

Predicting pKa Values of Para-Substituted Aniline Radical Cations vs. Stable Anilinium Ions in Aqueous Media


 , and
, and 
Abstract

1. Introduction
2. Results and Discussion
2.1. Calculation of pKa
2.2. Numbers and Positions of Explicit Water Molecules in the Models
2.3. Impact of the Number of Explicit H2O Molecules on pKa Calculations
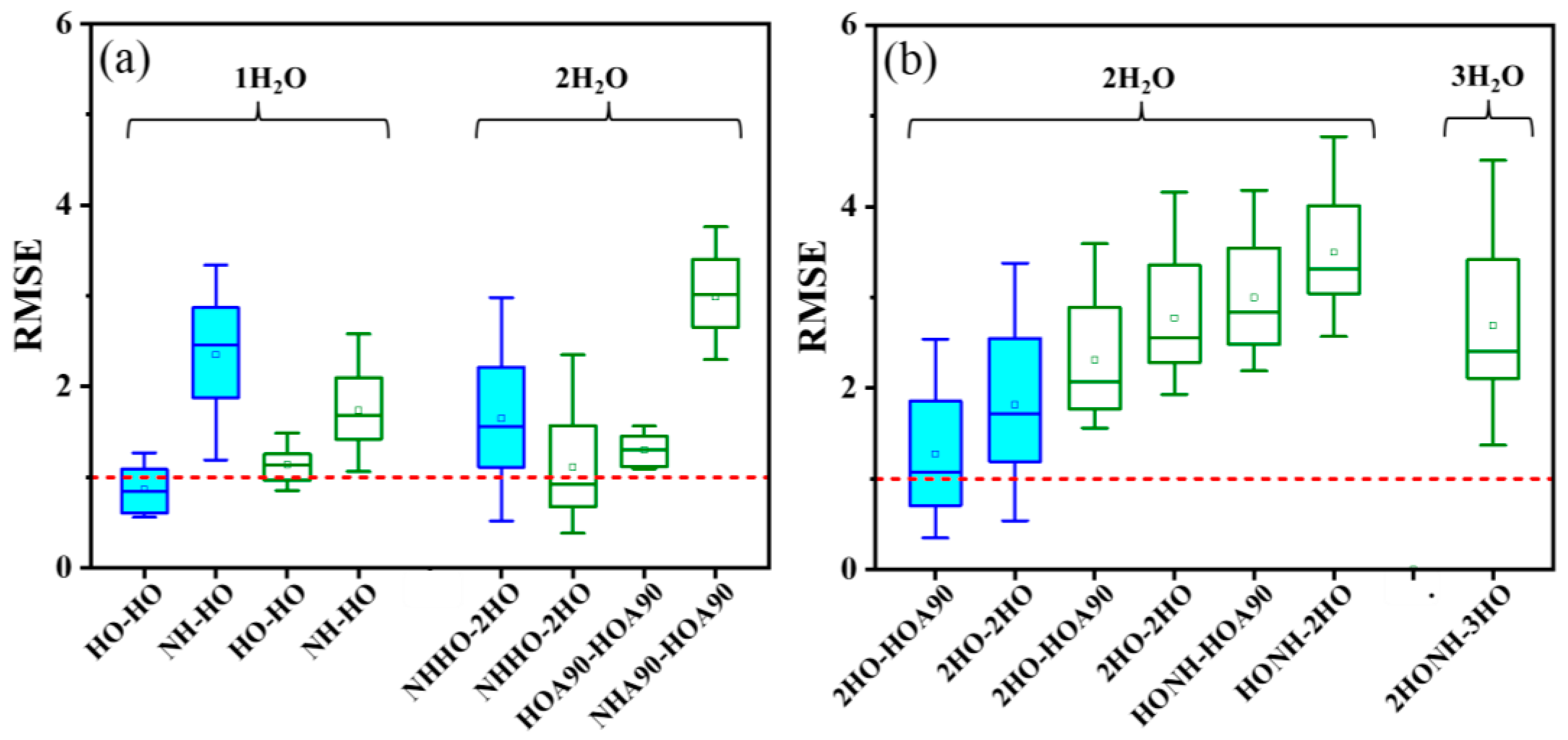
2.4. Impact of H2O Molecule Positions on pKa Calculation
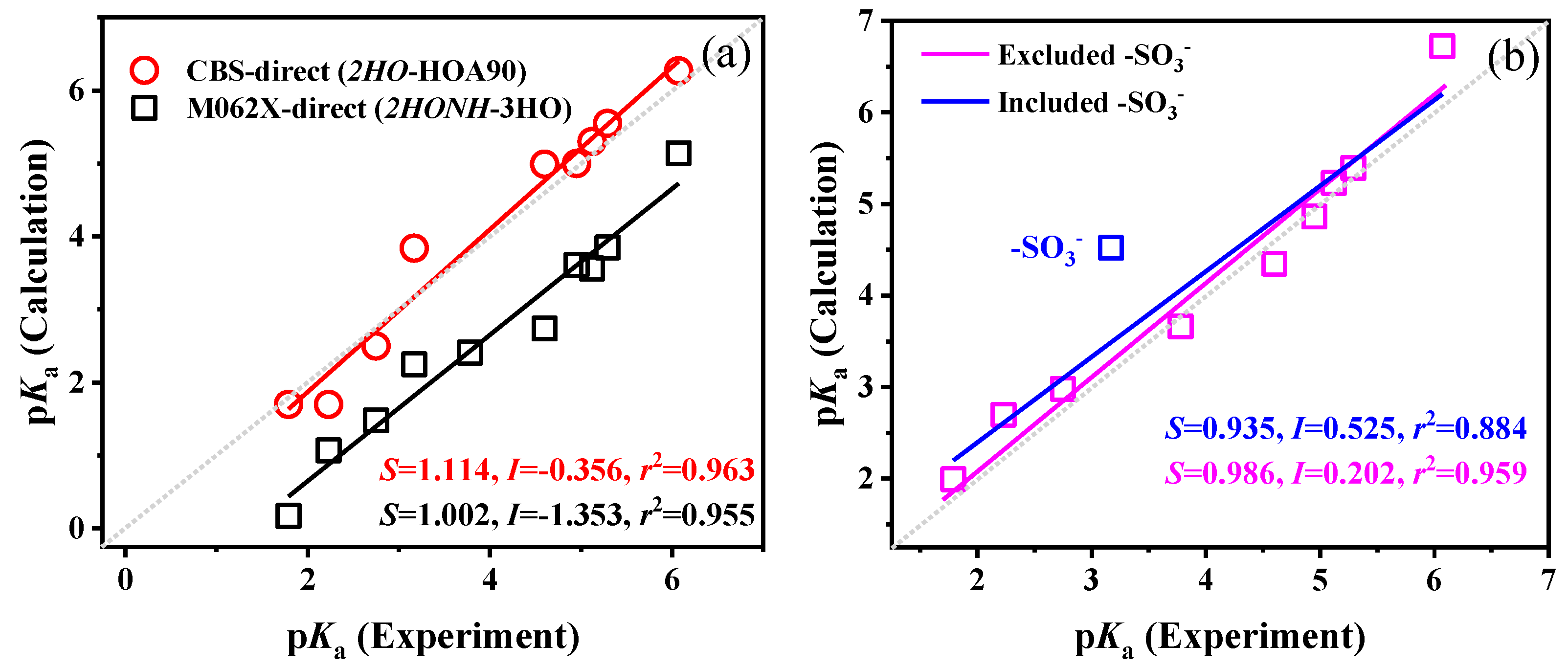
2.5. Combination of Optimal Models and Methods for pKa Calculation
2.6. Strategies for Further Improvements of pKa Calculations
3. Methods
3.1. Calculation of ΔGdeprot(sol)
3.1.1. Direct Approach
- (1)
- Methods and basis sets
- (2)
- Corrections of G(sol)
3.1.2. Indirect (Thermodynamic) Approach
- (1)
- Methods and basis sets
- (2)
- Corrections of G(gas)
3.1.3. Data Processing and Advanced Modification Method
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fang, H.S.; Gao, Y.P.; Wang, H.H.; Yin, H.; Li, G.Y.; An, T.C. Photo-induced oxidative damage to dissolved free amino acids by the photosensitizer polycyclic musk tonalide: Transformation kinetics and mechanisms. Water Res. 2017, 115, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Kolthoff, I.M.; Chantooni, M.K., Jr. Calibration of the glass electrode in acetonitrile. Shape of potentiometric titration curves. Dissociation constant of picric acid1. J. Am. Chem. Soc. 1965, 87, 4428–4436. [Google Scholar] [CrossRef]
- Zalipsky, J.J.; Patel, D.M.; Darnowski, R.J.; Reaveycantwell, N.H. pKa determination of methaqualone. J. Pharm. Sci. 1976, 65, 460–461. [Google Scholar] [CrossRef] [PubMed]
- Thapa, B.; Raghavachari, K. Accurate pKa evaluations for complex bio-organic molecules in aqueous media. J. Chem. Theory Comput. 2019, 15, 6025–6035. [Google Scholar] [CrossRef]
- Ruano, G.; Pedano, M.L.; Albornoz, M.; Fuhr, J.D.; Martiarena, M.L.; Zampieri, G. Deprotonation of the amine group of glyphosate studied by xps and dft. Appl. Surf. Sci. 2021, 567, 150753. [Google Scholar] [CrossRef]
- Zuo, W.L.; Li, N.; Chen, B.; Zhang, C.; Li, Q.; Yan, M. Investigation of the deprotonation of tetracycline using differential absorbance spectra: A comparative experimental and dft/td-dft study. Sci. Total Environ. 2020, 726, 138432. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, M.; Lind, J.; Eriksen, T.E.; Merenyi, G. Redox and acidity properties of 4-substituted aniline radical cations in water. J. Am. Chem. Soc. 1994, 116, 1423–1427. [Google Scholar] [CrossRef]
- Qian, T.T.; Kun, L.; Gao, B.; Zhu, R.; Wu, X.; Wang, S. Photo-ionization and photo-excitation of curcumin investigated by laser flash photolysis. Spectrochim. Acta A 2013, 116, 6–12. [Google Scholar] [CrossRef]
- Lang, B.; Mosquera-Vázquez, S.; Lovy, D.; Sherin, P.; Markovic, V.; Vauthey, E. Broadband ultraviolet-visible transient absorption spectroscopy in the nanosecond to microsecond time domain with sub-nanosecond time resolution. Rev. Sci. Instrum. 2013, 85, 73107. [Google Scholar] [CrossRef]
- Gangarapu, S.; Marcelis Antonius, T.M.; Zuilhof, H. Accurate pka calculation of the conjugate acids of alkanolamines, alkaloids and nucleotide bases by quantum chemical methods. Chem. Phy. Schem. 2013, 14, 990–995. [Google Scholar] [CrossRef]
- Pracht, P.; Wilcken, R.; Udvarhelyi, A.; Rodde, S.; Grimme, S. High accuracy quantum-chemistry-based calculation and blind prediction of macroscopic pka values in the context of the sampl6 challenge. J. Comput.-Aided Mol. Des. 2018, 32, 1139–1149. [Google Scholar] [CrossRef] [PubMed]
- Guerard, J.J.; Arey, J.S. Critical evaluation of implicit solvent models for predicting aqueous oxidation potentials of neutral organic compounds. J. Chem. Theory Comput. 2013, 9, 5046–5058. [Google Scholar] [CrossRef] [PubMed]
- Klamt, A. The cosmo and cosmo-rs solvation models. WIREs Comput. Mol. Sci. 2018, 8, 1338. [Google Scholar] [CrossRef]
- Marenich, V.A.; Cramer, J.C.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Ho, J.; Ertem, M.Z. Calculating free energy changes in continuum solvation models. J. Phys. Chem. B 2016, 120, 1319–1329. [Google Scholar] [CrossRef]
- Xu, T.; Chen, J.; Chen, X.; Xie, H.; Wang, Z.; Xia, D.; Tang, W.; Xie, H. Prediction models on pKa and base-catalyzed hydrolysis kinetics of parabens: Experimental and quantum chemical studies. Environ. Sci. Technol. 2021, 55, 6022–6031. [Google Scholar] [CrossRef] [PubMed]
- Thapa, B.; Schlegel, H.B. Density functional theory calculation of pKa’s of thiols in aqueous solution using explicit water molecules and the polarizable continuum model. J. Phys. Chem. A 2016, 120, 5726–5735. [Google Scholar] [CrossRef]
- Haworth, N.L.; Wang, Q.; Coote, M.L. Modeling flexible molecules in solution: A pKa case study. J. Phys. Chem. A 2017, 121, 5217–5225. [Google Scholar] [CrossRef] [PubMed]
- Hebert, S.P.; Schlegel, H.B. Computational study of the pH-dependent competition between carbonate and thymine addition to the guanine radical. Chem. Res. Toxicol. 2019, 32, 195–210. [Google Scholar] [CrossRef]
- Xu, L.K.; Coote, M.L. Methods to improve the calculations of solvation model density solvation free energies and associated aqueous pKa values: Comparison between choosing an optimal theoretical level, solute cavity scaling, and using explicit solvent molecules. J. Phys. Chem. A 2019, 123, 7430–7438. [Google Scholar] [CrossRef]
- Xu, L.K.; Coote, M.L. Improving the accuracy of pcm-uahf and pcm-uaks calculations using optimized electrostatic scaling factors. J. Chem. Theory Comput. 2019, 15, 6958–6967. [Google Scholar] [CrossRef] [PubMed]
- Barone, V.; Carnimeo, I.; Scalmani, G. Computational spectroscopy of large systems in solution: The dftb/pcm and td-dftb/pcm approach. J. Chem. Theory Comput. 2013, 9, 2052–2071. [Google Scholar] [CrossRef]
- Provorse, L.M.R.; Isborn, C.M. Combining explicit quantum solvent with a polarizable continuum model. J. Phys. Chem. B 2017, 121, 10105–10117. [Google Scholar] [CrossRef] [PubMed]
- Behjatmanesh-Ardakani, R.; Karimi, M.A.; Ebady, A. Cavity shape effect on pka prediction of small amines. J. Mol. Struct. THEOCHEM 2009, 910, 99–103. [Google Scholar] [CrossRef]
- Vyboishchikov, S.F.; Voityuk, A.A. Fast non-iterative calculation of solvation energies for water and non-aqueous solvents. J. Comput. Chem. 2021, 42, 1184–1194. [Google Scholar] [CrossRef]
- Trovó, A.G.; Nogueira, R.F.P.; Agüera, A.; Sirtori, C.; Fernández-Alba, A.R. Photodegradation of sulfamethoxazole in various aqueous media: Persistence, toxicity and photoproducts assessment. Chemosphere 2009, 77, 1292–1298. [Google Scholar] [CrossRef]
- Shi, C.; Qin, L.; Wu, S.; Kang, S.; Li, X. Highly sensitive sers detection and photocatalytic degradation of 4-aminothiophenol by a cost-effective cobalt metal–organic framework-based sandwich-like sheet. Chem. Eng. J. 2021, 422, 129970. [Google Scholar] [CrossRef]
- Zhang, C.J.; Chen, H.; Xue, G.; Liu, Y.; Chen, S.; Jia, C. A critical review of the aniline transformation fate in azo dye wastewater treatment. J. Cleaner Prod. 2021, 321, 128971. [Google Scholar] [CrossRef]
- İhsan, H.M.; İnce, U.; Gündüz, M.G.; Coşkun, G.P.; Birgül, K.; Doğan, Ş.D.; Küçükgüzel, Ş.G. Synthesis, antimicrobial evaluation and molecular modeling studies of novel thiosemicarbazides/semicarbazides derived from p-aminobenzoic acid. J. Mol. Struct. 2022, 1261, 132907. [Google Scholar] [CrossRef]
- Wang, D.; Lan, Y.; Chen, W.; Han, X.; Liu, S.; Cao, D.; Cheng, X.; Wang, Q.; Zhan, Z.; He, W. The six-year biochar retention interacted with fertilizer addition alters the soil organic nitrogen supply capacity in bulk and rhizosphere soil. J. Environ. Manag. 2023, 338, 117757. [Google Scholar] [CrossRef]
- Tentscher, P.R.; Eustis, S.N.; McNeill, K.; Arey, J.S. Aqueous oxidation of sulfonamide antibiotics: Aromatic nucleophilic substitution of an aniline radical cation. Chem.-Eur. J. 2013, 19, 11216–11223. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Wang, J.; Nguyen, T.H.; Zheng, N. The chemistry of amine radical cations produced by visible light photoredox catalysis. Beilstein J. Org. Chem. 2013, 9, 1977–2001. [Google Scholar] [CrossRef]
- Wenk, J.; Canonica, S. Phenolic antioxidants inhibit the triplet-induced transformation of anilines and sulfonamide antibiotics in aqueous solution. Environ. Sci. Technol. 2012, 46, 5455–5462. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.D.; Xue, X.S.; Ji, P. Internet Bond-Energy Databank (pKa and BDE): iBonD Home Page. Available online: http://ibond.nankai.edu.cn/ (accessed on 10 January 2024).
- Land, E.J.; Porter, G. Primary photochemical processes in aromatic molecules. Part 8—Absorption spectra and acidity constants of anilino radicals. Trans. Far. Soci. 1963, 59, 2027–2037. [Google Scholar] [CrossRef]
- Jovanovic, S.V.; Steenken, S.; Tosic, M.; Marjanovic, B.; Simic, M.G. Flavonoids as antioxidants. J. Am. Chem. Soc. 1994, 116, 4846–4851. [Google Scholar] [CrossRef]
- Hall, N.F.; Sprinkle, M.R. Relations between the structure and strength of certain organic bases in aqueous solution. J. Am. Chem. Soc. 1932, 54, 3469–3485. [Google Scholar] [CrossRef]
- Shapiro, S.L.; Isaacs, E.S.; Bandurco, V.; Freedman, L. Apparent dissociation constants of haloaralkylamines. J. Med. Pharm. Chem. 1962, 5, 793–799. [Google Scholar] [CrossRef]
- Sheppard, W.A. The electrical effect of the sulfur pentafluoride group. J. Am. Chem. Soc. 1962, 84, 3072–3076. [Google Scholar] [CrossRef]
- Sheppard, W.A. The electronic properties of fluoroalkyl groups. Fluorine p-π interaction1. J. Am. Chem. Soc. 1965, 87, 2410–2420. [Google Scholar] [CrossRef]
- Roberts, J.D.; Webb, R.L.; McElhill, E.A. The electrical effect of the trifluoromethyl group. J. Am. Chem. Soc. 1950, 72, 408–411. [Google Scholar] [CrossRef]
- Vandenbelt, J.M.; Henrich, C.; Vanden, B.S.G. Comparison of pKa values determined by electrometric titration and ultraviolet absorption methods. Anal. Chem. 1954, 26, 726–727. [Google Scholar] [CrossRef]
- Garcia, B.; Leal, J.M. The pkbh+ calculation of strong bases—A revision of various methods. Collect. Czech. Chem. C 1987, 52, 299–307. [Google Scholar] [CrossRef]
- Biggs, A.I.; Robinson, R.A. The ionisation constants of some substituted anilines and phenols: A test of the Hammett relation. J. Chem. Soc. 1961, 388–393. [Google Scholar] [CrossRef]
- Kalopissis, G. Structure-activity relationships of aromatic amines in the ames salmonella typhimurium assay. Mutat. Res.–Fund. Mol. M. 1991, 246, 45–66. [Google Scholar] [CrossRef] [PubMed]
- Garcia, B.; Domingo, I.; Domingo, P.L.; Leal, J.M. Determination of limiting molar conductivities of weak organic-acids in aqueous-solutions. Collect. Czech. Chem. C 1991, 56, 1184–1192. [Google Scholar] [CrossRef]
- Hargreaves, M.K.; Stevinson, E.A.; Evans, J. The apparent dissociation constants of various weak acids in mixed aqueous solvents. J. Chem. Soc. 1965, 4582–4583. [Google Scholar] [CrossRef]
- McCoy, R.D.; Swinehart, D.F. The ionization constant of metanilic acid from 0 to 50° by means of e.M.F. Measurements. J. Am. Chem. Soc. 1954, 76, 4708–4710. [Google Scholar] [CrossRef]
- Yu, A.; Liu, Y.H.; Li, Z.C.; Cheng, J.P. Computation of pka values of substituted aniline radical cations in dimethylsulfoxide solution. J. Phys. Chem. A 2007, 111, 9978–9987. [Google Scholar] [CrossRef]
- Bordwell, F.G.; Zhang, X.M.; Cheng, J.P. Bond dissociation energies of the nitrogen-hydrogen bonds in anilines and in the corresponding radical anions. Equilibrium acidities of aniline radical cations. J. Org. Chem. 1993, 58, 6410–6416. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. C.01; Fox, Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The m06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four m06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Clark, T.; Chandrasekhar, J.; Spitznagel, G.W.; Schleyer, P.V.R. Efficient diffuse function-augmented basis sets for anion calculations. Iii. The 3-21+g basis set for first-row elements, li–f. J. Comput. Chem. 1983, 4, 294–301. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. Xx. A basis set for correlated wave functions. J. Chem. Phys. 2008, 72, 650–654. [Google Scholar] [CrossRef]
- Cancès, E.; Mennucci, B.; Tomasi, J. A new integral equation formalism for the polarizable continuum model: Theoretical background and applications to isotropic and anisotropic dielectrics. J. Chem. Phys. 1997, 107, 3032–3041. [Google Scholar] [CrossRef]
- Mennucci, B.; Tomasi, J. Continuum solvation models: A new approach to the problem of solute’s charge distribution and cavity boundaries. J. Chem. Phys. 1997, 106, 5151–5158. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for h to rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.J.; Xu, X.F.; Truhlar, D.G. Minimally augmented karlsruhe basis sets. Theor. Chem. Acc. 2011, 128, 295–305. [Google Scholar] [CrossRef]
- Montgomery, J.A.; Frisch, M.J.; Ochterski, J.W.; Petersson, G.A. A complete basis set model chemistry. Vi. Use of density functional geometries and frequencies. J. Chem. Phys. 1999, 110, 2822–2827. [Google Scholar] [CrossRef]
- Teranishi, K.; Ishikawa, A.; Sato, H.; Nakai, H. Systematic investigation of the thermodynamic properties of amine solvents for CO2 chemical absorption using the cluster-continuum model. B. Chem. Soc. Jpn. 2017, 90, 451–460. [Google Scholar] [CrossRef]
- Banerjee, S.; Bhanja, S.K.; Kanti, C.P. Quantum chemical predictions of aqueous pKa values for oh groups of some α-hydroxycarboxylic acids based on ab initio and dft calculations. Comput. Theor. Chem. 2018, 1125, 29–38. [Google Scholar] [CrossRef]
- Casasnovas, R.; Frau, J.; Ortega-Castro, J.; Salvà, A.; Donoso, J.; Muñoz, F. Absolute and relative pKa calculations of mono and diprotic pyridines by quantum methods. J. Mol. Struc.-Theochem. 2009, 912, 5–12. [Google Scholar] [CrossRef]
- Najgebauer, P.; Staś, M.; Wrzalik, R.; Broda, M.A.; Wieczorek, P.P.; Andrushchenko, V.; Kupka, T. Muscimol hydration and vibrational spectroscopy—The impact of explicit and implicit water. J. Mol. Liq. 2022, 363, 119870. [Google Scholar] [CrossRef]
- Kato, M.; Izuka, S.; Fujihara, T.; Nagasawa, A.; Kawai, S.; Tanaka, T.; Takayanagi, T. Electronic structure calculation study of metal complexes with a phytosiderophore mugineic acid. Inorg. Chim. Acta. 2011, 370, 304–310. [Google Scholar] [CrossRef]
- Zhang, S.M. A reliable and efficient first principles-based method for predicting pka values. Iii. Adding explicit water molecules: Can the theoretical slope be reproduced and pka values predicted more accurately? J. Comput. Chem. 2012, 33, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Bao, J.W.; Lucas, Z.J.; Alecu, I.M.; Lynch, B.J.; Zhao, Y.; Truhlar, D.G. Database of Frequency Scale Factors for Electronic Model Chemistries. Available online: https://comp.chem.umn.edu/freqscale/version3b2.htm (accessed on 16 July 2023).
- Lu, T.; Chen, Q.X. Shermo: A general code for calculating molecular thermochemistry properties. Comput. Theor. Chem. 2021, 1200, 113249. [Google Scholar] [CrossRef]
- Ribeiro, R.F.; Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Use of solution-phase vibrational frequencies in continuum models for the free energy of solvation. J. Phys. Chem. B 2011, 115, 14556–14562. [Google Scholar] [CrossRef]
- Grimme, S. Supramolecular binding thermodynamics by dispersion-corrected density functional theory. Chem.-Eur. J. 2012, 18, 9955–9964. [Google Scholar] [CrossRef] [PubMed]
- Marenich, A.V.; Ding, W.D.; Cramer, C.J.; Truhlar, D.G. Resolution of a challenge for solvation modeling: Calculation of dicarboxylic acid dissociation constants using mixed discrete–continuum solvation models. J. Phys. Chem. Lett. 2012, 3, 1437–1442. [Google Scholar] [CrossRef]
- Sutton, C.C.R.; Franks, G.V.; da Silva, G. First principles pKa calculations on carboxylic acids using the smd solvation model: Effect of thermodynamic cycle, model chemistry, and explicit solvent molecules. J. Phys. Chem. B 2012, 116, 11999–12006. [Google Scholar] [CrossRef]
- Santra, G.; Sylvetsky, N.; Martin, J.M.L. Minimally empirical double-hybrid functionals trained against the gmtkn55 database: Revdsd-pbep86-d4, revdod-pbe-d4, and dod-scan-d4. J. Phys. Chem. A 2019, 123, 5129–5143. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| R | H | C4H9 | CF3 | CH3 | OCH3 | CN | COCH3 | I | NH2 | SO3− | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| pKa | R-PhNH2•+ | 7.05 [35] | 8.2 [7] | 4.8 [7] | 8.5 [36] | 9.6 [7] | 4 [7] | 6.1 [7] | 7.1 [7] | 12 [7] | 5.8 [7] |
| R-PhNH3+ a | 4.62 [37], 4.58 [38] | 4.95 [34] | 2.92 [39], 2.75 [40], 2.57 [41] | 5.12 [37] | 5.29 [37] | 1.75 [42], 1.82 [43] | 2.19 [42], 2.26 [43] | 3.78 [44] | 5.94 [45], 6.2 [42] | 3.25 [46], 2.93 [47], 3.32 [48] | |
| No. | Models | Methods a | Ref. |
|---|---|---|---|
| P1 b | Implicit (single) | M052X/6-31G(d) | [14] |
| P2 c | Implicit (mixed) | M052X/6-31G(d) for neutral, e.g., R-PhNH• and R-PhNH2 M052X/6-31+G(d,p) for cation, e.g., R-PhNH2•+ and R-PhNH3+ HF/6-31G(d) for anion, e.g., SO3−-PhNH• and SO3−-PhNH2 d HF/6-31G(d) for SO3−-PhNH•+ and SO3−-PhNH3+ | [14] |
| P3 e | Implicit (mixed) | M052X/6-31G(d) for radical cation, e.g., R-PhNH2•+ HF/6-31G(d) for neutral and anion radical, e.g., R-PhNH• d HF/6-31G(d) for SO3−-PhNH•+ | - |
| P4 | Explicit | M062X/6-31G(d) | [70] |
| P5 | Explicit | M052X/cc-pVTZ | [71] |
| P6 | Explicit | ωb97xd/6-31+g(d,p) | [17] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Fang, H.; Zhong, Z.; Huang, H.; Liang, X.; Yuan, Y.; Zhou, W.; Vione, D. Predicting pKa Values of Para-Substituted Aniline Radical Cations vs. Stable Anilinium Ions in Aqueous Media. Molecules 2024, 29, 4522. https://doi.org/10.3390/molecules29194522
Wang J, Fang H, Zhong Z, Huang H, Liang X, Yuan Y, Zhou W, Vione D. Predicting pKa Values of Para-Substituted Aniline Radical Cations vs. Stable Anilinium Ions in Aqueous Media. Molecules. 2024; 29(19):4522. https://doi.org/10.3390/molecules29194522
Chicago/Turabian StyleWang, Jingxin, Hansun Fang, Zixi Zhong, Huajun Huang, Ximei Liang, Yufan Yuan, Wenwen Zhou, and Davide Vione. 2024. "Predicting pKa Values of Para-Substituted Aniline Radical Cations vs. Stable Anilinium Ions in Aqueous Media" Molecules 29, no. 19: 4522. https://doi.org/10.3390/molecules29194522
APA StyleWang, J., Fang, H., Zhong, Z., Huang, H., Liang, X., Yuan, Y., Zhou, W., & Vione, D. (2024). Predicting pKa Values of Para-Substituted Aniline Radical Cations vs. Stable Anilinium Ions in Aqueous Media. Molecules, 29(19), 4522. https://doi.org/10.3390/molecules29194522