
Revolutionizing Molecular Design for Innovative Therapeutic Applications through Artificial Intelligence

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Machine Learning and AI Applications in Protein Design
2.1. Deep Learning Approaches
2.1.1. Convolutional Neural Networks for Structure Prediction
2.1.2. Recurrent Neural Networks for Sequence Optimization
2.1.3. Generative Adversarial Networks in De Novo Protein Design
2.2. Reinforcement Learning in Protein Engineering
2.2.1. Optimization of Protein Properties
2.2.2. Design of Protein–Protein Interactions
2.3. Transfer Learning and Few-Shot Learning
2.3.1. Leveraging Pre-Trained Models for Protein Design
2.3.2. Addressing the Challenge of Limited Data in Protein Engineering
2.4. Interpretable AI for Protein Design
2.4.1. Explainable AI Models for Rational Protein Engineering
2.4.2. Integration of Domain Knowledge with AI-Driven Approaches
3. Computational Methods in Enzyme Engineering
3.1. Structure-Based Design Strategies
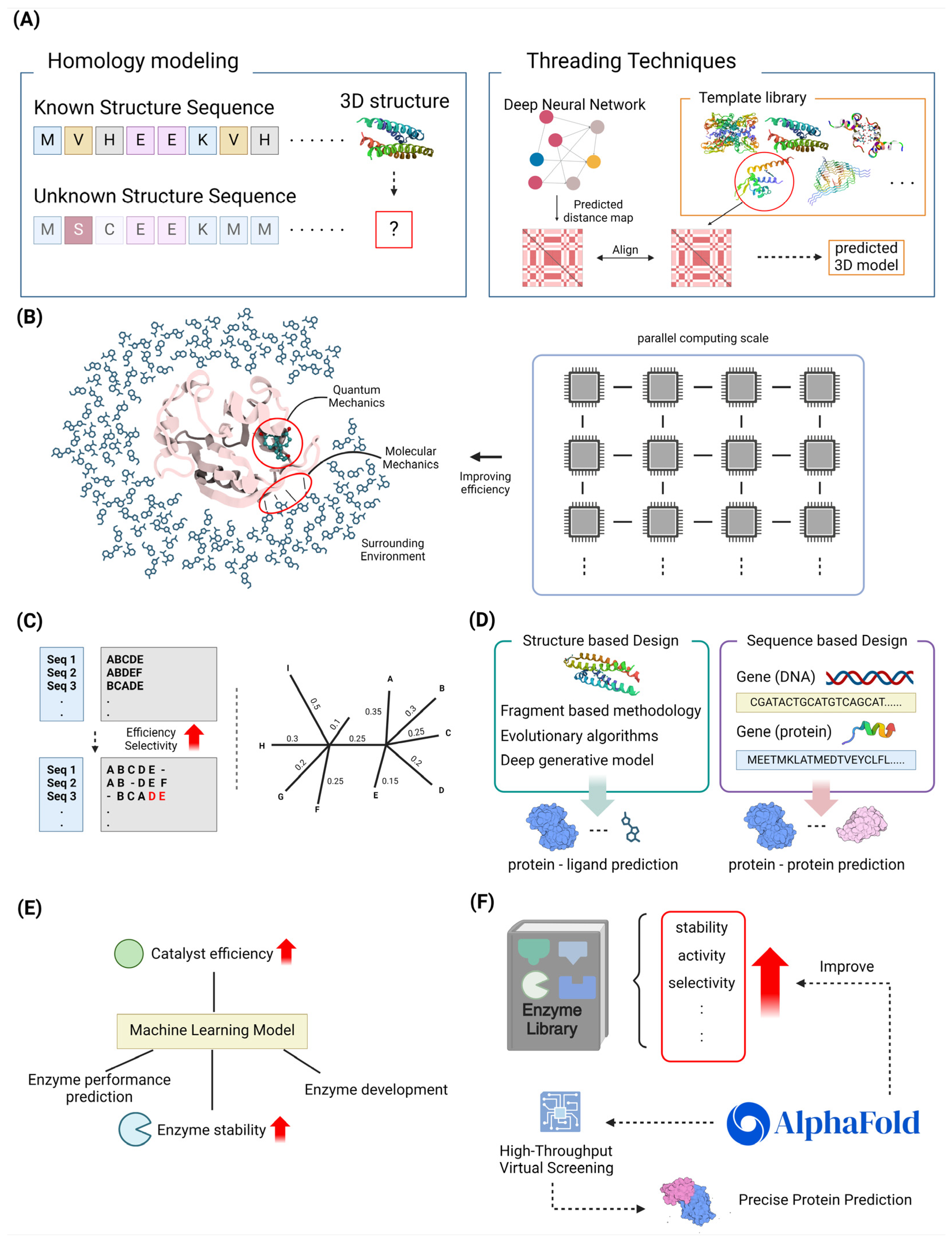
3.1.1. Homology Modeling and Threading Techniques
3.1.2. Quantum Mechanics/Molecular Mechanics Approaches
3.2. Sequence-Based Design Methods
3.2.1. Multiple Sequence Alignments and Phylogenetic Analysis
3.2.2. Coevolution-Based Approaches for Enzyme Design
3.3. Hybrid Methods
3.3.1. Integration of Structure and Sequence Information
3.3.2. Machine Learning-Assisted Enzyme Engineering
3.4. High-Throughput Virtual Screening
3.4.1. In Silico Directed Evolution
3.4.2. Computational Library Design for Enzyme Engineering
4. Molecular Dynamics Simulation Studies of Biomolecular Systems
4.1. Advanced Sampling Techniques
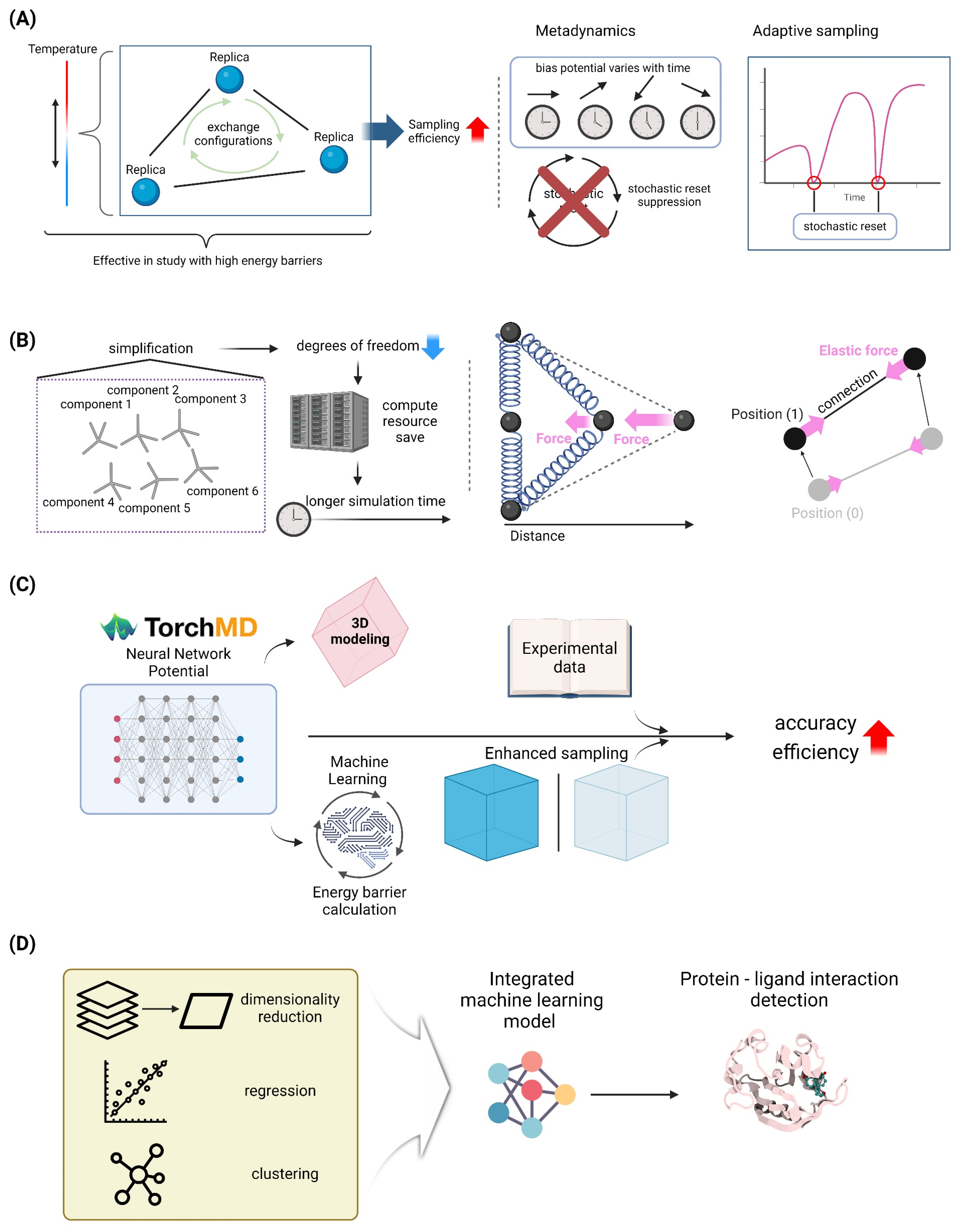
4.1.1. Replica Exchange Molecular Dynamics
4.1.2. Metadynamics and Adaptive Sampling Methods
4.2. Coarse-Grained Models
4.2.1. MARTINI Force Field and Its Applications
4.2.2. Elastic Network Models for Large-Scale Simulations
4.3. Long-Timescale Simulations
4.3.1. Specialized Hardware for MD Simulations
4.3.2. Enhanced Sampling Techniques for Accessing Biologically Relevant Timescales
4.4. Machine Learning-Enhanced MD Simulations
4.4.1. Neural Network Potentials for Accurate and Efficient Simulations
4.4.2. AI-Driven Analysis of MD Trajectories
5. Advances in Computational Docking and Drug Design
5.1. Protein–Ligand Docking
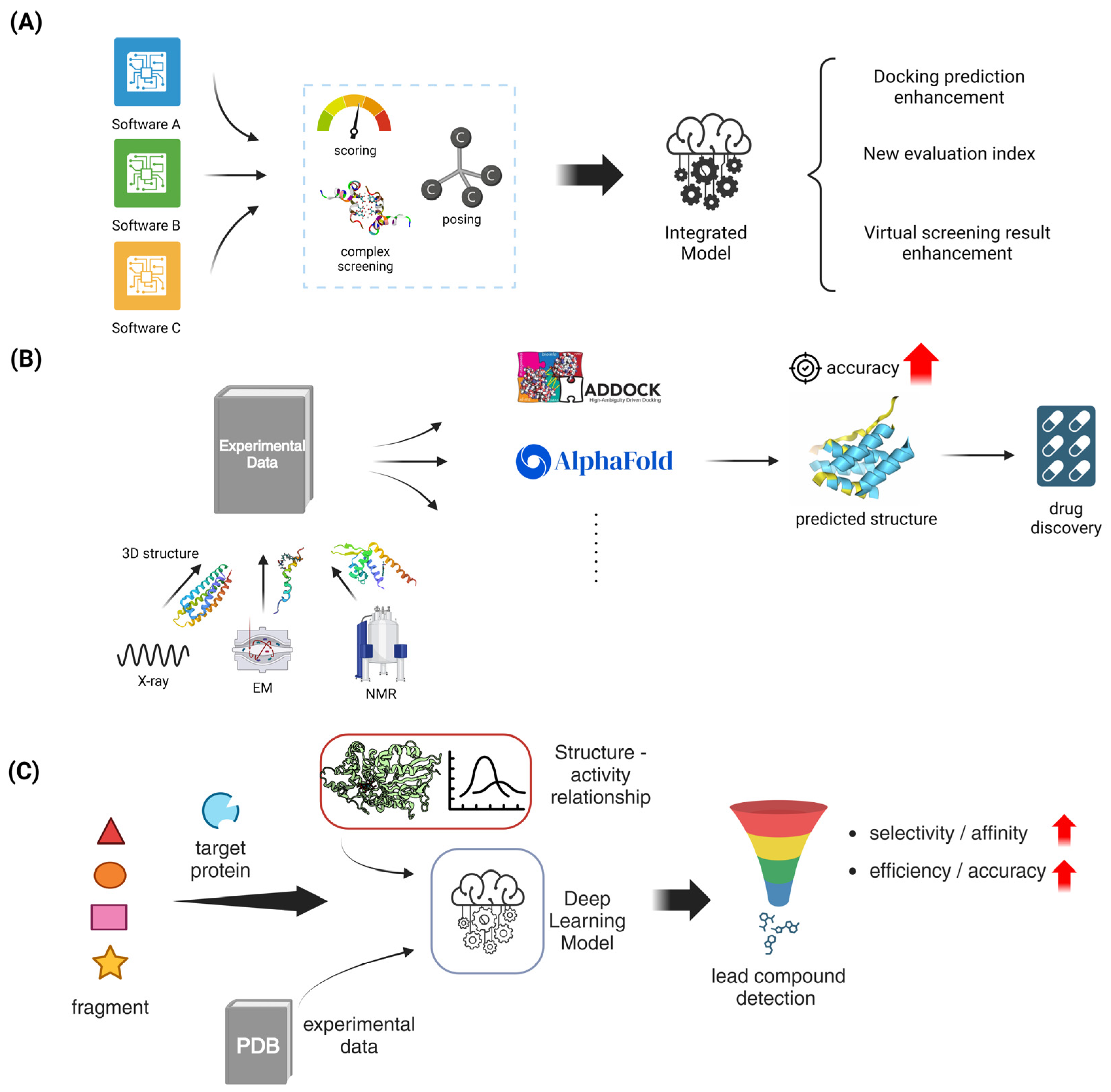
5.1.1. Flexible Docking Algorithms
5.1.2. Consensus Docking Approaches
5.2. Protein–Protein Docking
5.2.1. Template-Based Docking Methods
5.2.2. Integration of Experimental Data in Docking Protocols
5.3. Fragment-Based Drug Design
5.3.1. In Silico Fragment Growing and Linking Strategies
5.3.2. Machine Learning in Fragment-Based Approaches
5.4. Structure-Based Virtual Screening
5.4.1. Pharmacophore Modeling and Shape-Based Screening
5.4.2. AI-Driven Virtual Screening Pipelines
6. Design and Development of Novel Proteins with Enhanced Functionalities
6.1. De Novo Protein Design
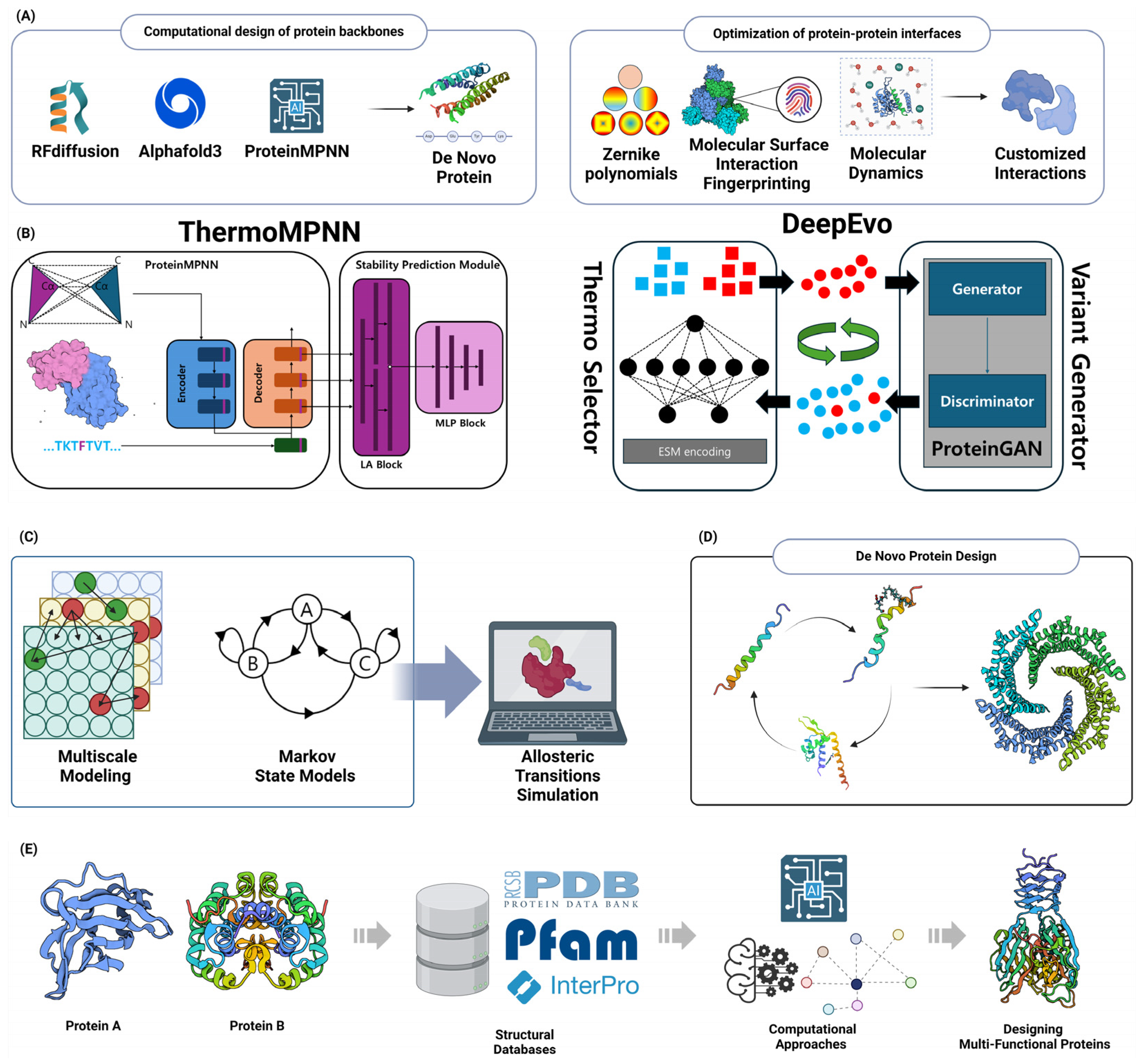
6.1.1. Computational Design of Protein Backbones
6.1.2. Optimization of Protein–Protein Interfaces
6.2. Protein Stability Engineering
6.2.1. Computational Prediction of Stabilizing Mutations
6.2.2. Design of Thermostable Proteins
6.3. Protein Functionalization
6.3.1. Computational Design of Allosteric Regulation
6.3.2. Engineering Proteins with Novel Binding Properties
6.4. Designing Multi-Functional Proteins
6.4.1. Computational Approaches for Domain Fusion
6.4.2. Rational Design of Chimeric Proteins
7. Case Studies and Applications in Biotechnology and Pharmaceuticals
7.1. Engineered Antibodies and Immunotherapeutics
7.1.1. Computational Design of Antibody–Antigen Interfaces
7.1.2. In Silico Optimization of Antibody Stability and Specificity
7.2. Biosensors and Diagnostics
7.2.1. Rational Design of Protein-Based Biosensors
7.2.2. Computational Approaches for Enhancing Sensor Sensitivity and Specificity
7.3. Industrial Enzymes
7.3.1. Computational Engineering of Enzymes for Biocatalysis
7.3.2. Design of Enzymes for Biodegradation and Environmental Applications
7.4. Therapeutic Protein Design
7.4.1. Computational Approaches for Improving Protein Drug Properties
7.4.2. In Silico Prediction of Immunogenicity and Optimization of Protein Therapeutics
8. Challenges and Future Perspectives
8.1. Integration of Multi-Scale Modeling Approaches
8.2. Addressing the Limitations of Current Force Fields
8.3. Bridging the Gap between Computation and Experiment
8.4. Ethical Considerations in AI-Driven Protein Engineering
8.5. Emerging Opportunities in Synthetic Biology and Protein Design
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sequeiros-Borja, C.E.; Surpeta, B.; Brezovsky, J. Recent advances in user-friendly computational tools to engineer protein function. Brief. Bioinform. 2021, 22, bbaa150. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Chen, Y.; Xue, W. Computational Protein Design—Where it goes? Curr. Med. Chem. 2024, 31, 2841–2854. [Google Scholar] [CrossRef] [PubMed]
- Derat, E.; Kamerlin, S.C.L. Computational Advances in Protein Engineering and Enzyme Design. J. Phys. Chem. B 2022, 126, 2449–2451. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.J.; Shao, Q.; Jiang, Y.; Jurich, C.; Ran, X.; Juarez, R.J.; Yan, B.; Stull, S.L.; Gollu, A.; Ding, N. Mutexa: A Computational Ecosystem for Intelligent Protein Engineering. J. Chem. Theory Comput. 2023, 19, 7459–7477. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Ben-Sasson, A.J. Precision materials: Computational design methods of accurate protein materials. Curr. Opin. Struct. Biol. 2022, 74, 102367. [Google Scholar] [CrossRef]
- Sadybekov, A.V.; Katritch, V. Computational approaches streamlining drug discovery. Nature 2023, 616, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Ai, H.; Huang, Y.; Tai, D.I.; Tsui, P.H.; Zhou, Z. Ultrasonic Assessment of Liver Fibrosis Using One-Dimensional Convolutional Neural Networks Based on Frequency Spectra of Radiofrequency Signals with Deep Learning Segmentation of Liver Regions in B-Mode Images: A Feasibility Study. Sensors 2024, 24, 5513. [Google Scholar] [CrossRef]
- Alzubaidi, L.; Zhang, J.; Humaidi, A.J.; Al-Dujaili, A.; Duan, Y.; Al-Shamma, O.; Santamaria, J.; Fadhel, M.A.; Al-Amidie, M.; Farhan, L. Review of deep learning: Concepts, CNN architectures, challenges, applications, future directions. J. Big Data 2021, 8, 53. [Google Scholar] [CrossRef] [PubMed]
- Gligorijevic, V.; Renfrew, P.D.; Kosciolek, T.; Leman, J.K.; Berenberg, D.; Vatanen, T.; Chandler, C.; Taylor, B.C.; Fisk, I.M.; Vlamakis, H.; et al. Structure-based protein function prediction using graph convolutional networks. Nat. Commun. 2021, 12, 3168. [Google Scholar] [CrossRef]
- Gao, W.; Mahajan, S.P.; Sulam, J.; Gray, J.J. Deep Learning in Protein Structural Modeling and Design. Patterns 2020, 1, 100142. [Google Scholar] [CrossRef]
- Saman Booy, M.; Ilin, A.; Orponen, P. RNA secondary structure prediction with convolutional neural networks. BMC Bioinform. 2022, 23, 58. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Zhang, D.; Chen, Y.; Zhang, Y.; Wang, Z.; Wang, X.; Li, S.; Guo, Y.; Webb, G.I.; Nguyen, A.T.N.; et al. GraphormerDTI: A graph transformer-based approach for drug-target interaction prediction. Comput. Biol. Med. 2024, 173, 108339. [Google Scholar] [CrossRef] [PubMed]
- Kulikova, A.V.; Diaz, D.J.; Loy, J.M.; Ellington, A.D.; Wilke, C.O. Learning the local landscape of protein structures with convolutional neural networks. J. Biol. Phys. 2021, 47, 435–454. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Srivastava, R. Deep learning in structural bioinformatics: Current applications and future perspectives. Brief. Bioinform. 2024, 25, bbae042. [Google Scholar] [CrossRef]
- Lalapura, V.S.; Bhimavarapu, V.R.; Amudha, J.; Satheesh, H.S. A Systematic Evaluation of Recurrent Neural Network Models for Edge Intelligence and Human Activity Recognition Applications. Algorithms 2024, 17, 104. [Google Scholar] [CrossRef]
- Asabuki, T.; Kokate, P.; Fukai, T. Neural circuit mechanisms of hierarchical sequence learning tested on large-scale recording data. PLoS Comput. Biol. 2022, 18, e1010214. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Jain, A.; Mauro, E.; LeShane, K.; Densmore, D. ICOR: Improving codon optimization with recurrent neural networks. BMC Bioinform. 2023, 24, 132. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.; Wang, Z.; Sun, Y.; Fan, L.; Yang, Y.; Guo, X.; Wang, Y.; Yan, S.; Qiao, Z.; Li, Y.; et al. Recurrent neural network for predicting absence of heterozygosity from low pass WGS with ultra-low depth. BMC Genom. 2024, 25, 470. [Google Scholar] [CrossRef]
- Das, S.; Tariq, A.; Santos, T.; Kantareddy, S.S.; Banerjee, I. Recurrent Neural Networks (RNNs): Architectures, Training Tricks, and Introduction to Influential Research. In Machine Learning for Brain Disorders; Colliot, O., Ed.; Humana: New York, NY, USA, 2023; pp. 117–138. [Google Scholar]
- Goodfellow, I.; Pouget-Abadie, J.; Mirza, M.; Xu, B.; Warde-Farley, D.; Ozair, S.; Courville, A.; Bengio, Y. Generative adversarial networks. Commun. ACM 2020, 63, 139–144. [Google Scholar] [CrossRef]
- Sharma, P.; Kumar, M.; Sharma, H.K.; Biju, S.M. Generative adversarial networks (GANs): Introduction, Taxonomy, Variants, Limitations, and Applications. Multimed. Tools Appl. 2024. [Google Scholar] [CrossRef]
- Lin, E.; Lin, C.H.; Lane, H.Y. De Novo Peptide and Protein Design Using Generative Adversarial Networks: An Update. J. Chem. Inf. Model. 2022, 62, 761–774. [Google Scholar] [CrossRef] [PubMed]
- Kucera, T.; Togninalli, M.; Meng-Papaxanthos, L. Conditional generative modeling for de novo protein design with hierarchical functions. Bioinformatics 2022, 38, 3454–3461. [Google Scholar] [CrossRef] [PubMed]
- Strokach, A.; Kim, P.M. Deep generative modeling for protein design. Curr. Opin. Struct. Biol. 2022, 72, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Held, L.K.; Vermeylen, L.; Dignath, D.; Notebaert, W.; Krebs, R.M.; Braem, S. Reinforcement learning of adaptive control strategies. Commun. Psychol. 2024, 2, 8. [Google Scholar] [CrossRef] [PubMed]
- Atz, K.; Cotos, L.; Isert, C.; Hakansson, M.; Focht, D.; Hilleke, M.; Nippa, D.F.; Iff, M.; Ledergerber, J.; Schiebroek, C.C.G.; et al. Prospective de novo drug design with deep interactome learning. Nat. Commun. 2024, 15, 3408. [Google Scholar] [CrossRef]
- Kim, H.; Choi, H.; Kang, D.; Lee, W.B.; Na, J. Materials discovery with extreme properties via reinforcement learning-guided combinatorial chemistry. Chem. Sci. 2024, 15, 7908–7925. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Huang, M. Navigating the landscape of enzyme design: From molecular simulations to machine learning. Chem. Soc. Rev. 2024. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, S.; Xing, M.; Yuan, Q.; He, H.; Sun, S. Universal Approach to De Novo Drug Design for Target Proteins Using Deep Reinforcement Learning. ACS Omega 2023, 8, 5464–5474. [Google Scholar] [CrossRef]
- Palukuri, M.V.; Patil, R.S.; Marcotte, E.M. Molecular complex detection in protein interaction networks through reinforcement learning. BMC Bioinform. 2023, 24, 306. [Google Scholar] [CrossRef]
- Dietrich, L.; Rathmer, B.; Ewan, K.; Bange, T.; Heinrichs, S.; Dale, T.C.; Schade, D.; Grossmann, T.N. Cell Permeable Stapled Peptide Inhibitor of Wnt Signaling that Targets beta-Catenin Protein-Protein Interactions. Cell Chem. Biol. 2017, 24, 958–968.e955. [Google Scholar] [CrossRef]
- Wang, G.; Liu, X.; Wang, K.; Gao, Y.; Li, G.; Baptista-Hon, D.T.; Yang, X.H.; Xue, K.; Tai, W.H.; Jiang, Z.; et al. Deep-learning-enabled protein-protein interaction analysis for prediction of SARS-CoV-2 infectivity and variant evolution. Nat. Med. 2023, 29, 2007–2018. [Google Scholar] [CrossRef] [PubMed]
- Iman, M.; Arabnia, H.R.; Rasheed, K. A Review of Deep Transfer Learning and Recent Advancements. Technologies 2023, 11, 40. [Google Scholar] [CrossRef]
- Wang, J.; Liu, K.; Zhang, Y.; Leng, B.; Lu, J. Recent advances of few-shot learning methods and applications. Sci. China Technol. Sci. 2023, 66, 920–944. [Google Scholar] [CrossRef]
- Zhou, Z.; Zhang, L.; Yu, Y.; Wu, B.; Li, M.; Hong, L.; Tan, P. Enhancing efficiency of protein language models with minimal wet-lab data through few-shot learning. Nat. Commun. 2024, 15, 5566. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, R.; Bouatta, N.; Biswas, S.; Floristean, C.; Kharkar, A.; Roy, K.; Rochereau, C.; Ahdritz, G.; Zhang, J.; Church, G.M.; et al. Single-sequence protein structure prediction using a language model and deep learning. Nat. Biotechnol. 2022, 40, 1617–1623. [Google Scholar] [CrossRef]
- Khakzad, H.; Igashov, I.; Schneuing, A.; Goverde, C.; Bronstein, M.; Correia, B. A new age in protein design empowered by deep learning. Cell Syst. 2023, 14, 925–939. [Google Scholar] [CrossRef]
- Listov, D.; Goverde, C.A.; Correia, B.E.; Fleishman, S.J. Opportunities and challenges in design and optimization of protein function. Nat. Rev. Mol. Cell Biol. 2024, 25, 639–653. [Google Scholar] [CrossRef]
- Kafri, M.; Metzl-Raz, E.; Jona, G.; Barkai, N. The Cost of Protein Production. Cell Rep. 2016, 14, 22–31. [Google Scholar] [CrossRef]
- Ao, Y.F.; Dorr, M.; Menke, M.J.; Born, S.; Heuson, E.; Bornscheuer, U.T. Data-Driven Protein Engineering for Improving Catalytic Activity and Selectivity. Chembiochem 2024, 25, e202300754. [Google Scholar] [CrossRef]
- Derry, A.; Carpenter, K.A.; Altman, R.B. Training data composition affects performance of protein structure analysis algorithms. Pac. Symp. Biocomput. 2022, 27, 10–21. [Google Scholar]
- Illig, A.M.; Siedhoff, N.E.; Davari, M.D.; Schwaneberg, U. Evolutionary Probability and Stacked Regressions Enable Data-Driven Protein Engineering with Minimized Experimental Effort. J. Chem. Inf. Model. 2024, 64, 6350–6360. [Google Scholar] [CrossRef] [PubMed]
- Medl, M.; Leisch, F.; Durauer, A.; Scharl, T. Explainable deep learning enhances robust and reliable real-time monitoring of a chromatographic protein A capture step. Biotechnol. J. 2024, 19, e2300554. [Google Scholar] [CrossRef] [PubMed]
- Lee, M. Recent Advances in Deep Learning for Protein-Protein Interaction Analysis: A Comprehensive Review. Molecules 2023, 28, 5169. [Google Scholar] [CrossRef]
- Kim, M.; Kang, D.; Kim, M.S.; Choe, J.C.; Lee, S.H.; Ahn, J.H.; Oh, J.H.; Choi, J.H.; Lee, H.C.; Cha, K.S.; et al. Acute myocardial infarction prognosis prediction with reliable and interpretable artificial intelligence system. J. Am. Med. Inform. Assoc. 2024, 31, 1540–1550. [Google Scholar] [CrossRef]
- Malinverno, L.; Barros, V.; Ghisoni, F.; Visona, G.; Kern, R.; Nickel, P.J.; Ventura, B.E.; Simic, I.; Stryeck, S.; Manni, F.; et al. A historical perspective of biomedical explainable AI research. Patterns 2023, 4, 100830. [Google Scholar] [CrossRef] [PubMed]
- Dash, T.; Chitlangia, S.; Ahuja, A.; Srinivasan, A. A review of some techniques for inclusion of domain-knowledge into deep neural networks. Sci. Rep. 2022, 12, 1040. [Google Scholar] [CrossRef]
- Sirocchi, C.; Bogliolo, A.; Montagna, S. Medical-informed machine learning: Integrating prior knowledge into medical decision systems. BMC Med. Inform. Decis. Mak. 2024, 24, 186. [Google Scholar] [CrossRef]
- Laxmi, B.; Devi, P.U.M.; Thanjavur, N.; Buddolla, V. The Applications of Artificial Intelligence (AI)-Driven Tools in Virus-Like Particles (VLPs) Research. Curr. Microbiol. 2024, 81, 234. [Google Scholar] [CrossRef] [PubMed]
- Khlaif, Z.N.; Mousa, A.; Hattab, M.K.; Itmazi, J.; Hassan, A.A.; Sanmugam, M.; Ayyoub, A. The Potential and Concerns of Using AI in Scientific Research: ChatGPT Performance Evaluation. JMIR Med. Educ. 2023, 9, e47049. [Google Scholar] [CrossRef] [PubMed]
- Musa, N.; Gital, A.Y.; Aljojo, N.; Chiroma, H.; Adewole, K.S.; Mojeed, H.A.; Faruk, N.; Abdulkarim, A.; Emmanuel, I.; Folawiyo, Y.Y.; et al. A systematic review and Meta-data analysis on the applications of Deep Learning in Electrocardiogram. J. Ambient. Intell. Humaniz. Comput. 2023, 14, 9677–9750. [Google Scholar] [CrossRef]
- Dikmen, M.; Burns, C. The effects of domain knowledge on trust in explainable AI and task performance: A case of peer-to-peer lending. Int. J. Hum.-Comput. Stud. 2022, 162, 102792. [Google Scholar] [CrossRef]
- Wodak, S.J.; Vajda, S.; Lensink, M.F.; Kozakov, D.; Bates, P.A. Critical Assessment of Methods for Predicting the 3D Structure of Proteins and Protein Complexes. Annu. Rev. Biophys. 2023, 52, 183–206. [Google Scholar] [CrossRef] [PubMed]
- Wuyun, Q.; Chen, Y.; Shen, Y.; Cao, Y.; Hu, G.; Cui, W.; Gao, J.; Zheng, W. Recent Progress of Protein Tertiary Structure Prediction. Molecules 2024, 29, 832. [Google Scholar] [CrossRef]
- Bertoline, L.M.F.; Lima, A.N.; Krieger, J.E.; Teixeira, S.K. Before and after AlphaFold2: An overview of protein structure prediction. Front. Bioinform. 2023, 3, 1120370. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Roche, R.; Shuvo, M.H.; Bhattacharya, D. Recent Advances in Protein Homology Detection Propelled by Inter-Residue Interaction Map Threading. Front. Mol. Biosci. 2021, 8, 643752. [Google Scholar] [CrossRef]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef]
- Raghavan, B.; Paulikat, M.; Ahmad, K.; Callea, L.; Rizzi, A.; Ippoliti, E.; Mandelli, D.; Bonati, L.; De Vivo, M.; Carloni, P. Drug Design in the Exascale Era: A Perspective from Massively Parallel QM/MM Simulations. J. Chem. Inf. Model. 2023, 63, 3647–3658. [Google Scholar] [CrossRef] [PubMed]
- Rossetti, G.; Mandelli, D. How exascale computing can shape drug design: A perspective from multiscale QM/MM molecular dynamics simulations and machine learning-aided enhanced sampling algorithms. Curr. Opin. Struct. Biol. 2024, 86, 102814. [Google Scholar] [CrossRef] [PubMed]
- Ginex, T.; Vazquez, J.; Estarellas, C.; Luque, F.J. Quantum mechanical-based strategies in drug discovery: Finding the pace to new challenges in drug design. Curr. Opin. Struct. Biol. 2024, 87, 102870. [Google Scholar] [CrossRef]
- Kubar, T.; Elstner, M.; Cui, Q. Hybrid Quantum Mechanical/Molecular Mechanical Methods For Studying Energy Transduction in Biomolecular Machines. Annu. Rev. Biophys. 2023, 52, 525–551. [Google Scholar] [CrossRef]
- Giese, T.J.; Zeng, J.; Lerew, L.; McCarthy, E.; Tao, Y.; Ekesan, S.; York, D.M. Software Infrastructure for Next-Generation QM/MM-DeltaMLP Force Fields. J. Phys. Chem. B 2024, 128, 6257–6271. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Liu, B.; Williams, K.P.; Warnow, T. EMMA: A new method for computing multiple sequence alignments given a constraint subset alignment. Algorithms Mol. Biol. 2023, 18, 21. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.K.; Yusof, U.K.; Eisa, T.A.E.; Nasser, M. Bioinspired Algorithms for Multiple Sequence Alignment: A Systematic Review and Roadmap. Appl. Sci. 2024, 14, 2433. [Google Scholar] [CrossRef]
- Zou, Y.; Zhang, Z.; Zeng, Y.; Hu, H.; Hao, Y.; Huang, S.; Li, B. Common Methods for Phylogenetic Tree Construction and Their Implementation in R. Bioengineering 2024, 11, 480. [Google Scholar] [CrossRef] [PubMed]
- Kapli, P.; Kotari, I.; Telford, M.J.; Goldman, N.; Yang, Z. DNA Sequences Are as Useful as Protein Sequences for Inferring Deep Phylogenies. Syst. Biol. 2023, 72, 1119–1135. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Wu, L.Y.; Xia, X.Y.; Chen, X.; Wang, Z.X.; Pan, X.M. A sequence-based evolutionary distance method for Phylogenetic analysis of highly divergent proteins. Sci. Rep. 2023, 13, 20304. [Google Scholar] [CrossRef]
- Chao, J.; Tang, F.; Xu, L. Developments in Algorithms for Sequence Alignment: A Review. Biomolecules 2022, 12, 546. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Noh, M.H.; Park, M.; Kim, I.; Ahn, H.; Ye, D.Y.; Jung, G.Y.; Kim, S. Enzyme activity engineering based on sequence co-evolution analysis. Metab. Eng. 2022, 74, 49–60. [Google Scholar] [CrossRef]
- Xie, J.; Zhang, W.; Zhu, X.; Deng, M.; Lai, L. Coevolution-based prediction of key allosteric residues for protein function regulation. Elife 2023, 12, e81850. [Google Scholar] [CrossRef] [PubMed]
- Hossack, E.J.; Hardy, F.J.; Green, A.P. Building Enzymes through Design and Evolution. ACS Catal. 2023, 13, 12436–12444. [Google Scholar] [CrossRef]
- Pinto, G.P.; Corbella, M.; Demkiv, A.O.; Kamerlin, S.C.L. Exploiting enzyme evolution for computational protein design. Trends Biochem. Sci. 2022, 47, 375–389. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Moretti, R.; Meiler, J. Recent Advances in Automated Structure-Based De Novo Drug Design. J. Chem. Inf. Model. 2024, 64, 1794–1805. [Google Scholar] [CrossRef] [PubMed]
- Isert, C.; Atz, K.; Schneider, G. Structure-based drug design with geometric deep learning. Curr. Opin. Struct. Biol. 2023, 79, 102548. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Saha, S.; Tvedt, N.C.; Yang, L.W.; Bahar, I. Mutually beneficial confluence of structure-based modeling of protein dynamics and machine learning methods. Curr. Opin. Struct. Biol. 2023, 78, 102517. [Google Scholar] [CrossRef]
- Kinshuk, S.; Li, L.; Meckes, B.; Chan, C.T.Y. Sequence-Based Protein Design: A Review of Using Statistical Models to Characterize Coevolutionary Traits for Developing Hybrid Proteins as Genetic Sensors. Int. J. Mol. Sci. 2024, 25, 8320. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Basu, S.; Kurgan, L. HybridDBRpred: Improved sequence-based prediction of DNA-binding amino acids using annotations from structured complexes and disordered proteins. Nucleic Acids Res. 2024, 52, e10. [Google Scholar] [CrossRef]
- Hummer, A.M.; Abanades, B.; Deane, C.M. Advances in computational structure-based antibody design. Curr. Opin. Struct. Biol. 2022, 74, 102379. [Google Scholar] [CrossRef] [PubMed]
- Siedhoff, N.E.; Schwaneberg, U.; Davari, M.D. Machine learning-assisted enzyme engineering. Methods Enzymol. 2020, 643, 281–315. [Google Scholar] [CrossRef]
- Gantz, M.; Neun, S.; Medcalf, E.J.; van Vliet, L.D.; Hollfelder, F. Ultrahigh-Throughput Enzyme Engineering and Discovery in In Vitro Compartments. Chem. Rev. 2023, 123, 5571–5611. [Google Scholar] [CrossRef]
- Ding, K.; Chin, M.; Zhao, Y.; Huang, W.; Mai, B.K.; Wang, H.; Liu, P.; Yang, Y.; Luo, Y. Machine learning-guided co-optimization of fitness and diversity facilitates combinatorial library design in enzyme engineering. Nat. Commun. 2024, 15, 6392. [Google Scholar] [CrossRef]
- Atomwise, A.P. AI is a viable alternative to high throughput screening: A 318-target study. Sci. Rep. 2024, 14, 7526. [Google Scholar] [CrossRef]
- Carlsson, J.; Luttens, A. Structure-based virtual screening of vast chemical space as a starting point for drug discovery. Curr. Opin. Struct. Biol. 2024, 87, 102829. [Google Scholar] [CrossRef] [PubMed]
- Goudy, O.J.; Nallathambi, A.; Kinjo, T.; Randolph, N.Z.; Kuhlman, B. In silico evolution of autoinhibitory domains for a PD-L1 antagonist using deep learning models. Proc. Natl. Acad. Sci. USA 2023, 120, e2307371120. [Google Scholar] [CrossRef]
- McLure, R.J.; Radford, S.E.; Brockwell, D.J. High-throughput directed evolution: A golden era for protein science. Trends Chem. 2022, 4, 378–391. [Google Scholar] [CrossRef]
- Shao, Q.; Jiang, Y.; Yang, Z.J. EnzyHTP Computational Directed Evolution with Adaptive Resource Allocation. J. Chem. Inf. Model. 2023, 63, 5650–5659. [Google Scholar] [CrossRef] [PubMed]
- Orsi, E.; Schada von Borzyskowski, L.; Noack, S.; Nikel, P.I.; Lindner, S.N. Automated in vivo enzyme engineering accelerates biocatalyst optimization. Nat. Commun. 2024, 15, 3447. [Google Scholar] [CrossRef]
- Scherer, M.; Fleishman, S.J.; Jones, P.R.; Dandekar, T.; Bencurova, E. Computational Enzyme Engineering Pipelines for Optimized Production of Renewable Chemicals. Front. Bioeng. Biotechnol. 2021, 9, 673005. [Google Scholar] [CrossRef]
- Vanella, R.; Kovacevic, G.; Doffini, V.; Fernandez de Santaella, J.; Nash, M.A. High-throughput screening, next generation sequencing and machine learning: Advanced methods in enzyme engineering. Chem. Commun. 2022, 58, 2455–2467. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Tao, C.; Shen, X.; Sun, X.; Wang, J.; Yuan, Q. Unlocking the potential of enzyme engineering via rational computational design strategies. Biotechnol. Adv. 2024, 73, 108376. [Google Scholar] [CrossRef]
- Bernardi, R.C.; Melo, M.C.R.; Schulten, K. Enhanced sampling techniques in molecular dynamics simulations of biological systems. Biochim. Biophys. Acta 2015, 1850, 872–877. [Google Scholar] [CrossRef]
- Gong, X.; Zhang, Y.; Chen, J. Advanced Sampling Methods for Multiscale Simulation of Disordered Proteins and Dynamic Interactions. Biomolecules 2021, 11, 1416. [Google Scholar] [CrossRef] [PubMed]
- Qi, R.; Wei, G.; Ma, B.; Nussinov, R. Replica Exchange Molecular Dynamics: A Practical Application Protocol with Solutions to Common Problems and a Peptide Aggregation and Self-Assembly Example. Methods Mol. Biol. 2018, 1777, 101–119. [Google Scholar] [CrossRef] [PubMed]
- Blumer, O.; Reuveni, S.; Hirshberg, B. Combining stochastic resetting with Metadynamics to speed-up molecular dynamics simulations. Nat. Commun. 2024, 15, 240. [Google Scholar] [CrossRef] [PubMed]
- Kleiman, D.E.; Nadeem, H.; Shukla, D. Adaptive Sampling Methods for Molecular Dynamics in the Era of Machine Learning. J. Phys. Chem. B 2023, 127, 10669–10681. [Google Scholar] [CrossRef] [PubMed]
- Brooks, C.L., 3rd; MacKerell, A.D., Jr.; Post, C.B.; Nilsson, L. Biomolecular dynamics in the 21st century. Biochim. Biophys. Acta Gen. Subj. 2024, 1868, 130534. [Google Scholar] [CrossRef] [PubMed]
- Marrink, S.J.; Monticelli, L.; Melo, M.N.; Alessandri, R.; Tieleman, D.P.; Souza, P.C.T. Two decades of Martini: Better beads, broader scope. WIREs Comput. Mol. Sci. 2022, 13, e1620. [Google Scholar] [CrossRef]
- Kjolbye, L.R.; Pereira, G.P.; Bartocci, A.; Pannuzzo, M.; Albani, S.; Marchetto, A.; Jimenez-Garcia, B.; Martin, J.; Rossetti, G.; Cecchini, M.; et al. Towards design of drugs and delivery systems with the Martini coarse-grained model. QRB Discov. 2022, 3, e19. [Google Scholar] [CrossRef] [PubMed]
- Periole, X.; Marrink, S.J. The Martini coarse-grained force field. Methods Mol. Biol. 2013, 924, 533–565. [Google Scholar] [CrossRef]
- MacCallum, J.L.; Hu, S.; Lenz, S.; Souza, P.C.T.; Corradi, V.; Tieleman, D.P. An implementation of the Martini coarse-grained force field in OpenMM. Biophys. J. 2023, 122, 2864–2870. [Google Scholar] [CrossRef]
- Togashi, Y.; Flechsig, H. Coarse-Grained Protein Dynamics Studies Using Elastic Network Models. Int. J. Mol. Sci. 2018, 19, 3899. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wang, Y.; Zhang, J.; Abdelmoneim, A.A.; Liang, Z.; Wang, L.; Jin, J.; Dai, Q.; Ye, F. Elastic network models and molecular dynamic simulations reveal the molecular basis of allosteric regulation in ubiquitin-specific protease 7 (USP7). Comput. Biol. Med. 2023, 162, 107068. [Google Scholar] [CrossRef] [PubMed]
- Leioatts, N.; Romo, T.D.; Grossfield, A. Elastic Network Models are Robust to Variations in Formalism. J. Chem. Theory Comput. 2012, 8, 2424–2434. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.E.; Hynninen, A.P.; Phillips, J.C.; Schulten, K. Early Experiences Porting the NAMD and VMD Molecular Simulation and Analysis Software to GPU-Accelerated OpenPOWER Platforms. High Perform. Comput. 2016, 9945, 188–206. [Google Scholar] [CrossRef]
- Ahmed, M.; Maldonado, A.M.; Durrant, J.D. From byte to bench to bedside: Molecular dynamics simulations and drug discovery. BMC Biol. 2023, 21, 299. [Google Scholar] [CrossRef] [PubMed]
- Chiu, M.; Herbordt, M.C. Molecular Dynamics Simulations on High-Performance Reconfigurable Computing Systems. ACM Trans. Reconfigurable Technol. Syst. 2010, 3, 23. [Google Scholar] [CrossRef]
- Jones, D.; Allen, J.E.; Yang, Y.; Drew Bennett, W.F.; Gokhale, M.; Moshiri, N.; Rosing, T.S. Accelerators for Classical Molecular Dynamics Simulations of Biomolecules. J. Chem. Theory Comput. 2022, 18, 4047–4069. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, S.A.; Dror, R.O. Molecular Dynamics Simulation for All. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef]
- Rizzi, V.; Aureli, S.; Ansari, N.; Gervasio, F.L. OneOPES, a Combined Enhanced Sampling Method to Rule Them All. J. Chem. Theory Comput. 2023, 19, 5731–5742. [Google Scholar] [CrossRef] [PubMed]
- Doerr, S.; Majewski, M.; Perez, A.; Kramer, A.; Clementi, C.; Noe, F.; Giorgino, T.; De Fabritiis, G. TorchMD: A Deep Learning Framework for Molecular Simulations. J. Chem. Theory Comput. 2021, 17, 2355–2363. [Google Scholar] [CrossRef]
- Pelaez, R.P.; Simeon, G.; Galvelis, R.; Mirarchi, A.; Eastman, P.; Doerr, S.; Tholke, P.; Markland, T.E.; De Fabritiis, G. TorchMD-Net 2.0: Fast Neural Network Potentials for Molecular Simulations. J. Chem. Theory Comput. 2024, 20, 4076–4087. [Google Scholar] [CrossRef]
- Thaler, S.; Zavadlav, J. Learning neural network potentials from experimental data via Differentiable Trajectory Reweighting. Nat. Commun. 2021, 12, 6884. [Google Scholar] [CrossRef] [PubMed]
- Jung, G.S.; Choi, J.Y.; Lee, S.M. Active learning of neural network potentials for rare events. Digit. Discov. 2024, 3, 514–527. [Google Scholar] [CrossRef]
- Duignan, T.T. The Potential of Neural Network Potentials. ACS Phys. Chem. Au 2024, 4, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Kaptan, S.; Vattulainen, I. Machine learning in the analysis of biomolecular simulations. Adv. Phys. X 2022, 7, 2006080. [Google Scholar] [CrossRef]
- Mustali, J.; Yasuda, I.; Hirano, Y.; Yasuoka, K.; Gautieri, A.; Arai, N. Unsupervised deep learning for molecular dynamics simulations: A novel analysis of protein-ligand interactions in SARS-CoV-2 M(pro). RSC Adv. 2023, 13, 34249–34261. [Google Scholar] [CrossRef] [PubMed]
- Han, R.; Luber, S. Trajectory-based machine learning method and its application to molecular dynamics. Mol. Phys. 2020, 118, e1788189. [Google Scholar] [CrossRef]
- Prašnikar, E.; Ljubič, M.; Perdih, A.; Borišek, J. Machine learning heralding a new development phase in molecular dynamics simulations. Artif. Intell. Rev. 2024, 57, 102. [Google Scholar] [CrossRef]
- Huang, S.Y. Comprehensive assessment of flexible-ligand docking algorithms: Current effectiveness and challenges. Brief. Bioinform. 2018, 19, 982–994. [Google Scholar] [CrossRef]
- Yang, C.; Chen, E.A.; Zhang, Y. Protein-Ligand Docking in the Machine-Learning Era. Molecules 2022, 27, 4568. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.Y.; Zou, X. Advances and challenges in protein-ligand docking. Int. J. Mol. Sci. 2010, 11, 3016–3034. [Google Scholar] [CrossRef]
- Torres, P.H.M.; Sodero, A.C.R.; Jofily, P.; Silva-Jr, F.P. Key Topics in Molecular Docking for Drug Design. Int. J. Mol. Sci. 2019, 20, 4574. [Google Scholar] [CrossRef] [PubMed]
- Palacio-Rodriguez, K.; Lans, I.; Cavasotto, C.N.; Cossio, P. Exponential consensus ranking improves the outcome in docking and receptor ensemble docking. Sci. Rep. 2019, 9, 5142. [Google Scholar] [CrossRef] [PubMed]
- Blanes-Mira, C.; Fernandez-Aguado, P.; de Andres-Lopez, J.; Fernandez-Carvajal, A.; Ferrer-Montiel, A.; Fernandez-Ballester, G. Comprehensive Survey of Consensus Docking for High-Throughput Virtual Screening. Molecules 2022, 28, 175. [Google Scholar] [CrossRef]
- Kamal, I.M.; Chakrabarti, S. MetaDOCK: A Combinatorial Molecular Docking Approach. ACS Omega 2023, 8, 5850–5860. [Google Scholar] [CrossRef]
- Xue, L.C.; Rodrigues, J.; Dobbs, D.; Honavar, V.; Bonvin, A. Template-based protein-protein docking exploiting pairwise interfacial residue restraints. Brief. Bioinform. 2017, 18, 458–466. [Google Scholar] [CrossRef]
- Meng, Q.; Guo, F.; Wang, E.; Tang, J. ComDock: A novel approach for protein-protein docking with an efficient fusing strategy. Comput. Biol. Med. 2023, 167, 107660. [Google Scholar] [CrossRef]
- Bryant, P.; Pozzati, G.; Elofsson, A. Improved prediction of protein-protein interactions using AlphaFold2. Nat. Commun. 2022, 13, 1265. [Google Scholar] [CrossRef]
- Schneidman-Duhovny, D.; Rossi, A.; Avila-Sakar, A.; Kim, S.J.; Velazquez-Muriel, J.; Strop, P.; Liang, H.; Krukenberg, K.A.; Liao, M.; Kim, H.M.; et al. A method for integrative structure determination of protein-protein complexes. Bioinformatics 2012, 28, 3282–3289. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, Y.; Yamamori, Y.; Tomii, K. Protein-protein interaction prediction methods: From docking-based to AI-based approaches. Biophys. Rev. 2022, 14, 1341–1348. [Google Scholar] [CrossRef]
- de Souza Neto, L.R.; Moreira-Filho, J.T.; Neves, B.J.; Maidana, R.; Guimaraes, A.C.R.; Furnham, N.; Andrade, C.H.; Silva, F.P., Jr. In silico Strategies to Support Fragment-to-Lead Optimization in Drug Discovery. Front. Chem. 2020, 8, 93. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.X.; Wang, Z.Z.; Wang, F.; Hao, G.F.; Yang, G.F. ACFIS 2.0: An improved web-server for fragment-based drug discovery via a dynamic screening strategy. Nucleic Acids Res. 2023, 51, W25–W32. [Google Scholar] [CrossRef] [PubMed]
- Mouchlis, V.D.; Afantitis, A.; Serra, A.; Fratello, M.; Papadiamantis, A.G.; Aidinis, V.; Lynch, I.; Greco, D.; Melagraki, G. Advances in de Novo Drug Design: From Conventional to Machine Learning Methods. Int. J. Mol. Sci. 2021, 22, 1676. [Google Scholar] [CrossRef] [PubMed]
- Powers, A.S.; Yu, H.H.; Suriana, P.; Koodli, R.V.; Lu, T.; Paggi, J.M.; Dror, R.O. Geometric Deep Learning for Structure-Based Ligand Design. ACS Cent. Sci. 2023, 9, 2257–2267. [Google Scholar] [CrossRef]
- Mukaidaisi, M.; Vu, A.; Grantham, K.; Tchagang, A.; Li, Y. Multi-Objective Drug Design Based on Graph-Fragment Molecular Representation and Deep Evolutionary Learning. Front. Pharmacol. 2022, 13, 920747. [Google Scholar] [CrossRef] [PubMed]
- Opo, F.; Rahman, M.M.; Ahammad, F.; Ahmed, I.; Bhuiyan, M.A.; Asiri, A.M. Structure based pharmacophore modeling, virtual screening, molecular docking and ADMET approaches for identification of natural anti-cancer agents targeting XIAP protein. Sci. Rep. 2021, 11, 4049. [Google Scholar] [CrossRef]
- Giordano, D.; Biancaniello, C.; Argenio, M.A.; Facchiano, A. Drug Design by Pharmacophore and Virtual Screening Approach. Pharmaceuticals 2022, 15, 646. [Google Scholar] [CrossRef]
- Moyano-Gomez, P.; Lehtonen, J.V.; Pentikainen, O.T.; Postila, P.A. Building shape-focused pharmacophore models for effective docking screening. J. Cheminform 2024, 16, 97. [Google Scholar] [CrossRef]
- Cieslak, M.; Danel, T.; Krzysztynska-Kuleta, O.; Kalinowska-Tluscik, J. Machine learning accelerates pharmacophore-based virtual screening of MAO inhibitors. Sci. Rep. 2024, 14, 8228. [Google Scholar] [CrossRef]
- Visan, A.I.; Negut, I. Integrating Artificial Intelligence for Drug Discovery in the Context of Revolutionizing Drug Delivery. Life 2024, 14, 233. [Google Scholar] [CrossRef]
- Turon, G.; Hlozek, J.; Woodland, J.G.; Kumar, A.; Chibale, K.; Duran-Frigola, M. First fully-automated AI/ML virtual screening cascade implemented at a drug discovery centre in Africa. Nat. Commun. 2023, 14, 5736. [Google Scholar] [CrossRef]
- Qureshi, R.; Irfan, M.; Gondal, T.M.; Khan, S.; Wu, J.; Hadi, M.U.; Heymach, J.; Le, X.; Yan, H.; Alam, T. AI in drug discovery and its clinical relevance. Heliyon 2023, 9, e17575. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, J.T.; Freemont, P.S. Computational protein design with backbone plasticity. Biochem. Soc. Trans. 2016, 44, 1523–1529. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Kortemme, T. Recent advances in de novo protein design: Principles, methods, and applications. J. Biol. Chem. 2021, 296, 100558. [Google Scholar] [CrossRef] [PubMed]
- Watson, J.L.; Juergens, D.; Bennett, N.R.; Trippe, B.L.; Yim, J.; Eisenach, H.E.; Ahern, W.; Borst, A.J.; Ragotte, R.J.; Milles, L.F.; et al. De novo design of protein structure and function with RFdiffusion. Nature 2023, 620, 1089–1100. [Google Scholar] [CrossRef]
- Bennett, N.R.; Coventry, B.; Goreshnik, I.; Huang, B.; Allen, A.; Vafeados, D.; Peng, Y.P.; Dauparas, J.; Baek, M.; Stewart, L.; et al. Improving de novo protein binder design with deep learning. Nat. Commun. 2023, 14, 2625. [Google Scholar] [CrossRef] [PubMed]
- Kortemme, T. De novo protein design-From new structures to programmable functions. Cell 2024, 187, 526–544. [Google Scholar] [CrossRef]
- Di Rienzo, L.; Milanetti, E.; Testi, C.; Montemiglio, L.C.; Baiocco, P.; Boffi, A.; Ruocco, G. A novel strategy for molecular interfaces optimization: The case of Ferritin-Transferrin receptor interaction. Comput. Struct. Biotechnol. J. 2020, 18, 2678–2686. [Google Scholar] [CrossRef]
- Gainza, P.; Wehrle, S.; Van Hall-Beauvais, A.; Marchand, A.; Scheck, A.; Harteveld, Z.; Buckley, S.; Ni, D.; Tan, S.; Sverrisson, F.; et al. De novo design of protein interactions with learned surface fingerprints. Nature 2023, 617, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, S.; Mittal, N.; Bhat, A.; Adiga, R.S.; Ganesan, A.; Nagarajan, D.; Varadarajan, R. Improved Prediction of Stabilizing Mutations in Proteins by Incorporation of Mutational Effects on Ligand Binding. Proteins 2024. online ahead of print. [Google Scholar] [CrossRef]
- Zheng, F.; Liu, Y.; Yang, Y.; Wen, Y.; Li, M. Assessing computational tools for predicting protein stability changes upon missense mutations using a new dataset. Protein Sci. 2024, 33, e4861. [Google Scholar] [CrossRef]
- Blaabjerg, L.M.; Kassem, M.M.; Good, L.L.; Jonsson, N.; Cagiada, M.; Johansson, K.E.; Boomsma, W.; Stein, A.; Lindorff-Larsen, K. Rapid protein stability prediction using deep learning representations. Elife 2023, 12, e82593. [Google Scholar] [CrossRef] [PubMed]
- Musil, M.; Stourac, J.; Bendl, J.; Brezovsky, J.; Prokop, Z.; Zendulka, J.; Martinek, T.; Bednar, D.; Damborsky, J. FireProt: Web server for automated design of thermostable proteins. Nucleic Acids Res. 2017, 45, W393–W399. [Google Scholar] [CrossRef] [PubMed]
- Musil, M.; Jezik, A.; Horackova, J.; Borko, S.; Kabourek, P.; Damborsky, J.; Bednar, D. FireProt 2.0: Web-based platform for the fully automated design of thermostable proteins. Brief. Bioinform. 2023, 25, bbad425. [Google Scholar] [CrossRef]
- Gonzalez, N.A.; Li, B.A.; McCully, M.E. The stability and dynamics of computationally designed proteins. Protein Eng. Des. Sel. 2022, 35, gzac001. [Google Scholar] [CrossRef] [PubMed]
- Thomson, R.E.S.; Carrera-Pacheco, S.E.; Gillam, E.M.J. Engineering functional thermostable proteins using ancestral sequence reconstruction. J. Biol. Chem. 2022, 298, 102435. [Google Scholar] [CrossRef]
- Sumida, K.H.; Nunez-Franco, R.; Kalvet, I.; Pellock, S.J.; Wicky, B.I.M.; Milles, L.F.; Dauparas, J.; Wang, J.; Kipnis, Y.; Jameson, N.; et al. Improving Protein Expression, Stability, and Function with ProteinMPNN. J. Am. Chem. Soc. 2024, 146, 2054–2061. [Google Scholar] [CrossRef] [PubMed]
- Ni, D.; Liu, Y.; Kong, R.; Yu, Z.; Lu, S.; Zhang, J. Computational elucidation of allosteric communication in proteins for allosteric drug design. Drug Discov. Today 2022, 27, 2226–2234. [Google Scholar] [CrossRef]
- Verkhivker, G.M.; Agajanian, S.; Hu, G.; Tao, P. Allosteric Regulation at the Crossroads of New Technologies: Multiscale Modeling, Networks, and Machine Learning. Front. Mol. Biosci. 2020, 7, 136. [Google Scholar] [CrossRef]
- Sheik Amamuddy, O.; Veldman, W.; Manyumwa, C.; Khairallah, A.; Agajanian, S.; Oluyemi, O.; Verkhivker, G.; Tastan Bishop, O. Integrated Computational Approaches and Tools forAllosteric Drug Discovery. Int. J. Mol. Sci. 2020, 21, 847. [Google Scholar] [CrossRef]
- Chen, J.; Vishweshwaraiah, Y.L.; Dokholyan, N.V. Design and engineering of allosteric communications in proteins. Curr. Opin. Struct. Biol. 2022, 73, 102334. [Google Scholar] [CrossRef]
- Ebrahimi, S.B.; Samanta, D. Engineering protein-based therapeutics through structural and chemical design. Nat. Commun. 2023, 14, 2411. [Google Scholar] [CrossRef] [PubMed]
- Alvisi, N.; de Vries, R. Biomedical applications of solid-binding peptides and proteins. Mater. Today Bio 2023, 19, 100580. [Google Scholar] [CrossRef] [PubMed]
- Vymetal, J.; Mertova, K.; Bousova, K.; Sulc, J.; Tripsianes, K.; Vondrasek, J. Fusion of two unrelated protein domains in a chimera protein and its 3D prediction: Justification of the x-ray reference structures as a prediction benchmark. Proteins 2022, 90, 2067–2079. [Google Scholar] [CrossRef] [PubMed]
- Truong, K.; Ikura, M. Domain fusion analysis by applying relational algebra to protein sequence and domain databases. BMC Bioinform. 2003, 4, 16. [Google Scholar] [CrossRef]
- Chen, X.; Zaro, J.L.; Shen, W.C. Fusion protein linkers: Property, design and functionality. Adv. Drug Deliv. Rev. 2013, 65, 1357–1369. [Google Scholar] [CrossRef]
- Xia, Y.; Zhao, K.; Liu, D.; Zhou, X.; Zhang, G. Multi-domain and complex protein structure prediction using inter-domain interactions from deep learning. Commun. Biol. 2023, 6, 1221. [Google Scholar] [CrossRef]
- Ferruz, N.; Noske, J.; Hocker, B. Protlego: A Python package for the analysis and design of chimeric proteins. Bioinformatics 2021, 37, 3182–3189. [Google Scholar] [CrossRef]
- Garcia-Paz, F.M.; Del Moral, S.; Morales-Arrieta, S.; Ayala, M.; Trevino-Quintanilla, L.G.; Olvera-Carranza, C. Multidomain chimeric enzymes as a promising alternative for biocatalysts improvement: A minireview. Mol. Biol. Rep. 2024, 51, 410. [Google Scholar] [CrossRef]
- Norman, R.A.; Ambrosetti, F.; Bonvin, A.; Colwell, L.J.; Kelm, S.; Kumar, S.; Krawczyk, K. Computational approaches to therapeutic antibody design: Established methods and emerging trends. Brief. Bioinform. 2020, 21, 1549–1567. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; McFee, M.; Fang, Q.; Abdin, O.; Kim, P.M. Computational and artificial intelligence-based methods for antibody development. Trends Pharmacol. Sci. 2023, 44, 175–189. [Google Scholar] [CrossRef]
- Madsen, A.V.; Mejias-Gomez, O.; Pedersen, L.E.; Preben Morth, J.; Kristensen, P.; Jenkins, T.P.; Goletz, S. Structural trends in antibody-antigen binding interfaces: A computational analysis of 1833 experimentally determined 3D structures. Comput. Struct. Biotechnol. J. 2024, 23, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, M.; Ruffolo, J.A.; Haskins, N.; Iannotti, M.; Vozza, G.; Pham, T.; Mehzabeen, N.; Shandilya, H.; Rickert, K.; Croasdale-Wood, R.; et al. Toward enhancement of antibody thermostability and affinity by computational design in the absence of antigen. MAbs 2024, 16, 2362775. [Google Scholar] [CrossRef] [PubMed]
- Kaczmarski, J.A.; Mitchell, J.A.; Spence, M.A.; Vongsouthi, V.; Jackson, C.J. Structural and evolutionary approaches to the design and optimization of fluorescence-based small molecule biosensors. Curr. Opin. Struct. Biol. 2019, 57, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Quijano-Rubio, A.; Yeh, H.W.; Park, J.; Lee, H.; Langan, R.A.; Boyken, S.E.; Lajoie, M.J.; Cao, L.; Chow, C.M.; Miranda, M.C.; et al. De novo design of modular and tunable protein biosensors. Nature 2021, 591, 482–487. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, Y.; Wang, M. Design, Optimization and Application of Small Molecule Biosensor in Metabolic Engineering. Front. Microbiol. 2017, 8, 2012. [Google Scholar] [CrossRef]
- Singh, A.; Sharma, A.; Ahmed, A.; Sundramoorthy, A.K.; Furukawa, H.; Arya, S.; Khosla, A. Recent Advances in Electrochemical Biosensors: Applications, Challenges, and Future Scope. Biosensors 2021, 11, 336. [Google Scholar] [CrossRef]
- Naresh, V.; Lee, N. A Review on Biosensors and Recent Development of Nanostructured Materials-Enabled Biosensors. Sensors 2021, 21, 1109. [Google Scholar] [CrossRef]
- Pham, C.; Stogios, P.J.; Savchenko, A.; Mahadevan, R. Computation-guided transcription factor biosensor specificity engineering for adipic acid detection. Comput. Struct. Biotechnol. J. 2024, 23, 2211–2219. [Google Scholar] [CrossRef]
- Markus, B.; Andreas, K.; Arkadij, K.; Stefan, L.; Gustav, O.; Elina, S.; Radka, S. Accelerating Biocatalysis Discovery with Machine Learning: A Paradigm Shift in Enzyme Engineering, Discovery, and Design. ACS Catal. 2023, 13, 14454–14469. [Google Scholar] [CrossRef] [PubMed]
- Bell, E.L.; Finnigan, W.; France, S.P.; Green, A.P.; Hayes, M.A.; Hepworth, L.J.; Lovelock, S.L.; Niikura, H.; Osuna, S.; Romero, E.; et al. Biocatalysis. Nat. Rev. Methods Primers 2021, 1, 46. [Google Scholar] [CrossRef]
- Radley, E.; Davidson, J.; Foster, J.; Obexer, R.; Bell, E.L.; Green, A.P. Engineering Enzymes for Environmental Sustainability. Angew. Chem. Weinheim Bergstr. Ger. 2023, 135, e202309305. [Google Scholar] [CrossRef]
- Qiu, J.; Chen, Y.; Zhang, L.; Wu, J.; Zeng, X.; Shi, X.; Liu, L.; Chen, J. A comprehensive review on enzymatic biodegradation of polyethylene terephthalate. Environ. Res. 2024, 240, 117427. [Google Scholar] [CrossRef]
- Mesbah, N.M. Industrial Biotechnology Based on Enzymes From Extreme Environments. Front. Bioeng. Biotechnol. 2022, 10, 870083. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, X.; Chen, X.; Huang, J.; Wang, C.; Wang, J.; Wang, Z. Accelerating therapeutic protein design with computational approaches toward the clinical stage. Comput. Struct. Biotechnol. J. 2023, 21, 2909–2926. [Google Scholar] [CrossRef]
- Ewaisha, R.; Anderson, K.S. Immunogenicity of CRISPR therapeutics-Critical considerations for clinical translation. Front. Bioeng. Biotechnol. 2023, 11, 1138596. [Google Scholar] [CrossRef]
- Harris, C.T.; Cohen, S. Reducing Immunogenicity by Design: Approaches to Minimize Immunogenicity of Monoclonal Antibodies. BioDrugs 2024, 38, 205–226. [Google Scholar] [CrossRef]
- Yin, R.; Ribeiro-Filho, H.V.; Lin, V.; Gowthaman, R.; Cheung, M.; Pierce, B.G. TCRmodel2: High-resolution modeling of T cell receptor recognition using deep learning. Nucleic Acids Res. 2023, 51, W569–W576. [Google Scholar] [CrossRef]
- Sidhom, J.W.; Larman, H.B.; Pardoll, D.M.; Baras, A.S. DeepTCR is a deep learning framework for revealing sequence concepts within T-cell repertoires. Nat. Commun. 2021, 12, 1605. [Google Scholar] [CrossRef]
- Katayama, Y.; Yokota, R.; Akiyama, T.; Kobayashi, T.J. Machine Learning Approaches to TCR Repertoire Analysis. Front. Immunol. 2022, 13, 858057. [Google Scholar] [CrossRef]
- Leary, A.Y.; Scott, D.; Gupta, N.T.; Waite, J.C.; Skokos, D.; Atwal, G.S.; Hawkins, P.G. Designing meaningful continuous representations of T cell receptor sequences with deep generative models. Nat. Commun. 2024, 15, 4271. [Google Scholar] [CrossRef] [PubMed]
- Ingolfsson, H.I.; Bhatia, H.; Aydin, F.; Oppelstrup, T.; Lopez, C.A.; Stanton, L.G.; Carpenter, T.S.; Wong, S.; Di Natale, F.; Zhang, X.; et al. Machine Learning-Driven Multiscale Modeling: Bridging the Scales with a Next-Generation Simulation Infrastructure. J. Chem. Theory Comput. 2023, 19, 2658–2675. [Google Scholar] [CrossRef]
- Qiu, Y.; Wei, G.W. Artificial intelligence-aided protein engineering: From topological data analysis to deep protein language models. Brief. Bioinform. 2023, 24, bbad289. [Google Scholar] [CrossRef] [PubMed]
- Poleto, M.D.; Lemkul, J.A. Integration of Experimental Data and Use of Automated Fitting Methods in Developing Protein Force Fields. Commun. Chem. 2022, 5, 38. [Google Scholar] [CrossRef] [PubMed]
- Kamenik, A.S.; Handle, P.H.; Hofer, F.; Kahler, U.; Kraml, J.; Liedl, K.R. Polarizable and non-polarizable force fields: Protein folding, unfolding, and misfolding. J. Chem. Phys. 2020, 153, 185102. [Google Scholar] [CrossRef]
- Lopes, P.E.; Guvench, O.; MacKerell, A.D., Jr. Current status of protein force fields for molecular dynamics simulations. Methods Mol. Biol. 2015, 1215, 47–71. [Google Scholar] [CrossRef]
- Bamezai, S.; Maresca di Serracapriola, G.; Morris, F.; Hildebrandt, R.; Amil, M.A.S.; Sporadicate iGEM Team; Xin, H.; Montogomery-Johnson, J.; Bi, Y.; Ding, Y.; et al. Protein engineering in the computational age: An open source framework for exploring mutational landscapes in silico. Eng. Biol. 2023, 7, 29–38. [Google Scholar] [CrossRef]
- Barrozo, A.; Borstnar, R.; Marloie, G.; Kamerlin, S.C. Computational protein engineering: Bridging the gap between rational design and laboratory evolution. Int. J. Mol. Sci. 2012, 13, 12428–12460. [Google Scholar] [CrossRef]
- Verma, R.; Schwaneberg, U.; Roccatano, D. Computer-Aided Protein Directed Evolution: A Review of Web Servers, Databases and other Computational Tools for Protein Engineering. Comput. Struct. Biotechnol. J. 2012, 2, e201209008. [Google Scholar] [CrossRef]
- Carobene, A.; Padoan, A.; Cabitza, F.; Banfi, G.; Plebani, M. Rising adoption of artificial intelligence in scientific publishing: Evaluating the role, risks, and ethical implications in paper drafting and review process. Clin. Chem. Lab. Med. 2024, 62, 835–843. [Google Scholar] [CrossRef]
- Kargl, M.; Plass, M.; Muller, H. A Literature Review on Ethics for AI in Biomedical Research and Biobanking. Yearb. Med. Inform. 2022, 31, 152–160. [Google Scholar] [CrossRef]
- Holzinger, A.; Keiblinger, K.; Holub, P.; Zatloukal, K.; Muller, H. AI for life: Trends in artificial intelligence for biotechnology. N. Biotechnol. 2023, 74, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Resnik, D.B.; Hosseini, M. The ethics of using artificial intelligence in scientific research: New guidance needed for a new tool. AI Ethics 2024. [Google Scholar] [CrossRef]
- Maccaro, A.; Stokes, K.; Statham, L.; He, L.; Williams, A.; Pecchia, L.; Piaggio, D. Clearing the Fog: A Scoping Literature Review on the Ethical Issues Surrounding Artificial Intelligence-Based Medical Devices. J. Pers. Med. 2024, 14, 443. [Google Scholar] [CrossRef] [PubMed]
- Kohyama, S.; Frohn, B.P.; Babl, L.; Schwille, P. Machine learning-aided design and screening of an emergent protein function in synthetic cells. Nat. Commun. 2024, 15, 2010. [Google Scholar] [CrossRef] [PubMed]
- Yue, K.; Chen, J.; Li, Y.; Kai, L. Advancing synthetic biology through cell-free protein synthesis. Comput. Struct. Biotechnol. J. 2023, 21, 2899–2908. [Google Scholar] [CrossRef] [PubMed]
- Levin, D.B.; Budisa, N. Synthetic biology encompasses metagenomics, ecosystems, and biodiversity sustainability within its scope. Front. Synth. Biol. 2023, 1, 1255472. [Google Scholar] [CrossRef]
- Yamagata, M. SynBio: A Journal for Advancing Solutions to Global Challenges. SynBio 2023, 1, 190–193. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Son, A.; Park, J.; Kim, W.; Yoon, Y.; Lee, S.; Park, Y.; Kim, H. Revolutionizing Molecular Design for Innovative Therapeutic Applications through Artificial Intelligence. Molecules 2024, 29, 4626. https://doi.org/10.3390/molecules29194626
Son A, Park J, Kim W, Yoon Y, Lee S, Park Y, Kim H. Revolutionizing Molecular Design for Innovative Therapeutic Applications through Artificial Intelligence. Molecules. 2024; 29(19):4626. https://doi.org/10.3390/molecules29194626
Chicago/Turabian StyleSon, Ahrum, Jongham Park, Woojin Kim, Yoonki Yoon, Sangwoon Lee, Yongho Park, and Hyunsoo Kim. 2024. "Revolutionizing Molecular Design for Innovative Therapeutic Applications through Artificial Intelligence" Molecules 29, no. 19: 4626. https://doi.org/10.3390/molecules29194626
APA StyleSon, A., Park, J., Kim, W., Yoon, Y., Lee, S., Park, Y., & Kim, H. (2024). Revolutionizing Molecular Design for Innovative Therapeutic Applications through Artificial Intelligence. Molecules, 29(19), 4626. https://doi.org/10.3390/molecules29194626