
Curcumin-Based Molecularly Imprinted Polymer Electropolymerized on Single-Use Graphite Electrode for Dipyridamole Analysis

 , , , and
, , , and 
Abstract
:1. Introduction
2. Results
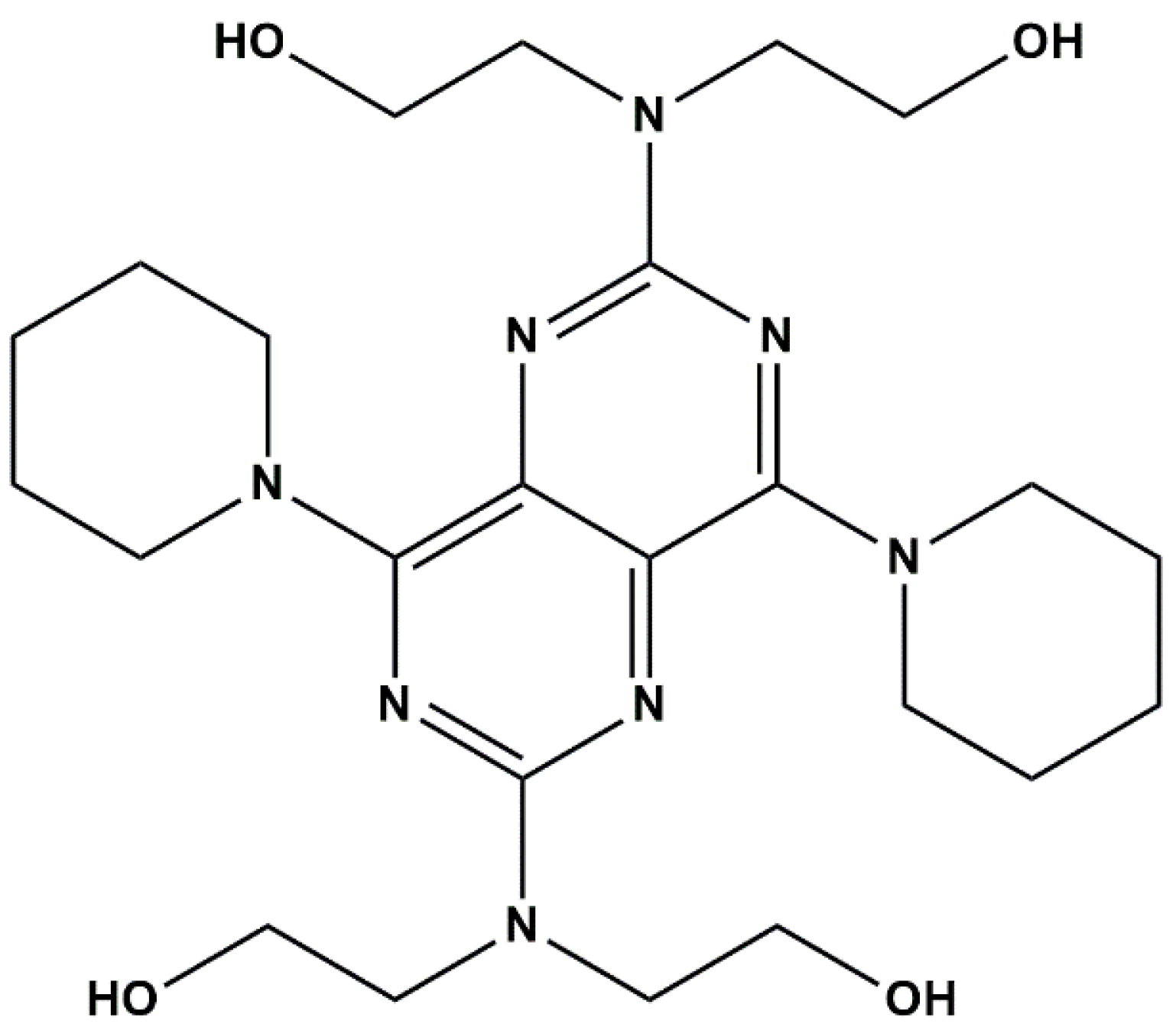
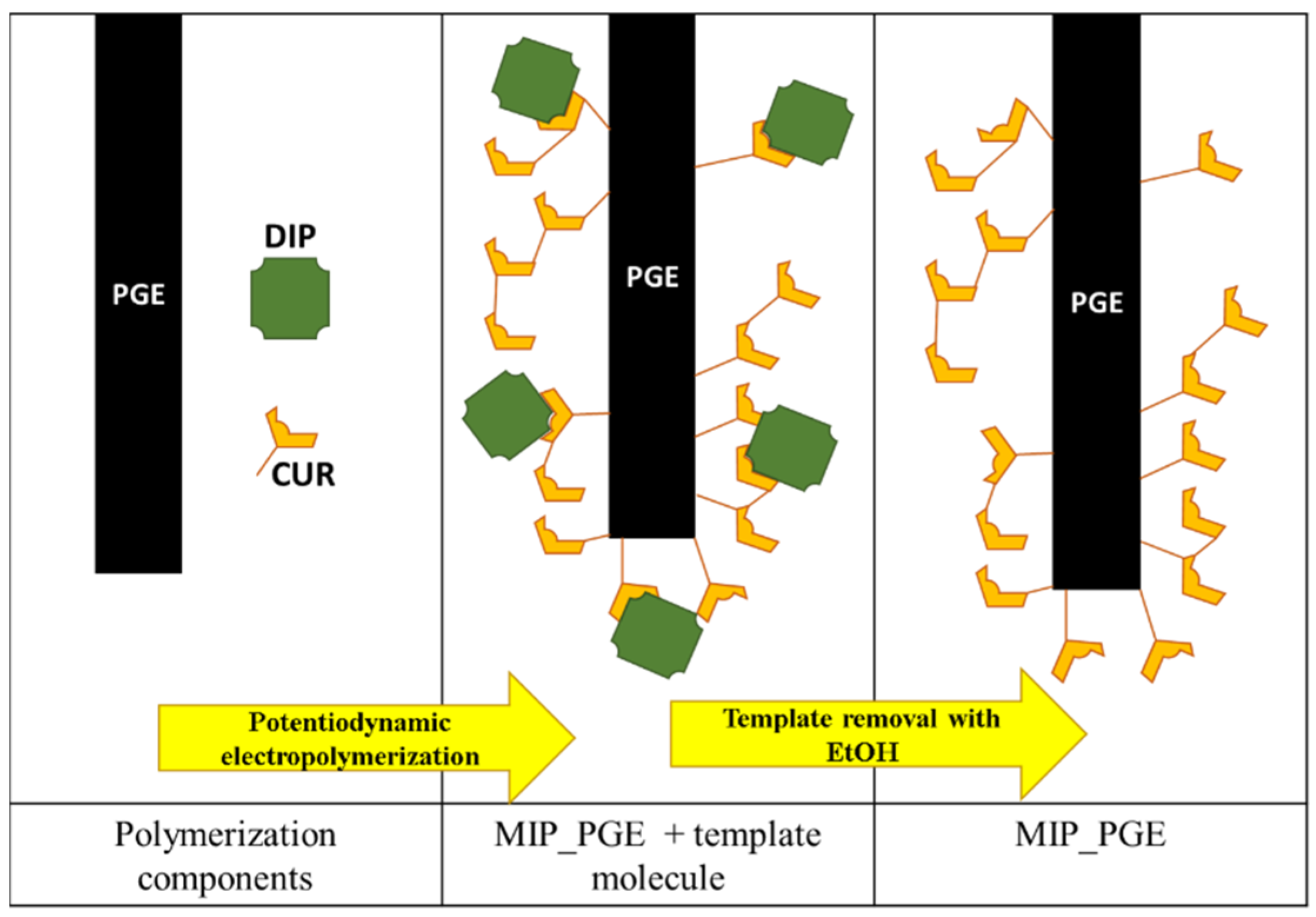
2.1. PGE Coating by Electrogenerated Polymeric Film
2.1.1. MIP Preparation—Experimental Conditions
- The influence of the supporting electrolyte employed in the electro-polymerization process
- The influence of the CUR/DIP ratio employed in the electro-polymerization solution
- The influence of the number of cyclic voltammetric scans involved in the electro-polymerization process
- The influence of scan rate applied in the e lectro-polymerization process
2.1.2. Template Removal Conditions
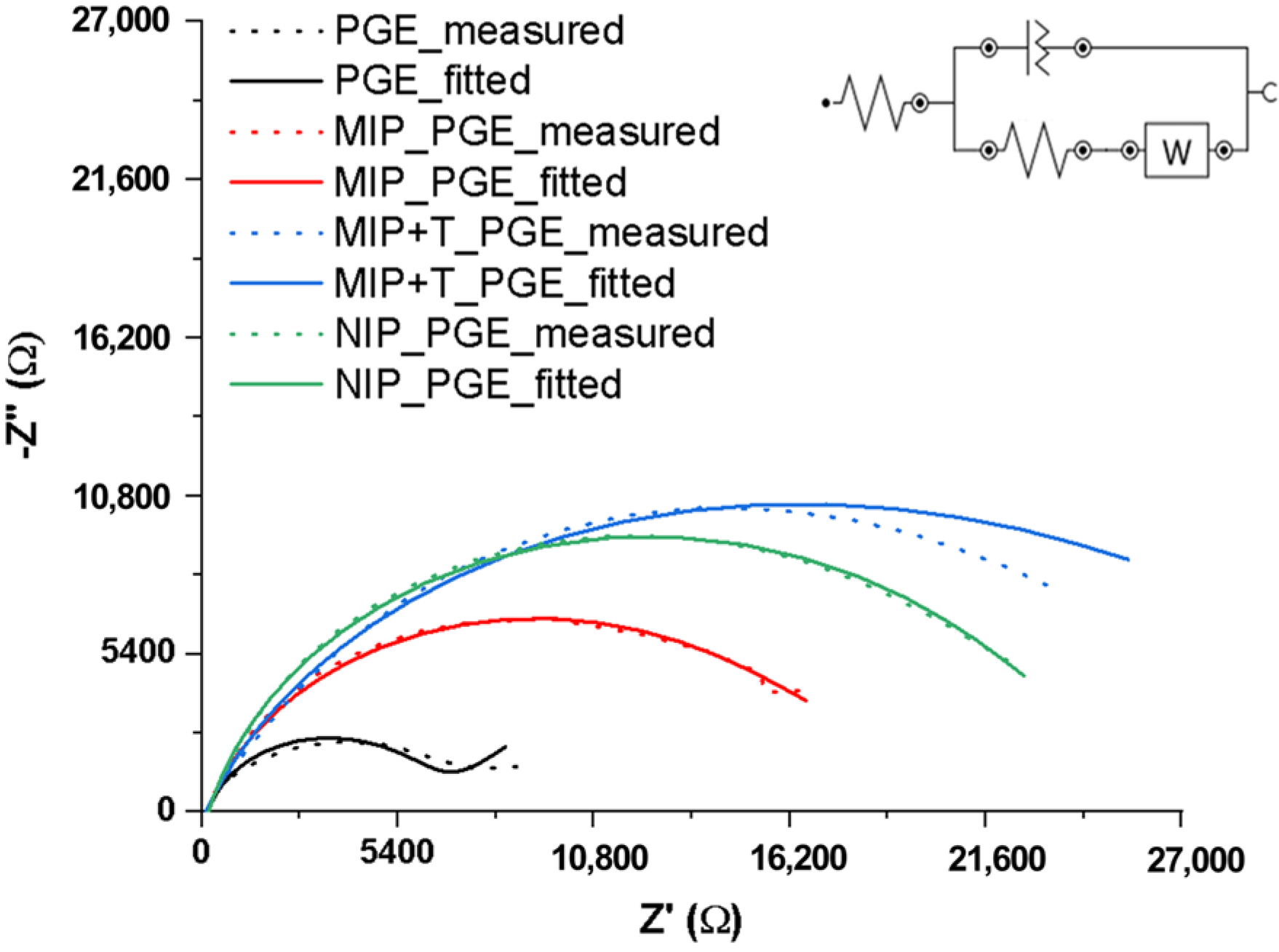
2.2. Characterization of the Electrode Surface
2.3. DIP Voltammetric Analysis at MIP_PGE
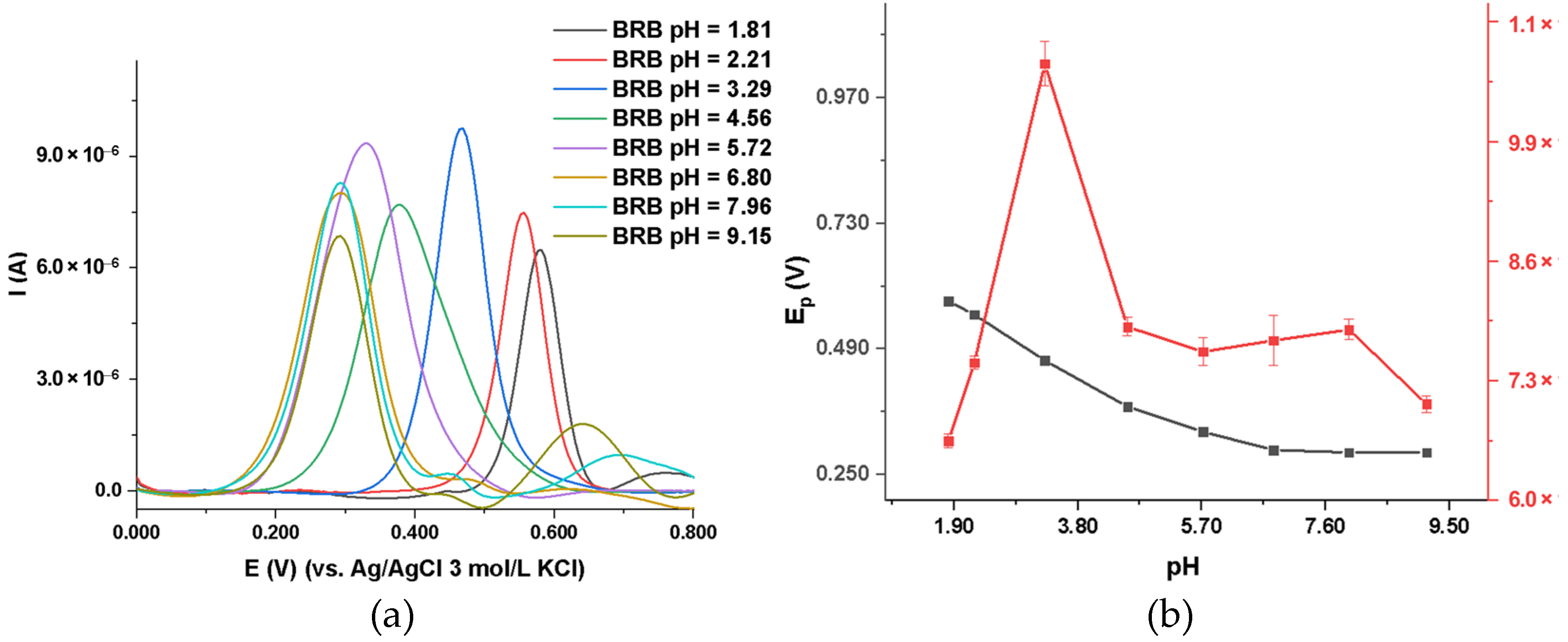
2.3.1. DIP Voltammetric Behavior at MIP_PGE in Supporting Electrolytes with Different pH Values
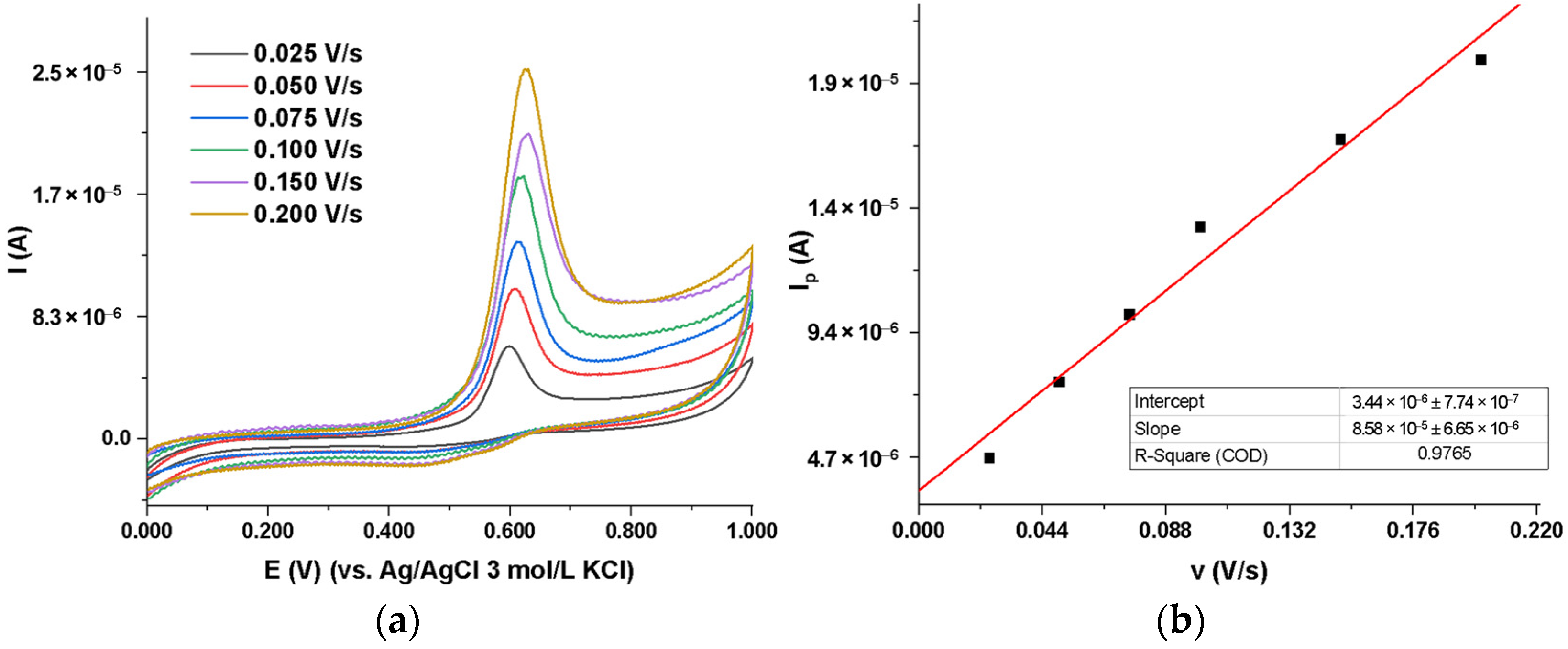
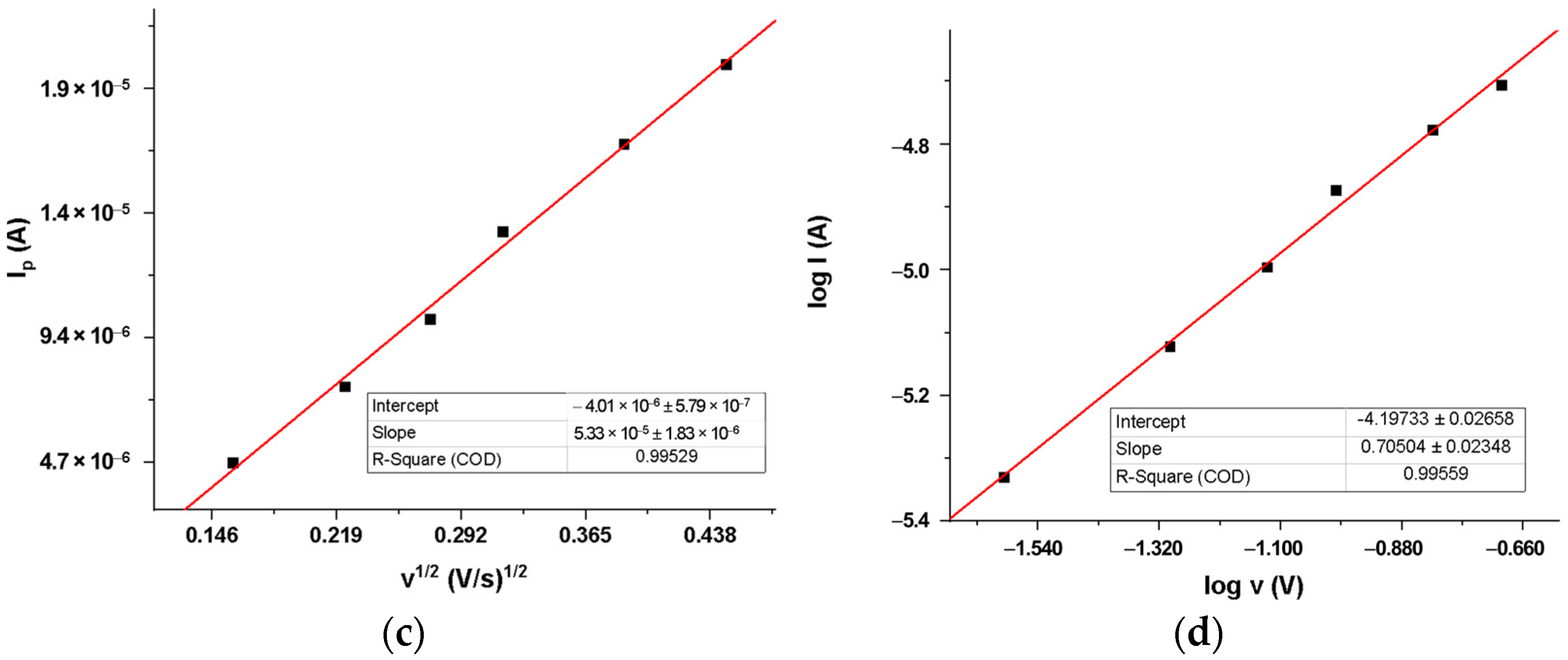
2.3.2. DIP Voltammetric Behavior at Different Potential Scan Rates at MIP_PGE
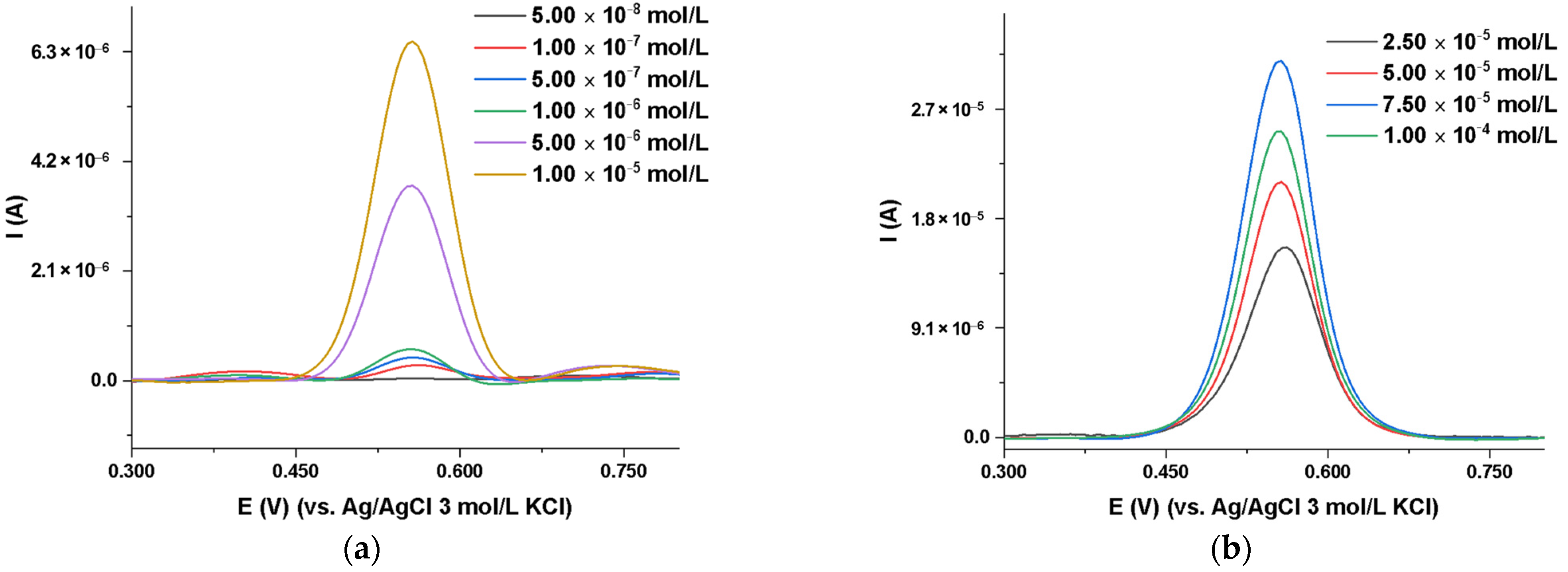
2.3.3. MIP_PGE Voltammetric Response to DIP Concentration
2.3.4. Performance Characteristics of the Developed Voltammetric Methods at MIP_PGE
2.3.5. Reproducibility
2.3.6. Stability
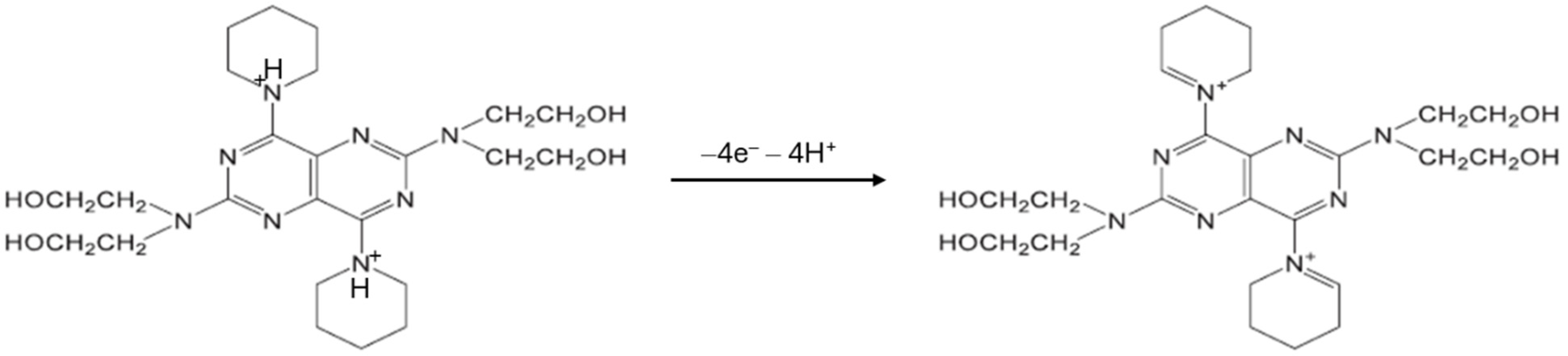
2.3.7. Determination of the Imprinting Factor
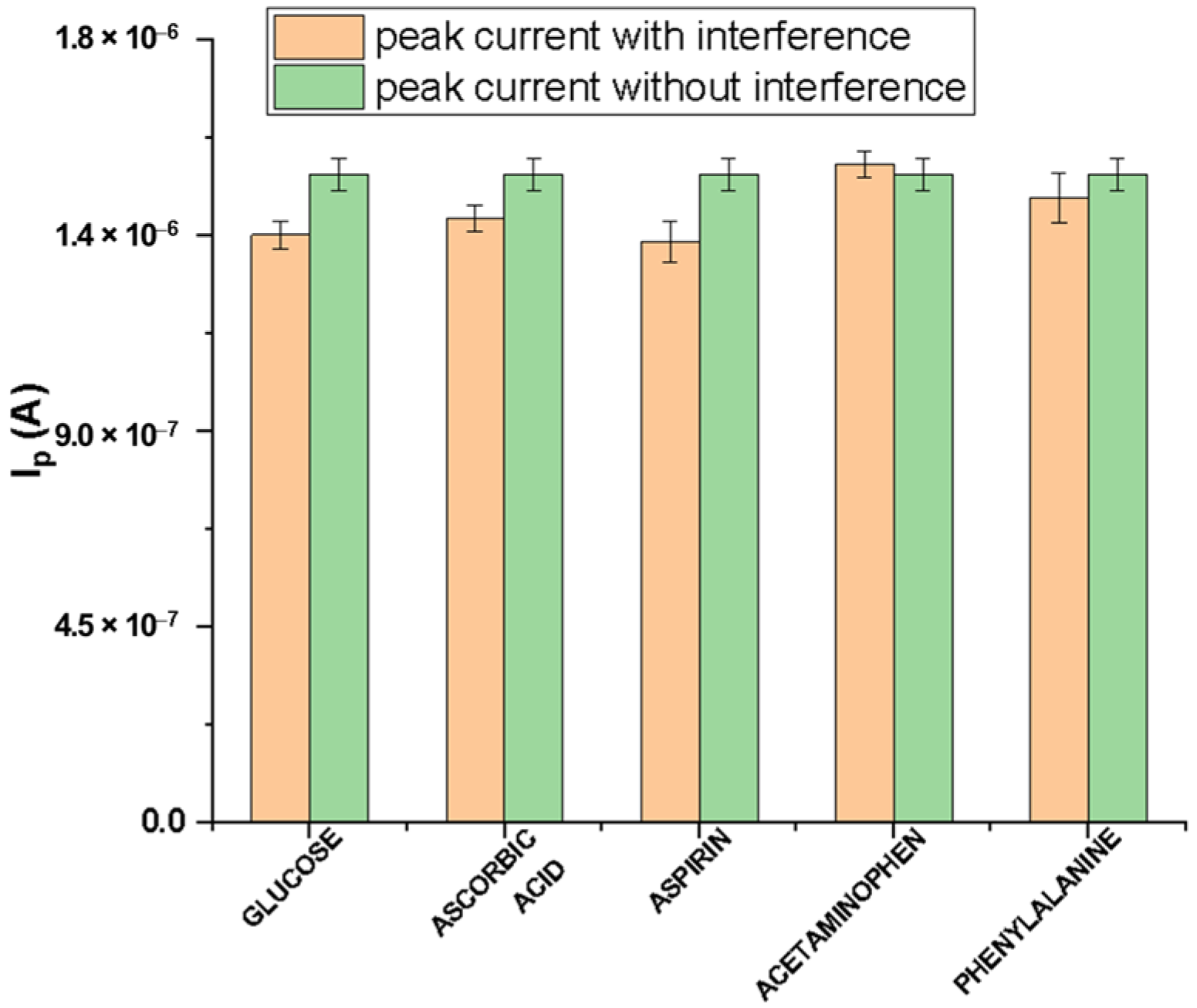
2.3.8. Interferences
2.3.9. DIP Quantification in Tablets/Tap Water Samples Using the MIP-PGE
3. Experimental
3.1. Reagents
3.2. Solutions
3.3. Instrumentation
3.4. Procedures
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Allahham, M.; Lerman, A.; Atar, D.; Birnbaum, Y. Why Not Dipyridamole: A Review of Current Guidelines and Re-Evaluation of Utility in the Modern Era. Cardiovasc. Drugs Ther. 2022, 1, 3. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Zhang, N.Q.; Xu, C.J.; Huang, W.Q.; Li, D.X.; Li, J.; Yao, L.L.; Sundquist, K.; Sundquist, J.; Jiang, S.H.; et al. Dipyridamole Enhances the Anti-Cancer Ability of Aspirin against Colorectal Cancer by Inducing Apoptosis in an Unfolded Protein Response-Dependent Manner. Cell. Oncol. 2023, 46, 953–967. [Google Scholar] [CrossRef] [PubMed]
- Irie, N.; Mizoguchi, K.; Warita, T.; Nakano, M.; Sasaki, K.; Tashiro, J.; Osaki, T.; Ishikawa, T.; Oltvai, Z.N.; Warita, K. Repurposing of the Cardiovascular Drug Statin for the Treatment of Cancers: Efficacy of Statin–Dipyridamole Combination Treatment in Melanoma Cell Lines. Biomedicines 2024, 12, 698. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Shu, L.; Zheng, J. Effects of Flunarizine Combined with Ginkgo Leaf Extract and Dipyridamole Injection on Hemorheology in Elderly Patients with Vertigo. Pak. J. Med. Sci. 2024, 40, 337. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-D.; Huang, W.-C.; Peng, N.-J.; Hu, C. Dipyridamole-Induced Adverse Effects in Myocardial Perfusion Scans: Dynamic Evaluation. IJC Heart Vasc. 2017, 14, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Li, C. Electrochemical Determination of Dipyridamole at a Carbon Paste Electrode Using Cetyltrimethyl Ammonium Bromide as Enhancing Element. Colloids Surf. B Biointerfaces 2007, 55, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Yi, D.; Dong, Y.; Yao, Y.; Hong, M.; Zhu, B.; Ren, G.B.; Qi, M.H. Enhancement of Solubility and Dissolution Rate of Dipyridamole by Salifying: Preparation, Characterization, and Theoretical Calculation. J. Mol. Struct. 2024, 1296, 136838. [Google Scholar] [CrossRef]
- Thangaraj, M.; Kuber, R. UPLC-Q-TOF-MS Method Development and Validation for Simultaneous Analysis of Dipyridamole and Its Related Impurities. J. Appl. Pharm. Sci. 2023, 13, 201–211. [Google Scholar] [CrossRef]
- Hammud, H.H.; El Yazbi, F.A.; Mahrous, M.E.; Sonji, G.M.; Sonji, N.M. Stability-Indicating Spectrofluorimetric and RP-HPLC Methods for the Determination of Aspirin and Dipyridamole in Their Combination~!2008-02-12~!2008-04-14~!2008-06-12~! Open Spectrosc. J. 2008, 2, 19–28. [Google Scholar] [CrossRef]
- Zhou, L.; Ping, Q.; Yang, L. HPLC Determination of Dipyridamole, Aspirin and Salicylic Acid in Compound Preparation. Chin. J. Pharm. Anal. 2003, 23, 199–201. [Google Scholar]
- De Toledo, R.A.; Castilho, M.; Mazo, L.H. Determination of Dipyridamole in Pharmaceutical Preparations Using Square Wave Voltammetry. J. Pharm. Biomed. Anal. 2005, 36, 1113–1117. [Google Scholar] [CrossRef] [PubMed]
- Barghash, S.; Abd El-Razeq, S.; El -Awady, M.; Belal, F. Validated Spectrophotometric Method for Analysis of Dipyridamole and Lamivudine Using Eosin Y. Azhar Int. J. Pharm. Med. Sci. 2021, 1, 92–101. [Google Scholar] [CrossRef]
- Yari, A.; Shams, A. Fabrication of a Renewable Syringe Carbon Paste Electrode with a Motor Rotary Blade and Using for Voltammetric Measurement of Dipyridamole. J. Iran. Chem. Soc. 2018, 15, 1861–1869. [Google Scholar] [CrossRef]
- Salinas-Castillo, A.; Carretero, A.S.; Fernández-Gutiérrez, A. Sensitive and Simple Determination of the Vasodilator Agent Dipyridamole in Pharmaceutical Preparations by Phosphorimetry. Anal. Bioanal. Chem. 2003, 376, 1111–1114. [Google Scholar] [CrossRef]
- Wang, L.; Tang, Y. Determination of Dipyridamole Using TCPO-H2O2 Chemiluminescence in the Presence of Silver Nanoparticles. Luminescence 2011, 26, 703–709. [Google Scholar] [CrossRef]
- Ankitha, M.; Shabana, N.; Rasheed, P.A. A Novel ReS2–Nb2CTx Composite as a Sensing Platform for Ultrasensitive and Selective Electrochemical Detection of Dipyramidole from Human Serum. Graphene 2D Mater. 2022, 8, 27–37. [Google Scholar] [CrossRef]
- Liu, X.; Zhong, J.; Rao, H.; Lu, Z.; Ge, H.; Chen, B.; Zou, P.; Wang, X.; He, H.; Zeng, X.; et al. Electrochemical Dipyridamole Sensor Based on Molecularly Imprinted Polymer on Electrode Modified with Fe3O4@Au/Amine-Multi-Walled Carbon Nanotubes. J. Solid State Electrochem. 2017, 21, 3071–3082. [Google Scholar] [CrossRef]
- Javanbakht, M.; Fathollahi, F.; Divsar, F.; Ganjali, M.R.; Norouzi, P. A Selective and Sensitive Voltammetric Sensor Based on Molecularly Imprinted Polymer for the Determination of Dipyridamole in Pharmaceuticals and Biological Fluids. Sens. Actuators B Chem. 2013, 182, 362–367. [Google Scholar] [CrossRef]
- Meng, F.; Duan, M.; Wu, W.; Shao, S.; Qin, Y.; Zhang, M. Enzymatic Construction Au NPs-RGO Based MIP Electrochemical Sensor for Adulteration Detection of Bovine-Derived Allergen in Camel Milk. Food Chem. 2024, 436, 137638. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Qi, X.; Wu, J.; Wan, X.; Wang, T.; Liu, Y.; Chen, Y.; Xia, Y. Highly Stable Electrochemical Sensing Platform for the Selective Determination of Pefloxacin in Food Samples Based on a Molecularly Imprinted-Polymer-Coated Gold Nanoparticle/Black Phosphorus Nanocomposite. Food Chem. 2024, 436, 137753. [Google Scholar] [CrossRef]
- Zhou, B.; Sheng, X.; Cao, J.; Xie, H.; Li, X.; Huang, L.; Yang, M.; Zhong, M.; Liu, Y.N. A Novel Electrochemical Sensor Based on Dual-Functional MMIP-CuMOFs for Both Target Recognition and Signal Reporting and Its Application for Sensing Bisphenol A in Milk. Food Chem. 2024, 437, 137756. [Google Scholar] [CrossRef]
- Geng, L.; Sun, J.; Liu, M.; Huang, J.; Dong, J.; Guo, Z.; Guo, Y.; Sun, X. Molecularly Imprinted Polymers-Aptamer Electrochemical Sensor Based on Dual Recognition Strategy for High Sensitivity Detection of Chloramphenicol. Food Chem. 2024, 437, 137933. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Gao, R.; Sun, Y.; Waterhouse, G.I.N.; Qiao, X.; Xu, Z. Development of ZrO(OH)2@HCS Co-Modified Molecularly Imprinted Gel-Based Electrochemical Sensing Platform for Sensitive and Selective Detection of Tert-Butylhydroquinone in Foods. Food Chem. 2024, 460, 140600. [Google Scholar] [CrossRef] [PubMed]
- Xue, S.; Zou, J.; Li, J.; Xu, J.; Chen, H.; Wang, L.; Gao, Y.; Duan, X.; Lu, L. Electrochemical Detection of Carbendazim Using Molecularly Imprinted Poly(3,4-Ethylenedioxythiophene) on Co,N Co-Doped Hollow Carbon Nanocage@CNTs-Modified Electrode. Food Chem. 2024, 456, 140063. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Han, S.; Zong, W.; Chu, H.; Zhang, X.; Jiang, H. Ultrasensitive Detection of Chlortetracycline in Animal-Origin Food Using Molecularly Imprinted Electrochemical Sensor Based on SnS2/ZnCo-MOF and AuNPs. Food Chem. 2024, 452, 139537. [Google Scholar] [CrossRef] [PubMed]
- Xue, C.; Jamal, R.; Abdiryim, T.; Liu, X.; Liu, F.; Xu, F.; Cheng, Q.; Tang, X.; Fan, N. An Ionic Liquid-Modified PEDOT/Ti3C2TX Based Molecularly Imprinted Electrochemical Sensor for Pico-Molar Sensitive Detection of L-Tryptophan in Milk. Food Chem. 2024, 449, 139114. [Google Scholar] [CrossRef]
- Nazari, Z.; Hashemi, M.; Noshirvani, N.; Zohdijamil, Z. Magnetic Perlite Based Molecularly Imprinted Polymer on Screen Printed Carbon Electrode as a New Tyramine Electrochemical Sensor. Microchem. J. 2024, 196, 109539. [Google Scholar] [CrossRef]
- Frigoli, M.; Caldara, M.; Royakkers, J.; Lowdon, J.W.; Cleij, T.J.; Diliën, H.; Eersels, K.; van Grinsven, B. Gold Screen-Printed Electrodes Coupled with Molecularly Imprinted Conjugated Polymers for Ultrasensitive Detection of Streptomycin in Milk. Microchem. J. 2024, 200, 110433. [Google Scholar] [CrossRef]
- Karthika, P.; Shanmuganathan, S.; Subramanian, V.; Delerue-Matos, C. Selective Detection of Salivary Cortisol Using Screen-Printed Electrode Coated with Molecularly Imprinted Polymer. Talanta 2024, 272, 125823. [Google Scholar] [CrossRef]
- Gagliani, F.; Di Giulio, T.; Grecchi, S.; Benincori, T.; Arnaboldi, S.; Malitesta, C.; Mazzotta, E. Green Synthesis of a Molecularly Imprinted Polymer Based on a Novel Thiophene-Derivative for Electrochemical Sensing. Molecules 2024, 29, 1632. [Google Scholar] [CrossRef]
- Shama, N.A.; Aşır, S.; Göktürk, I.; Yılmaz, F.; Türkmen, D.; Denizli, A. Electrochemical Detection of Cortisol by Silver Nanoparticle-Modified Molecularly Imprinted Polymer-Coated Pencil Graphite Electrodes. ACS Omega 2023, 8, 29202–29212. [Google Scholar] [CrossRef] [PubMed]
- Yulianti, E.S.; Rahman, S.F.; Rizkinia, M.; Zakiyuddin, A. Low-Cost Electrochemical Biosensor Based on a Multi-Walled Carbon Nanotube-Doped Molecularly Imprinted Polymer for Uric Acid Detection. Arab. J. Chem. 2024, 17, 105692. [Google Scholar] [CrossRef]
- Medhat, P.M.; Mohamed Fouad, M.; Mahmoud, A.M.; Ghoniem, N.S.; Monir, H.H. Implementation of Quality by Design Approach for Optimization of the Green Voltammetric Analysis of a Brain Doping Agent (Piracetam) Using a Novel Molecular Imprinted Polymeric Sensor. Microchem. J. 2024, 205, 111347. [Google Scholar] [CrossRef]
- Yamani, H.Z.; El Azab, N.F. First Electropolymerized Carbidopa-Based Molecularly Imprinted Film: Disposable Electrochemical Sensor for Monitoring of Anti-COVID-19 Drug Favipiravir in Human Plasma. Microchem. J. 2024, 196, 109572. [Google Scholar] [CrossRef]
- Alahmad, W.; Cetinkaya, A.; Kaya, S.I.; Varanusupakul, P.; Ozkan, S.A. Molecularly Imprinted Polymer Paper-Based Analytical Devices for Biomarkers Detection. TrAC Trends Anal. Chem. 2024, 170, 117475. [Google Scholar] [CrossRef]
- Mostafiz, B.; Bigdeli, S.A.; Banan, K.; Afsharara, H.; Hatamabadi, D.; Mousavi, P.; Hussain, C.M.; Keçili, R.; Ghorbani-Bidkorbeh, F. Molecularly Imprinted Polymer-Carbon Paste Electrode (MIP-CPE)-Based Sensors for the Sensitive Detection of Organic and Inorganic Environmental Pollutants: A Review. Trends Environ. Anal. Chem. 2021, 32, e00144. [Google Scholar] [CrossRef]
- Afsharara, H.; Asadian, E.; Mostafiz, B.; Banan, K.; Bigdeli, S.A.; Hatamabadi, D.; Keshavarz, A.; Hussain, C.M.; Keçili, R.; Ghorbani-Bidkorpeh, F. Molecularly Imprinted Polymer-Modified Carbon Paste Electrodes (MIP-CPE): A Review on Sensitive Electrochemical Sensors for Pharmaceutical Determinations. TrAC Trends Anal. Chem. 2023, 160, 116949. [Google Scholar] [CrossRef]
- Preda, D.; David, I.G.; Popa, D.E.; Buleandra, M.; Radu, G.L. Recent Trends in the Development of Carbon-Based Electrodes Modified with Molecularly Imprinted Polymers for Antibiotic Electroanalysis. Chemosensors 2022, 10, 243. [Google Scholar] [CrossRef]
- Ramanavicius, S.; Ramanavicius, A. Development of Molecularly Imprinted Polymer Based Phase Boundaries for Sensors Design (Review). Adv. Colloid Interface Sci. 2022, 305, 102693. [Google Scholar] [CrossRef]
- Ovezova, M.; Yılmaz, F.; Göktürk, I.; Güler, K.Ç.; Denizli, A. Molecularly Imprinted Polymers for Pharmaceutical and BIomedical Analysis. J. Pharm. Biomed. Anal. Open 2024, 4, 100038. [Google Scholar] [CrossRef]
- Yan, X.; Almajidi, Y.Q.; Uinarni, H.; Bokov, D.O.; Mansouri, S.; Fenjan, M.N.; Saxena, A.; Zabibah, R.S.; Hamzah, H.F.; Oudah, S.K. Bio(Sensors) Based on Molecularly Imprinted Polymers and Silica Materials Used for Food Safety and Biomedical Analysis: Recent Trends and Future Prospects. Talanta 2024, 276, 126292. [Google Scholar] [CrossRef] [PubMed]
- Geleta, G.S. Recent Advances in Electrochemical Sensors Based on Molecularly Imprinted Polymers and Nanomaterials for Detection of Ascorbic Acid, Dopamine, and Uric Acid: A Review. Sens. Bio-Sens. Res. 2024, 43, 100610. [Google Scholar] [CrossRef]
- Hu, B.; Peng, L.; Liang, P.; Li, X.; Cai, M.; Liu, B.; Jia, Y.; Jing, Y.; Li, Z.; Sun, S. Advances in Molecularly Imprinted Polymers-Based Electrochemical Sensors for the Detection of Gonadal Steroid Hormones. TrAC Trends Anal. Chem. 2024, 171, 117485. [Google Scholar] [CrossRef]
- Mohammad Mayet, A.; Ebrahimi, S.; Shukhratovich Abdullaev, S.; Alsaab, H.O.; Mansouri, S.; Malviya, J.; Hussien Alawadi, A.; Alsaalamy, A.; Kadhem Abid, M.; Thakur, G. Molecularly Imprinted Polymers for Biosensing of Hormones in Food Safety and Biomedical Analysis: Progress and Perspectives. Mater. Today Chem. 2024, 35, 101899. [Google Scholar] [CrossRef]
- Sala, A.; Brisset, H.; Margaillan, A.; Mullot, J.U.; Branger, C. Electrochemical Sensors Modified with Ion-Imprinted Polymers for Metal Ion Detection. TrAC Trends Anal. Chem. 2022, 148, 116536. [Google Scholar] [CrossRef]
- Preda, D.; Jinga, M.L.; David, I.G.; Radu, G.L. Determination of Dipyridamole Using a MIP-Modified Disposable Pencil Graphite Electrode. Chemosensors 2023, 11, 400. [Google Scholar] [CrossRef]
- Bučević Popović, V.; Karahmet Farhat, E.; Banjari, I.; Jeličić Kadić, A.; Puljak, L. Bioavailability of Oral Curcumin in Systematic Reviews: A Methodological Study. Pharmaceuticals 2024, 17, 164. [Google Scholar] [CrossRef]
- Tabei, P.; Yari, A. Modification of a Hollow Fiber Membrane with Silver/Multiwall Carbon Nanotube/Polyaniline Nanocomposite as a Highly Sensitive Sensor for Voltammetric Measurement of Antioxidant Drug Dipyridamole. Mater. Chem. Phys. 2024, 311, 128534. [Google Scholar] [CrossRef]
- Bernabé-Pineda, M.; Ramírez-Silva, M.T.; Romero-Romo, M.; González-Vergara, E.; Rojas-Hernández, A. Determination of Acidity Constants of Curcumin in Aqueous Solution and Apparent Rate Constant of Its Decomposition. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2004, 60, 1091–1097. [Google Scholar] [CrossRef]
- Martínez-Guerra, J.; Palomar-Pardavé, M.; Romero-Romo, M.; Corona-Avendaño, S.; Rojas-Hernández, A.; Ramírez-Silva, M.T. New Insights on the Chemical Stability of Curcumin in Aqueous Media at Different PH: Influence of the Experimental Conditions. Int. J. Electrochem. Sci. 2019, 14, 5373–5385. [Google Scholar] [CrossRef]
- David, I.G.; Iordache, L.; Popa, D.E.; Buleandra, M.; David, V.; Iorgulescu, E.E. Novel Voltammetric Investigation of Dipyridamole at a Disposable Pencil Graphite Electrode. Turk. J. Chem. 2019, 43, 1109–1122. [Google Scholar] [CrossRef]
- Konopka, S.J.; McDuffie, B. Diffusion Coefficients of Ferri- and Ferrocyanide Ions in Aqueous Media, Using Twin-Electrode Thin-Layer Electrochemistry. Anal. Chem. 1970, 42, 1741–1746. [Google Scholar] [CrossRef]
- Xi, Z.; Sharma, N.; Paprikar, A.; Lin, S. Development and Evaluation of Dipyridamole Sustained Release Tablets Containing Micro-Environmental PH Modifiers. J. Drug Deliv. Sci. Technol. 2019, 54, 101231. [Google Scholar] [CrossRef]
- Laviron, E. General Expression of the Linear Potential Sweep Voltammogram in the Case of Diffusionless Electrochemical Systems. J. Electroanal. Chem. Interfacial Electrochem. 1979, 101, 19–28. [Google Scholar] [CrossRef]
- Afkhami, A.; Moosavi, R.; Madrakian, T. Maghemite-Nanoparticles Enhanced Effects in Electrochemical Determination of Dipyridamole Utilizing Simultaneous Statistical Based Experimental Design Optimization. J. Electrochem. Soc. 2013, 160, H775–H781. [Google Scholar] [CrossRef]
- Sarakhman, O.; Pysarevska, S.; Dubenska, L.; Stanković, D.M.; Otřísal, P.; Planková, A.; Kianičková, K.; Švorc, Ľ. Voltammetric Protocol for Reliable Determination of a Platelet Aggregation Inhibitor Dipyridamole on a Bare Miniaturized Boron-Doped Diamond Electrochemical Sensor. J. Electrochem. Soc. 2019, 166, B219–B226. [Google Scholar] [CrossRef]
- Salandari-Jolge, N.; Ensafi, A.A.; Rezaei, B. Metal–Organic Framework Derived Metal Oxide/Reduced Graphene Oxide Nanocomposite, a New Tool for the Determination of Dipyridamole. New J. Chem. 2021, 45, 2781–2790. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, H.; Zhou, S. Determination of Trace Amounts of Dipyridamole by Stripping Voltammetry Using a Nafion Modified Electrode. Talanta 1997, 44, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Ghoneim, M.M.; Tawfik, A.; Radi, A. Assay of Dipyridamole in Human Serum Using Cathodic Adsorptive Square-Wave Stripping Voltammetry. Anal. Bioanal. Chem. 2002, 374, 289–293. [Google Scholar] [CrossRef]
- Zeng, X.; Lin, S.; Hu, N. Trace Measurement of Dipyridamole by Adsorptive Stripping Voltammetry. Talanta 1993, 40, 1183–1187. [Google Scholar] [CrossRef]
- AOAC International. Guidelines for Standard Method Performance Requirements. Available online: https://www.aoac.org/wp-content/uploads/2019/08/app_f.pdf (accessed on 25 May 2023).
- Lipton, R.B.; Bigal, M.E.; Kolodner, K.B.; Gorelick, P.B.; Wilks, K.; Schoebelock, M.; Davidai, G. Acetaminophen in the Treatment of Headaches Associated with Dipyridamole-Aspirin Combination. Neurology 2004, 63, 1099–1101. [Google Scholar] [CrossRef] [PubMed]
- Khorasanchi, A.; Arabi, M.; Akhavein, A.; Seyedabadi, M.; Eftekhari, M.; Javadi, H.; Nabipour, I.; Assadi, M. Effect of Dipyridamole Injected for Myocardial Perfusion Imaging on Blood Glucose Concentration; A Preliminary Study. J. Clin. Diagn. Res. 2016, 10, TC24. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhang, Y.; Zhao, D.; Yu, X.; Shen, X.; Zhou, Y.; Wang, S.; Qiu, Y.; Chen, Y.; Zhu, F. Phenylalanine. Nucleic Acids Res. 2024, 52, D1465–D1477. [Google Scholar] [CrossRef]
- David, I.G.; Litescu, S.C.; Popa, D.E.; Buleandra, M.; Iordache, L.; Albu, C.; Alecu, A.; Penu, R.L. Voltammetric Analysis of Naringenin at a Disposable Pencil Graphite Electrode—Application to Polyphenol Content Determination in Citrus Juice. Anal. Methods 2018, 10, 5763–5772. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Supporting Electrolyte Employed in the Electro-Polymerization Process | Ip (A) |
|---|---|
| 0.1 mol/L HCl Phosphate buffer solution (PBS) pH = 7.00 0.2 mol/L NaOH | 5.76 × 10−6 |
| 6.98 × 10−6 | |
| 7.51 × 10−6 |
| Dependence | Regression Equation |
|---|---|
| Ip = f(v) | Ip = 8.581 × 10−5 × v + 3.4420 (R2 = 0.9765) |
| Ip = f (v1/2) | Ip = 5.327 × 10−5 × v1/2 − 4.0140 × 10−6 (R2 = 0.9953) |
| log Ip = f (log v) | log Ip = 0.705 × log v − 4.1973 (R2 = 0.9956) |
| Concentration Range (mol/L) | Regression Equation | Correlation Coefficient |
|---|---|---|
| 5.00 × 10−8–1.00 × 10−5 | Ip (A) = 0.7341 × CDIP (mol/L) + 2.000 × 10−8 | (R2 = 0.9994) |
| 2.50 × 10−5–1.00 × 10−4 | Ip (A) = 0.2016 × CDIP (mol/L) + 1.000 × 10−5 | (R2 = 0.9997) |
| Technique | LOD (mol/L) | LOQ (mol/L) |
|---|---|---|
| DPV | 1.47 × 10−8 | 6.80 × 10−8 |
| AdSDPV | 3.96 × 10−9 | 2.89 × 10−8 |
| Electrode | Technique/Conditions | Linear Range (mol/L) | LOD (mol/L) | Sample | Mean Recovery (%) | Ref. |
|---|---|---|---|---|---|---|
| HMDE | SWV/PBS pH = 3.00 | 1.28 × 10−6–7.02 × 10−6 | 1.88 × 10−8 | Tablets | 100.8 | [11] |
| Injections | 103.4 | |||||
| HMDE | CAdSSWV/BRB pH = 8.00; Eacc = −1.000 V; tacc = 300 s | 9.00 × 10−9–5.00 × 10−6 | 4.00 × 10−11 | Human serum | 98.38 | [59] |
| SME | AdSV/0.05 mol/L NaOH pH = 12.70 + 10% (v/v) ethanol; Eacc = 1.200 V; tacc = 60 s | 5.00 × 10−9–1.00 × 10−8 | tacc = 300 s; 1.00 × 10−9 | Tablets | 105.00 | [60] |
| Injections | 92.00 | |||||
| BDDE | DPV/BRB pH = 3.00 SWV/BRB pH = 3.00 | 1.00 × 10−7–5.00 × 10−6 | 4.00 × 10−8 | Capsules | DPV: 95.80 SWV: 96.70 | [56] |
| 1.00 × 10−7–6.00 × 10−6 | 6.00 × 10−8 | Human urine | DPV: 103.85 SWV: 97.35 | |||
| CPE | DPV/6.00 × 10−5 mol/L CTAB; tacc = 120 s | 5.95 × 10−8–2.38 × 10−7 | 1.98 × 10−8 | Tablets | 102.23 | [6] |
| SCPE | SWV/PBS pH = 5.20; Eacc = 0.580 V; tacc = 100 s | 8.00 × 10−8–3.00 × 10−5 | 2.00 × 10−8 | Tablets | 98.80 | [13] |
| PGE | DPV/PBS pH = 7.00 | 5.00 × 10−7–2.50 × 10−4 | 1.21 × 10−7 | Tablets | 200.67 | [51] |
| Nafion-GCE | AdSV/BRB pH = 1.70; Eacc = 0.000 V; tacc = 60 s | 1.00 × 10−9–8.00 × 10−8 | tacc = 240 s 8.00 × 10−11 | Human serum | 100.70 | [58] |
| NiCo2O4/NiO@MOF-5/rGO/GCE | DPV | 2.00 × 10−8–5.50 × 10−4 | 2.80 × 10−9 | Free-drug plasma | 100.10 | [57] |
| Urine | 98.10 | |||||
| AgNP/MWCNT/PANI/HFME | DPV/PBS pH = 6.50 | 1.00 × 10−8–5.00 × 10−5 | 1.00 × 10−9 | Tablets | 100.30 | [48] |
| Human serum | 103.00 | |||||
| MIP_Fe3O4@Au-H2N-MWCNT/MGCE | DPV/PBS pH = 3.00 | 9.91 × 10−10–3.76 × 10−6 | 5.95 × 10−11 | Human serum | 99.23 | [17] |
| γ-Fe2O3_CPE | DPV/BRB pH = 2.00; tacc = 75 s | 1.00 × 10−8–5.70 × 10−6 | 5.00 × 10−9 | Tablets | 98.10 | [55] |
| 5.70 × 10−6–5.00 × 10−5 | Human serum | 99.30 | ||||
| MIP_CPE | AdSDPV/PBS pH = 3.00; Eacc = −0.200 V; tacc = 120 s; | 1.98 × 10−9–2.18 ×10−7 | 9.90 × 10−10 | Tablets | 98.60 | [18] |
| Human serum | 97.77 | |||||
| MIP_PGE | DPV/PBS pH = 7.00 AdSDPV/PBS pH = 7.00; Eacc = 0.200 V; tacc = 30 s | 1.00 × 10−7–1.00 × 10−5 | 2.04 × 10−8 | Tablets | 105.16 | [46] |
| 1.00 × 10−8–5.00 × 10−7 | 8.67 × 10−9 | |||||
| MIP_PGE | DPV/BRB pH = 3.29 AdSDPV BRB pH = 3.29; Eacc = −0.400 V; tacc = 30 s | 5.00 × 10−8–1.00 × 10−5 | 1.47 × 10−8 | Tablets Tap water | 104.67 105.78 | This work |
| 5.00 × 10−9–1.00 × 10−7 | 3.96 × 10−9 |
| DPV | AdSDPV | ||
|---|---|---|---|
| DIP Concentration (mol/L) | RSD% * | DIP Concentration (mol/L) | RSD% * |
| 1.00 × 10−7 | 7.90 | 1.00 × 10−8 | 8.88 |
| 1.00 × 10−6 | 3.72 | 7.50 × 10−8 | 5.91 |
| 1.00 × 10−5 | 3.09 | 5.00 × 10−7 | 4.06 |
| Technique | Tablet’s DIP Content (mg) | ||
|---|---|---|---|
| Claimed by Manufacturer | Found ± SD * | Mean Recovery ± SD * (%) | |
| DPV at MIP_PGE | 25.00 | 26.16 ± 0.47 | 104.66 ± 0.025 |
| Spectrofluorometry | 25.00 | 26.17 ± 0.77 | 104.67 ± 3.06 |
| DIP Concentration (mol/L) | ||||
|---|---|---|---|---|
| Added | Found by | Mean Recovery % | ||
| DPV | Spectrofluorometry | DPV | Spectrofluorometry | |
| 2.50 × 10−7 | 2.64 × 10−7 | 2.58 × 10−7 | 105.55 | 103.20 |
| 5.00 × 10−7 | 5.31 × 10−7 | 5.25 × 10−7 | 106.14 | 105.00 |
| 7.50 × 10−7 | 7.92 × 10−7 | 7.44 × 10−7 | 105.65 | 99.20 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Preda, D.; Radu, G.L.; Iorgulescu, E.-E.; Cheregi, M.-C.; David, I.G. Curcumin-Based Molecularly Imprinted Polymer Electropolymerized on Single-Use Graphite Electrode for Dipyridamole Analysis. Molecules 2024, 29, 4630. https://doi.org/10.3390/molecules29194630
Preda D, Radu GL, Iorgulescu E-E, Cheregi M-C, David IG. Curcumin-Based Molecularly Imprinted Polymer Electropolymerized on Single-Use Graphite Electrode for Dipyridamole Analysis. Molecules. 2024; 29(19):4630. https://doi.org/10.3390/molecules29194630
Chicago/Turabian StylePreda, Daniel, Gabriel Lucian Radu, Emilia-Elena Iorgulescu, Mihaela-Carmen Cheregi, and Iulia Gabriela David. 2024. "Curcumin-Based Molecularly Imprinted Polymer Electropolymerized on Single-Use Graphite Electrode for Dipyridamole Analysis" Molecules 29, no. 19: 4630. https://doi.org/10.3390/molecules29194630
APA StylePreda, D., Radu, G. L., Iorgulescu, E.-E., Cheregi, M.-C., & David, I. G. (2024). Curcumin-Based Molecularly Imprinted Polymer Electropolymerized on Single-Use Graphite Electrode for Dipyridamole Analysis. Molecules, 29(19), 4630. https://doi.org/10.3390/molecules29194630