
Insights into the Structural Modification of Selenium-Doped Derivatives with Narrowband Emissions: A Theory Study

 , , and
, , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
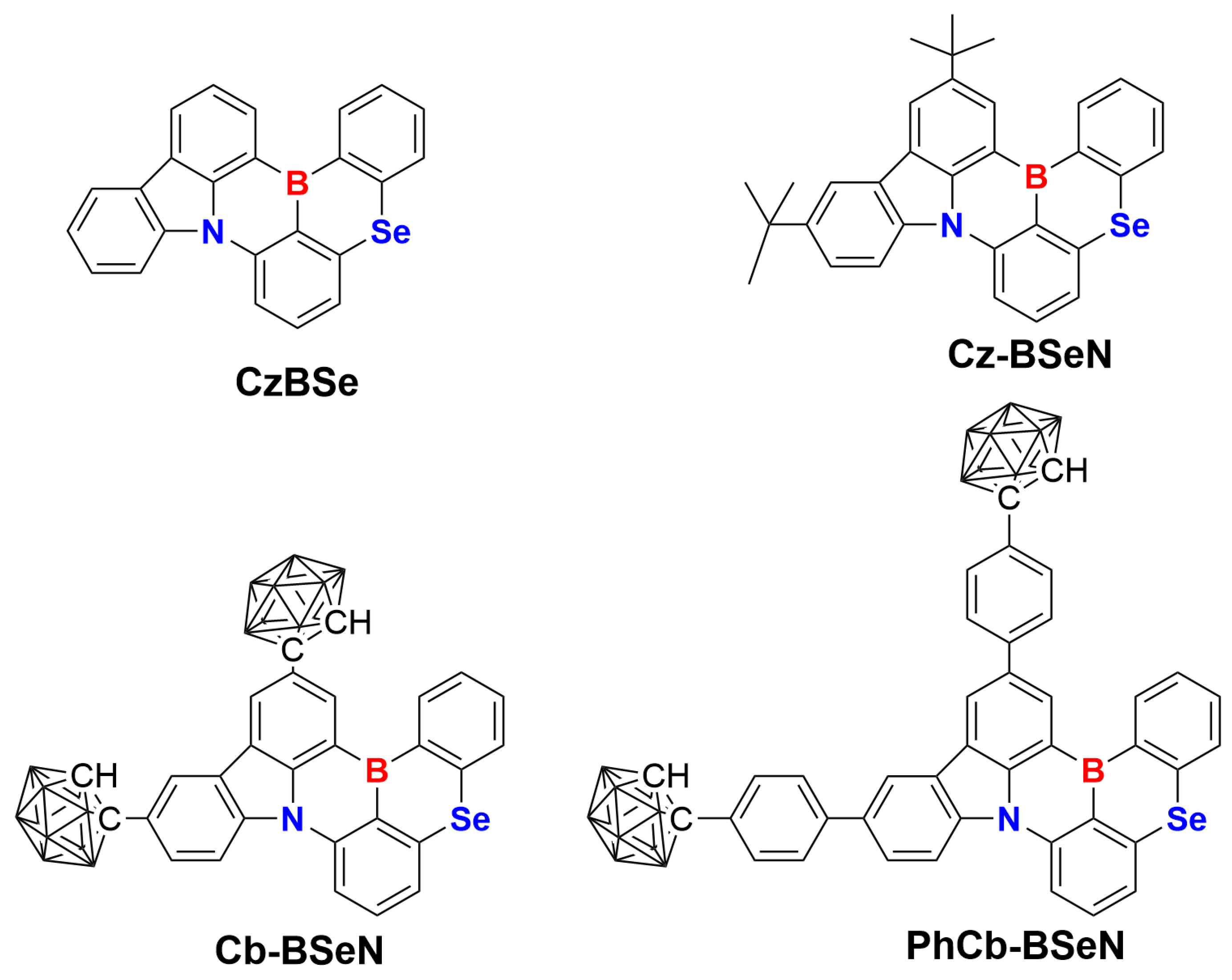
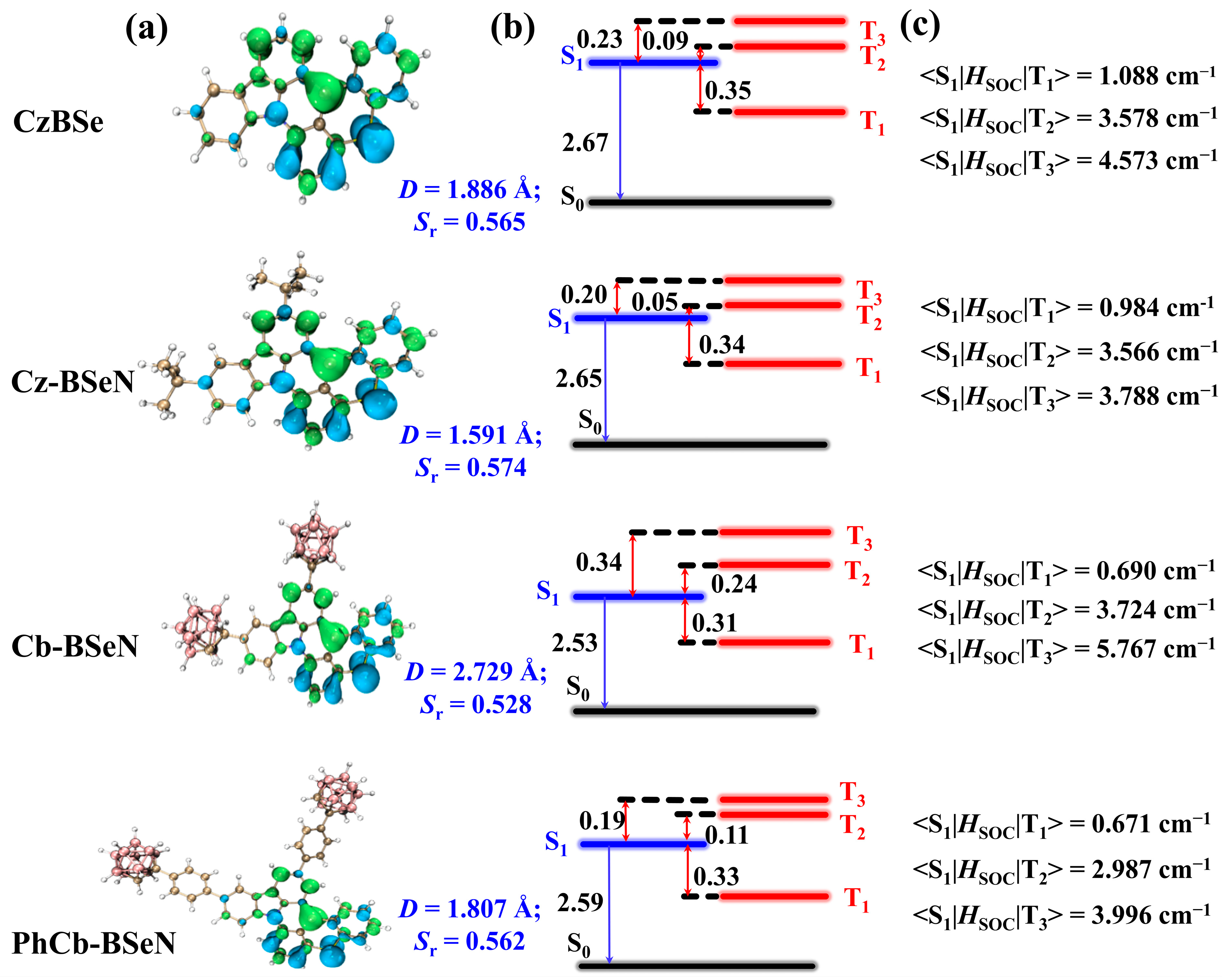
2.1. Nature of Low-Lying Excited States
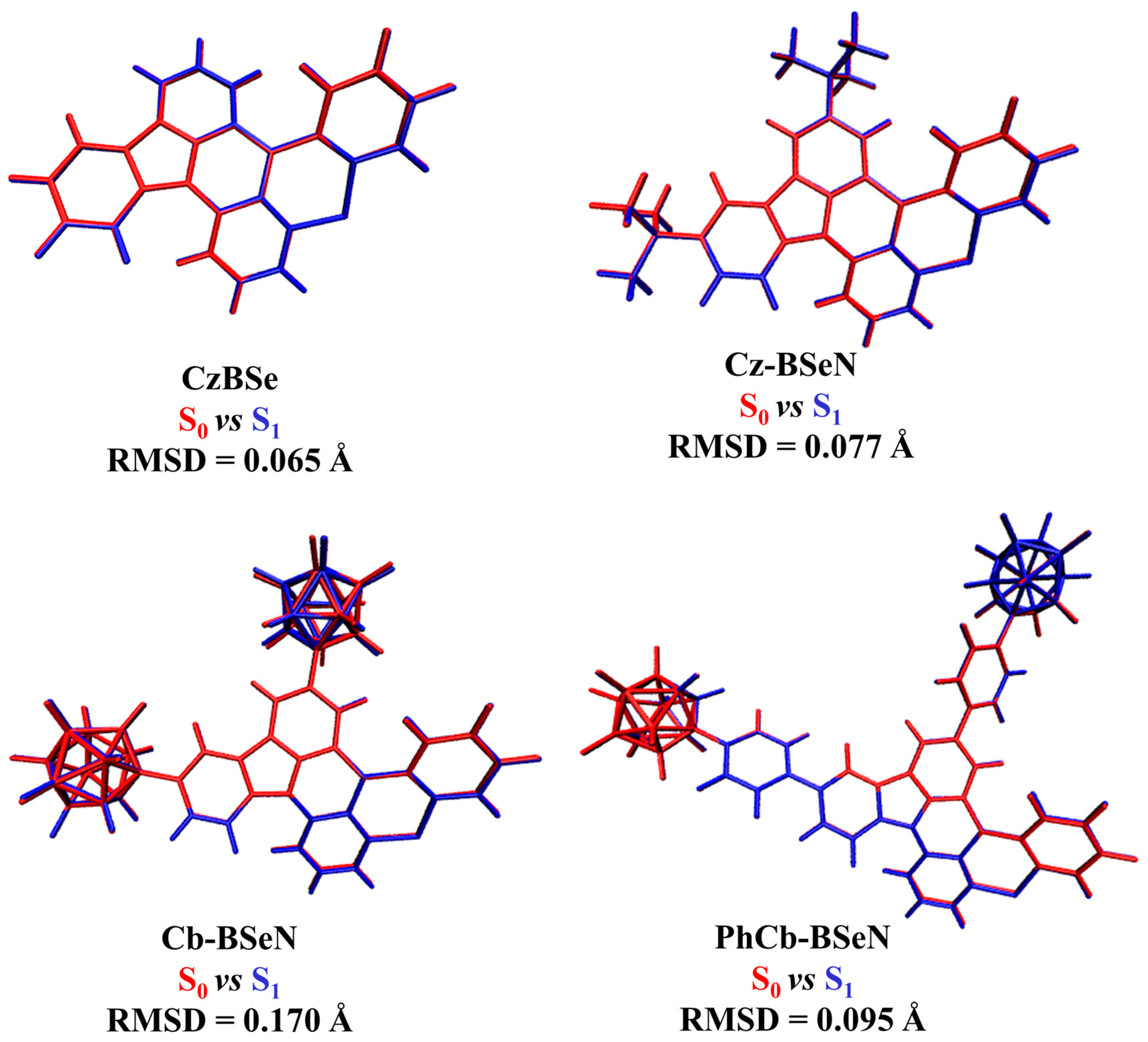
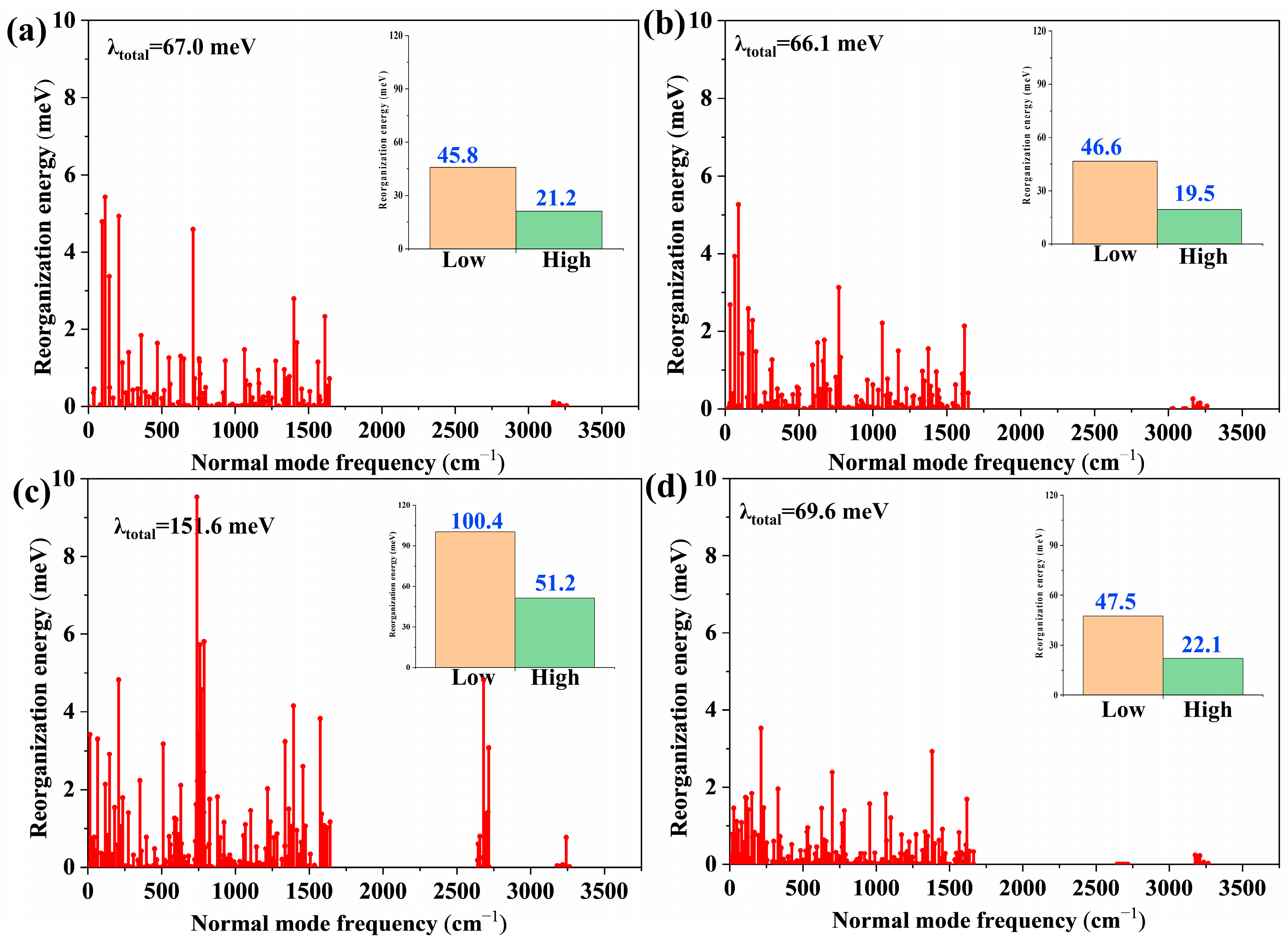
2.2. Structural Changes and Vibronic Analyses
2.3. Pressure Effects on Photophysical Properties
3. Methods
3.1. Calculations for Solvated Molecule
3.2. Crystal Structure Optimization and ONIOM Simulation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Uoyama, H.; Goushi, K.; Shizu, K.; Nomura, H.; Adachi, C. Highly Efficient Organic Light-Emitting Diodes from Delayed Fluorescence. Nature 2012, 492, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Shirota, Y.; Kageyama, H. Charge Carrier Transporting Molecular Materials and Their Applications in Devices. Chem. Rev. 2007, 107, 953–1010. [Google Scholar] [CrossRef]
- Ostroverkhova, O. Organic Optoelectronic Materials: Mechanisms and Applications. Chem. Rev. 2016, 116, 13279–13412. [Google Scholar] [CrossRef]
- Liu, Y.; Li, C.; Ren, Z.; Yan, S.; Bryce, M.R. All-Organic Thermally Activated Delayed Fluorescence Materials for Organic Light-Emitting Diodes. Nat. Rev. Mater. 2018, 3, 18020. [Google Scholar] [CrossRef]
- Tao, Y.; Yuan, K.; Chen, T.; Xu, P.; Li, H.; Chen, R.; Zheng, C.; Zhang, L.; Huang, W. Thermally Activated Delayed Fluorescence Materials towards the Breakthrough of Organoelectronics. Adv. Mater. 2014, 26, 7931–7958. [Google Scholar] [CrossRef]
- Chen, X.K.; Kim, D.; Brédas, J.L. Thermally Activated Delayed Fluorescence (TADF) Path toward Efficient Electroluminescence in Purely Organic Materials: Molecular Level Insight. Acc. Chem. Res. 2018, 51, 2215–2224. [Google Scholar] [CrossRef]
- Yang, Z.; Mao, Z.; Xie, Z.; Zhang, Y.; Liu, S.; Zhao, J.; Xu, J.; Chi, Z.; Aldred, M.P. Recent Advances in Organic Thermally Activated Delayed Fluorescence Materials. Chem. Soc. Rev. 2017, 46, 915–1016. [Google Scholar] [CrossRef] [PubMed]
- Ansari, R.; Shao, W.; Yoon, S.J.; Kim, J.; Kieffer, J. Charge Transfer as the Key Parameter Affecting the Color Purity of Thermally Activated Delayed Fluorescence Emitters. ACS Appl. Mater. Interfaces 2021, 13, 28529–28537. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.Y.; Liu, W.; Zheng, C.J.; Wang, K.; Li, F.; Tao, S.L.; Ou, X.M.; Zhang, X.H. Isomeric Thermally Activated Delayed Fluorescence Emitters for Color Purity-Improved Emission in Organic Light-Emitting Devices. ACS Appl. Mater. Interfaces 2016, 8, 16791–16798. [Google Scholar] [CrossRef]
- Hatakeyama, T.; Shiren, K.; Nakajima, K.; Nomura, S.; Nakatsuka, S.; Kinoshita, K.; Ni, J.; Ono, Y.; Ikuta, T. Ultrapure Blue Thermally Activated Delayed Fluorescence Molecules: Efficient HOMO-LUMO Separation by the Multiple Resonance Effect. Adv. Mater. 2016, 28, 2777–2781. [Google Scholar] [CrossRef]
- Lv, C.; Wang, X.; Zhang, Q.; Zhang, Y. Narrowband Emission: Organic Thermally-Activated Delayed Fluorescence Materials and Underlying Mechanisms. Mater. Chem. Front. 2023, 7, 2809–2827. [Google Scholar] [CrossRef]
- Kim, H.J.; Yasuda, T. Narrowband Emissive Thermally Activated Delayed Fluorescence Materials. Adv. Opt. Mater. 2022, 10, 2201714. [Google Scholar] [CrossRef]
- Xie, M.; Sun, M.; Xue, S.; Yang, W. Recent Progress of Blue Fluorescent Organic Light-Emitting Diodes with Narrow Full Width at Half Maximum. Dyes Pigments 2022, 208, 110799. [Google Scholar] [CrossRef]
- Murawski, C.; Leo, K.; Gather, M.C. Efficiency Roll-off in Organic Light-Emitting Diodes. Adv. Mater. 2013, 25, 6801–6827. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, D.; Huang, T.; Gillett, A.J.; Liu, Y.; Hu, D.; Cui, L.; Bin, Z.; Li, G.; Wei, J.; et al. Multi-Resonance Deep-Red Emitters with Shallow Potential-Energy Surfaces to Surpass Energy-Gap Law**. Angew. Chem. Int. Ed. 2021, 60, 20498–20503. [Google Scholar] [CrossRef]
- Einzinger, M.; Zhu, T.; de Silva, P.; Belger, C.; Swager, T.M.; Van Voorhis, T.; Baldo, M.A. Shorter Exciton Lifetimes via an External Heavy-Atom Effect: Alleviating the Effects of Bimolecular Processes in Organic Light-Emitting Diodes. Adv. Mater. 2017, 29, 1701987. [Google Scholar] [CrossRef] [PubMed]
- Park, I.S.; Min, H.; Yasuda, T. Ultrafast Triplet–Singlet Exciton Interconversion in Narrowband Blue Organoboron Emitters Doped with Heavy Chalcogens. Angew. Chem. Int. Ed. 2022, 61, e202205684. [Google Scholar] [CrossRef]
- Li, Q.; Wu, Y.; Yang, Q.; Wang, S.; Shao, S.; Wang, L. Selenium-Doped Polycyclic Aromatic Hydrocarbon Multiresonance Emitters with Fast Reverse Intersystem Crossing for Narrowband Blue Emission. ACS Appl. Mater. Interfaces 2022, 14, 49995–50003. [Google Scholar] [CrossRef]
- Yang, M.; Park, I.S.; Yasuda, T. Full-Color, Narrowband, and High-Efficiency Electroluminescence from Boron and Carbazole Embedded Polycyclic Heteroaromatics. J. Am. Chem. Soc. 2020, 142, 19468–19472. [Google Scholar] [CrossRef]
- Jin, J.; Wang, S.; Jiang, H.; Wang, L.; Wong, W. Peripheral Selenium Modification of Multi-Resonance Thermally Activated Delayed Fluorescence Molecules for High-Performance Blue Organic Light-Emitting Diodes. Adv. Opt. Mater. 2024, 12, 2302354. [Google Scholar] [CrossRef]
- Hu, Y.; Miao, J.; Zhong, C.; Zeng, Y.; Gong, S.; Cao, X. Peripherally Heavy-Atom-Decorated Strategy Towards High-Performance Pure Green Electroluminescence with External Quantum Efficiency over 40%. Angew. Chem. 2023, 135, e202302478. [Google Scholar] [CrossRef]
- Jin, J.; Chen, M.; Jiang, H.; Zhang, B.; Xie, Z.; Wong, W. Fusion of Selenium-Embedded Multi-Resonance Units Toward Narrowband Emission and Fast Triplet-Singlet Exciton Conversion. Adv. Opt. Mater. 2024, 12, 2400794. [Google Scholar] [CrossRef]
- Cao, X.; Pan, K.; Miao, J.; Lv, X.; Huang, Z.; Ni, F.; Yin, X.; Wei, Y.; Yang, C. Manipulating Exciton Dynamics toward Simultaneous High- Efficiency Narrowband Electroluminescence and Photon Upconversion by a Selenium-Incorporated Multiresonance Delayed Fluorescence Emitter. J. Am. Chem. Soc. 2022, 144, 22976–22984. [Google Scholar] [CrossRef] [PubMed]
- Núñez, R.; Romero, I.; Teixidor, F.; Viñas, C. Icosahedral Boron Clusters: A Perfect Tool for the Enhancement of Polymer Features. Chem. Soc. Rev. 2016, 45, 5147–5173. [Google Scholar] [CrossRef]
- Wee, K.; Cho, Y.; Song, J.K.; Kang, S.O. Multiple Photoluminescence from 1,2-Dinaphthyl-Ortho-Carborane. Angew. Chem. Int. Ed. 2013, 52, 9682–9685. [Google Scholar] [CrossRef]
- Naito, H.; Nishino, K.; Morisaki, Y.; Tanaka, K.; Chujo, Y. Solid-State Emission of the Anthracene-o-Carborane Dyad from the Twisted-Intramolecular Charge Transfer in the Crystalline State. Angew. Chem. Int. Ed. 2017, 56, 254–259. [Google Scholar] [CrossRef]
- Shen, Y.; Wang, L.; Fang, Y.; Sui, L.; Fu, Z.; Lv, C.; Wang, K.; Zhang, Q.; Guo, H.; Zhang, Y. Multistimuli-Responsive Luminescence of o-Carborane Dyads via Restriction of Electron Transfer and Molecular Motion. Adv. Opt. Mater. 2023, 11, 2300836. [Google Scholar] [CrossRef]
- Tu, D.; Leong, P.; Guo, S.; Yan, H.; Lu, C.; Zhao, Q. Highly Emissive Organic Single-Molecule White Emitters by Engineering o-Carborane-Based Luminophores. Angew. Chem. Int. Ed. 2017, 56, 11370–11374. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Tian, G.; Lin, C.; Pan, Y.; Ye, X.; Wang, B.; Ma, D.; Hu, D.; Luo, Y.; Ma, Y. Narrowband Emission from Organic Fluorescent Emitters with Dominant Low-Frequency Vibronic Coupling. Adv. Opt. Mater. 2021, 9, 2001845. [Google Scholar] [CrossRef]
- Pei, Z.; Ou, Q.; Mao, Y.; Yang, J.; De La Lande, A.; Plasser, F.; Liang, W.; Shuai, Z.; Shao, Y. Elucidating the Electronic Structure of a Delayed Fluorescence Emitter via Orbital Interactions, Excitation Energy Components, Charge-Transfer Numbers, and Vibrational Reorganization Energies. J. Phys. Chem. Lett. 2021, 12, 2712–2720. [Google Scholar] [CrossRef]
- Deng, C.; Niu, Y.; Peng, Q.; Shuai, Z. Electronic Structures and Spectroscopic Properties of Group-14 Metalloles MPh6 (M = Si, Ge, Sn). Acta Phys.-Chim. Sin. 2010, 35, 139–148. [Google Scholar]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Dutta, A.K.; Neese, F.; Izsák, R. Towards a Pair Natural Orbital Coupled Cluster Method for Excited States. J. Chem. Phys. 2016, 145, 034102. [Google Scholar] [CrossRef] [PubMed]
- Riplinger, C.; Neese, F. An Efficient and near Linear Scaling Pair Natural Orbital Based Local Coupled Cluster Method. J. Chem. Phys. 2013, 138, 034106. [Google Scholar] [CrossRef]
- Dutta, A.K.; Nooijen, M.; Neese, F.; Izsák, R. Automatic Active Space Selection for the Similarity Transformed Equations of Motion Coupled Cluster Method. J. Chem. Phys. 2017, 146, 074103. [Google Scholar] [CrossRef]
- Dutta, A.K.; Nooijen, M.; Neese, F.; Izsák, R. Exploring the Accuracy of a Low Scaling Similarity Transformed Equation of Motion Method for Vertical Excitation Energies. J. Chem. Theory Comput. 2018, 14, 72–91. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Li, W.; Lv, Q.; Chen, R.; Zheng, C. Design of High-Performance Circularly Polarized Multiple Resonance-Based TADF Materials via Participatory Chiral Perturbation. J. Mater. Chem. C 2023, 11, 4033–4041. [Google Scholar] [CrossRef]
- Li, P.; Zhang, Y.; Li, W.; Zhou, C.; Chen, R. Achieving Narrowband Emissions with Tunable Colors for Multiple Resonance-Thermally Activated Delayed Fluorescence Materials: Effect of Boron/Nitrogen Number and Position. Phys. Chem. Chem. Phys. 2023, 25, 27877–27884. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.J.; Refson, K.; Payne, M.C. First Principles Methods Using CASTEP. Z. Krist.-Cryst. Mater. 2005, 220, 567–570. [Google Scholar] [CrossRef]
- Chung, L.W.; Sameera, W.M.C.; Ramozzi, R.; Page, A.J.; Hatanaka, M.; Petrova, G.P.; Harris, T.V.; Li, X.; Ke, Z.; Liu, F.; et al. The ONIOM Method and Its Applications. Chem. Rev. 2015, 115, 5678–5796. [Google Scholar] [CrossRef] [PubMed]
- Rappé, A.K.; Casewit, C.J.; Colwell, K.S.; Goddard, W.A.; Skiff, W.M. UFF, a Full Periodic Table Force Field for Molecular Mechanics and Molecular Dynamics Simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. [Google Scholar] [CrossRef]
- Rappe, A.K.; Goddard, W.A. Charge Equilibration for Molecular Dynamics Simulations. J. Phys. Chem. 1991, 95, 3358–3363. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Al, E. Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Neese, F. Software Update: The ORCA Program System—Version 5.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2022, 12, e1606. [Google Scholar] [CrossRef]
- Lu, T.; Liu, Z.; Chen, Q. Comment on “18 and 12—Member Carbon Rings (Cyclo[n]Carbons)—A Density Functional Study”. Mater. Sci. Eng. B 2021, 273, 115425. [Google Scholar] [CrossRef]
- Chen, X.; Sakurai, H.; Wang, H.; Gao, S.; Bi, H.D.; Bai, F.Q. Theoretical Study on the Molecular Stacking Interactions and Charge Transport Properties of Triazasumanene Crystals—From Explanation to Prediction. Phys. Chem. Chem. Phys. 2021, 23, 4681–4689. [Google Scholar] [CrossRef]
- Zhu, S.; Gan, Q.; Feng, C. Multimolecular Complexes of CL-20 with Nitropyrazole Derivatives: Geometric, Electronic Structure, and Stability. ACS Omega 2019, 4, 13408–13417. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Aidas, K.; Angeli, C.; Bak, K.L.; Bakken, V.; Bast, R.; Boman, L.; Christiansen, O.; Cimiraglia, R.; Coriani, S.; Dahle, P.; et al. The Dalton Quantum Chemistry Program System. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 269–284. [Google Scholar] [CrossRef]
- Shuai, Z.G.; Peng, Q.; Niu, Y.L.; Geng, H. MOMAP, Revision 2020A (2.2.0); Tsinghua University: Beijing, China, 2014. [Google Scholar]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Q.; Liu, T.; Huang, X.; Wang, K.; Sun, F.; Wang, X.; Lv, C. Insights into the Structural Modification of Selenium-Doped Derivatives with Narrowband Emissions: A Theory Study. Molecules 2024, 29, 4589. https://doi.org/10.3390/molecules29194589
Zhang Q, Liu T, Huang X, Wang K, Sun F, Wang X, Lv C. Insights into the Structural Modification of Selenium-Doped Derivatives with Narrowband Emissions: A Theory Study. Molecules. 2024; 29(19):4589. https://doi.org/10.3390/molecules29194589
Chicago/Turabian StyleZhang, Qing, Tao Liu, Xin Huang, Kunyan Wang, Fangxiang Sun, Xin Wang, and Chunyan Lv. 2024. "Insights into the Structural Modification of Selenium-Doped Derivatives with Narrowband Emissions: A Theory Study" Molecules 29, no. 19: 4589. https://doi.org/10.3390/molecules29194589