Condition Monitoring and Diagnosis (CMD), Xi’an, China, 25–28 September 2016; IEEE: Piscataway, NJ, USA, 2016; pp. 376–379. [Google Scholar]
- Chen, R.; Zhao, Z.; Su, Z.; Huang, D.; Tang, C. Effect of Nano-Al2O3 on Water Molecular Diffusion in Natural Ester: A Molecular Dynamics Simulation. J. Mol. Liq. 2023, 375, 121326. [Google Scholar] [CrossRef]
- Yadav, N.; Jarial, R.K.; Rao, U.M. Characterization of Mineral Oil Based Fe3O4 Nanofluid for Application in Oil Filled Transformers. Int. J. Electr. Eng. Inform. 2018, 10, 338–349. [Google Scholar] [CrossRef]
- Yamuna, M.; Madavan, R. Impact of Nanoparticles Mixtures on Mineral and Pongamia Ester Oils Dielectric Strength. Biomass Convers. Biorefinery 2022, 14, 7167–7179. [Google Scholar] [CrossRef]
- Das, A.K. Exploring SiO2, TiO2, and ZnO Nanoparticles in Coconut Oil and Mineral Oil under Changing Environmental Conditions. J. Mol. Liq. 2024, 397, 124168. [Google Scholar] [CrossRef]
- Qiu, Q.; Tian, W.; Yang, L.; Ding, Z.; Zhang, J.; Tang, C. Simulation Effect of SiO2 Nanoparticles on the Water Molecules Diffusion inside Insulating Oil at Different Temperatures. Colloids Surf. Physicochem. Eng. Asp. 2021, 610, 125738. [Google Scholar] [CrossRef]
- Tian, W.; Tang, C.; Wang, Q.; Zhang, S.; Yang, Y. The Effect and Associate Mechanism of Nano SiO2 Particles on the Diffusion Behavior of Water in Insulating Oil. Materials 2018, 11, 2373. [Google Scholar] [CrossRef]
- Hua, X.; Wang, L.; Yang, S. Molecular Dynamics Simulation of Improving the Physical Properties of Polytetrafluoroethylene Cable Insulation Materials by Boron Nitride Nanoparticle under Moisture-Temperature-Electric Fields Conditions. Polymers 2019, 11, 971. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Ding, S.; Yuan, S.; Gou, X.; Cai, F.; Wang, D.; Zhao, H. Study on the Thermal Properties and Insulation Resistance of Epoxy Resin Modified by Hexagonal Boron Nitride. E-Polym. 2021, 21, 681–690. [Google Scholar] [CrossRef]
- Zhu, M.; Li, J.; Chen, J.; Song, H.; Zhang, H. Improving Thermal Conductivity of Epoxy Resin by Filling Boron Nitride Nanomaterials: A Molecular Dynamics Investigation. Comput. Mater. Sci. 2019, 164, 108–115. [Google Scholar] [CrossRef]
- Zimmermann-Ptacek, J.; Muggli, M.; Wildhack, S.; Hintzer, K.; Gerdes, T.; Willert-Porada, M.; Moos, R. Thermal, Dielectric, and Mechanical Properties of h-BN-filled PTFE Composites. J. Appl. Polym. Sci. 2018, 135, 46859. [Google Scholar] [CrossRef]
- Wu, K.; Wang, J.; Liu, D.; Lei, C.; Liu, D.; Lei, W.; Fu, Q. Highly Thermoconductive, Thermostable, and Super-Flexible Film by Engineering 1D Rigid Rod-Like Aramid Nanofiber/2D Boron Nitride Nanosheets. Adv. Mater. 2020, 32, 1906939. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Cox, M.; Li, B. Surface Modification of Hexagonal Boron Nitride Nanomaterials: A Review. J. Mater. Sci. 2018, 53, 66–99. [Google Scholar] [CrossRef]
- Li, M.; Wang, M.; Hou, X.; Zhan, Z.; Wang, H.; Fu, H.; Lin, C.-T.; Fu, L.; Jiang, N.; Yu, J. Highly Thermal Conductive and Electrical Insulating Polymer Composites with Boron Nitride. Compos. Part B Eng. 2020, 184, 107746. [Google Scholar] [CrossRef]
- Jiang, H.; Cai, Q.; Mateti, S.; Yu, Y.; Zhi, C.; Chen, Y. Boron Nitride Nanosheet Dispersion at High Concentrations. ACS Appl. Mater. Interfaces 2021, 13, 44751–44759. [Google Scholar] [CrossRef]
- Lei, W.; Mochalin, V.N.; Liu, D.; Qin, S.; Gogotsi, Y.; Chen, Y. Boron Nitride Colloidal Solutions, Ultralight Aerogels and Freestanding Membranes through One-Step Exfoliation and Functionalization. Nat. Commun. 2015, 6, 8849. [Google Scholar] [CrossRef]
- Bai, Z.; Yang, L.; Feng, Y.; Xu, C. Effect of boron nitride surface modification groups on thermal conductivity of boron nitride/polyimide composites. Insul. Mater. 2023, 56, 63–67. [Google Scholar] [CrossRef]
- Wu, Y.; Wagner, L.K.; Aluru, N.R. Hexagonal Boron Nitride and Water Interaction Parameters. J. Chem. Phys. 2016, 144, 164118. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, J.; Zhu, M.; Song, H.; Zhang, H. Interfacial Characteristics of Boron Nitride Nanosheet/Epoxy Resin Nanocomposites: A Molecular Dynamics Simulation. Appl. Sci. 2019, 9, 2832. [Google Scholar] [CrossRef]
- Du, D.; Tang, C.; Yang, L.; Guo, L.; Qiu, Q. Molecular Dynamics Simulation on the Distribution and Diffusion of Different Sulfides in Oil-Paper Insulation Systems. J. Mol. Liq. 2020, 314, 113678. [Google Scholar] [CrossRef]
- Tan, K.P.; Singh, K.; Hazra, A.; Madhusudhan, M.S. Peptide Bond Planarity Constrains Hydrogen Bond Geometry and Influences Secondary Structure Conformations. Curr. Res. Struct. Biol. 2021, 3, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.; Jung, H.; Bae, K.J.; Kim, Y.; Yu, J. Analysis of Mechanical and Thermal Characterization of Hexagonal Boron Nitride Using a Molecular Dynamics Simulation with the New Dreiding Force Field. Mech. Adv. Mater. Struct. 2022, 29, 6957–6965. [Google Scholar] [CrossRef]
- Qin, J.; Peng, X.; Qiu, Q.; Tang, C. A New Type of Nano APTES-hBN Modified Palm Oil as Natural Ester Insulating Oil with Upgraded Thermal Aging Characteristics. Renew. Energy 2022, 200, 743–750. [Google Scholar] [CrossRef]
- Zuo, H.; Wang, F.; Huang, Z.; Wang, Q.; Li, J.; Rozga, P. Synergistic Effect of Electric Field and Temperature on POSS Modified Natural Ester Insulating Oil: A Molecular Dynamics Study. J. Mol. Liq. 2022, 355, 118923. [Google Scholar] [CrossRef]
- Qiu, Q.; Zhang, J.; Yang, L.; Du, D.; Tang, C. Differences Analysis of Water Molecular Diffusion Behaviors in Vegetable Oil and Mineral Oil under Temperature Field. J. Mol. Liq. 2021, 323, 115030. [Google Scholar] [CrossRef]
- Ojha, D.; Karhan, K.; Kühne, T.D. On the Hydrogen Bond Strength and Vibrational Spectroscopy of Liquid Water. Sci. Rep. 2018, 8, 16888. [Google Scholar] [CrossRef]
- Jiang, T.; Yang, J.; Yang, X.; Li, Y.; Bi, M.; Zhou, X. Diffusion Mechanisms of Dissolved Gases in Transformer Oil Influenced with Moisture Based on Molecular Dynamics Simulation. ACS Omega 2022, 7, 39812–39821. [Google Scholar] [CrossRef]
- Fan, X.; Zhao, H.; Sun, T.; Liu, J.; Zhang, Y.; Zhang, Y. Investigation on Diffusion Mechanisms of Methanol in Paper/Oil Insulation Based on Molecular Dynamics. IEEE Access 2021, 9, 13368–13377. [Google Scholar] [CrossRef]
- Connolly, M.L. Analytical Molecular Surface Calculation. J. Appl. Crystallogr. 1983, 16, 548–558. [Google Scholar] [CrossRef]
- Fox, T.G.; Loshaek, S. Influence of Molecular Weight and Degree of Crosslinking on the Specific Volume and Glass Temperature of Polymers. J. Polym. Sci. 1955, 15, 371–390. [Google Scholar] [CrossRef]
- Hafner, J.; Kresse, G. The Vienna AB-Initio Simulation Program VASP: An Efficient and Versatile Tool for Studying the Structural, Dynamic, and Electronic Properties of Materials. In Properties of Complex Inorganic Solids; Gonis, A., Meike, A., Turchi, P.E.A., Eds.; Springer: Boston, MA, USA, 1997; pp. 69–82. ISBN 978-1-4613-7723-8. [Google Scholar]
- Wang, Z.L. From Contact Electrification to Triboelectric Nanogenerators. Rep. Prog. Phys. 2021, 84, 096502. [Google Scholar] [CrossRef] [PubMed]
- Castle, G.S.P.; Schein, L.B. General Model of Sphere-Sphere Insulator Contact Electrification. J. Electrost. 1995, 36, 165–173. [Google Scholar] [CrossRef]
- Xu, C.; Zi, Y.; Wang, A.C.; Zou, H.; Dai, Y.; He, X.; Wang, P.; Wang, Y.-C.; Feng, P.; Li, D.; et al. On the Electron-Transfer Mechanism in the Contact-Electrification Effect. Adv. Mater. 2018, 30, 1706790. [Google Scholar] [CrossRef]
- Köhler, L.; Kresse, G. Density Functional Study of CO on Rh(111). Phys. Rev. B 2004, 70, 165405. [Google Scholar] [CrossRef]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Hybrid Functionals Based on a Screened Coulomb Potential. J. Chem. Phys. 2003, 118, 8207–8215. [Google Scholar] [CrossRef]
- Mori-Sánchez, P.; Cohen, A.J.; Yang, W. Localization and Delocalization Errors in Density Functional Theory and Implications for Band-Gap Prediction. Phys. Rev. Lett. 2008, 100, 146401. [Google Scholar] [CrossRef]
- Van De Walle, C.G.; Martin, R.M. Theoretical Study of Band Offsets at Semiconductor Interfaces. Phys. Rev. B 1987, 35, 8154–8165. [Google Scholar] [CrossRef]
- Alkauskas, A.; Broqvist, P.; Devynck, F.; Pasquarello, A. Band Offsets at Semiconductor-Oxide Interfaces from Hybrid Density-Functional Calculations. Phys. Rev. Lett. 2008, 101, 106802. [Google Scholar] [CrossRef] [PubMed]
- Ye, W.; Hao, J.; Chen, Y.; Zhu, M.; Pan, Z.; Hou, F. Difference Analysis of Gas Molecules Diffusion Behavior in Natural Ester and Mineral Oil Based on Molecular Dynamic Simulation. Molecules 2019, 24, 4463. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Wang, Y.; Zhang, B.; Du, Y.; Teng, W.; Li, H. Molecular Simulation of the Effect and Mechanism of BN Doping on Local Morphology and Water Molecules Diffusion in Insulating Oil. In Proceedings of the 2024 IEEE 7th International Electrical and Energy Conference (CIEEC), Harbin, China, 10–12 May 2024; IEEE: Piscataway, NJ, USA, 2024; pp. 1207–1212. [Google Scholar]
- Feng, D.; Chen, G.; Lin, Y.; Liao, R.; Yuan, D. Microcosmic Mechanism and Correction of Moisture Effect on Furfural Diffusion and Equilibrium in Oil-paper Insulation. High Volt. 2023, 8, 772–780. [Google Scholar] [CrossRef]
- Liu, C.; Fu, K.; Wang, Z.; Cao, C.; Yang, J.; Zhai, Q.; Wang, B.; Zhou, Z.; Ji, J.; Li, M.; et al. Cavitating inside Spherical Boron Nitride Nanoparticles Dependent on Controllably Follow-up Treated Atmospheres. J. Nanoparticle Res. 2020, 22, 302. [Google Scholar] [CrossRef]
- Ren, Y.; Zhang, Q.; Wang, Y.; Li, J.; Yan, W.; Xu, H.; Cheng, C.; Wu, K. A First-Principles Study of the Formation and Regulation of the Electric Double Layers at Cu (0 0 1)/Mineral Oil Interfaces. J. Phys. Appl. Phys. 2024, 57, 345302. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab Initio Molecular Dynamics for Liquid Metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Hafner, J. Materials Simulations Using VASP—A Quantum Perspective to Materials Science. Comput. Phys. Commun. 2007, 177, 6–13. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; Di Nola, A.; Haak, J.R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Wei, Z.J.; Sun, C.H.; Chen, B.T.; Xu, Y.J.; Wu, S.W.; Wang, D.Q. Molecular Dynamics Simulation of Water Diffusion in Mixed Insulating Oil at Different Temperatures. J. Phys. Conf. Ser. 2021, 1750, 012079. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector Augmented-Wave Method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Lin, I.-C.; Seitsonen, A.P.; Tavernelli, I.; Rothlisberger, U. Structure and Dynamics of Liquid Water from Ab Initio Molecular Dynamics—Comparison of BLYP, PBE, and revPBE Density Functionals with and without van Der Waals Corrections. J. Chem. Theory Comput. 2012, 8, 3902–3910. [Google Scholar] [CrossRef] [PubMed]
- Cortese, R.; Campisi, D.; Prestianni, A.; Duca, D. Alkane Dehydrogenation on Defective BN Quasi-Molecular Nanoflakes: DFT Studies. Mol. Catal. 2020, 493, 110891. [Google Scholar] [CrossRef]
- Wang, V.; Xu, N.; Liu, J.-C.; Tang, G.; Geng, W.-T. VASPKIT: A User-Friendly Interface Facilitating High-Throughput Computing and Analysis Using VASP Code. Comput. Phys. Commun. 2021, 267, 108033. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | BN_NSheet | BN_NTube | BN_NSphere | W | A-BN_NSheet | A-BN_NTube | A-BN_NSphere |
|---|---|---|---|---|---|---|---|
| a | 1.1000 | 1.0723 | 1.0493 | 0.8799 | 0.7442 | 0.5752 | 0.4796 |
| D (Å2/ps) | 0.1833 | 0.1787 | 0.1749 | 0.1467 | 0.1240 | 0.0959 | 0.0799 |
| Model | BN_NSheet | BN_NTube | BN_NSphere | W | A-BN_NSheet | A-BN_NTube | A-BN_NSphere |
|---|---|---|---|---|---|---|---|
| * HBTotal | 16.1531 | 16.3117 | 17.2504 | 18.4509 | 19.1165 | 24.5957 | 30.0948 |
| * HBW-BN | 0 | 0 | 2.5674 | 0 | 6.8686 | 10.5308 | 14.1198 |
| Classification | Model | Vf | VO | FFV |
|---|---|---|---|---|
| Models without BN surface amination | BN_NSheet | 15,434.6428 | 31,525.2772 | 32.868% |
| BN_NTube | 14,728.8692 | 31,595.7208 | 31.876% | |
| BN_NSphere | 14,749.7695 | 31,522.2005 | 31.795% | |
| Models with BN surface amination | A-BN_NSheet | 15,211.6088 | 31,072.4812 | 32.866% |
| A-BN_NTube | 14,342.2594 | 31,750.8506 | 31.116% | |
| A-BN_NSphere | 14,035.2070 | 31,837.9530 | 30.596% |
| Temperature | 303 K | 323 K | 343 K | 363 K | 383 K |
|---|---|---|---|---|---|
| a | 0.38200 | 0.45153 | 0.47957 | 0.65631 | 0.85056 |
| D (Å2/ps) | 0.06367 | 0.07526 | 0.07993 | 0.10939 | 0.14176 |
| Temperature | 303 K | 323 K | 343 K | 363 K | 383 K |
|---|---|---|---|---|---|
| HBTotal | 33.7608 | 30.8239 | 30.0948 | 26.7774 | 23.8970 |
| HBW-BN | 19.3467 | 16.0300 | 14.1198 | 7.0067 | 5.3667 |
| Model | Average Potential (eV) | Potential Shift (eV) |
|---|---|---|
| h-BN/MO | 1.7621 | 0 |
| h-BN/H2O/MO | 1.6764 | −0.0857 |
| c-BN/MO | 2.7475 | 0.9854 |
| c-BN/H2O/MO | 2.6477 | 0.8856 |
| Model | h-BN/MO | h-BN/H2O/MO | c-BN/MO | c-BN/H2O/MO |
|---|---|---|---|---|
| Average potential (eV) | −2.3935 | −2.4274 | −6.4960 | −6.4638 |
| Structure | VBM (eV) | CBM (eV) | Eg (eV) | Average Potential (eV) |
|---|---|---|---|---|
| h-BN | 1.4057 | 6.8614 | 5.4557 | 0 |
| c-BN | 6.5584 | 12.9735 | 6.4151 | 0 |
| MO | −4.3345 | 3.3006 | 7.6351 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Yan, W.; Cui, K.; Cheng, C.; Ren, Y.; Wu, K. Molecular Simulation of the Water Diffusion Behavior and Electronic Properties of Boron-Nitride-Composited Mineral Oil. Molecules 2024, 29, 4500. https://doi.org/10.3390/molecules29184500
Wang Y, Yan W, Cui K, Cheng C, Ren Y, Wu K. Molecular Simulation of the Water Diffusion Behavior and Electronic Properties of Boron-Nitride-Composited Mineral Oil. Molecules. 2024; 29(18):4500. https://doi.org/10.3390/molecules29184500
Chicago/Turabian StyleWang, Yang, Wenchao Yan, Kunqi Cui, Chuanhui Cheng, Yuanyang Ren, and Kai Wu. 2024. "Molecular Simulation of the Water Diffusion Behavior and Electronic Properties of Boron-Nitride-Composited Mineral Oil" Molecules 29, no. 18: 4500. https://doi.org/10.3390/molecules29184500
APA StyleWang, Y., Yan, W., Cui, K., Cheng, C., Ren, Y., & Wu, K. (2024). Molecular Simulation of the Water Diffusion Behavior and Electronic Properties of Boron-Nitride-Composited Mineral Oil. Molecules, 29(18), 4500. https://doi.org/10.3390/molecules29184500