
Synthesis, Anti-Cancer Activity, Cell Cycle Arrest, Apoptosis Induction, and Docking Study of Fused Benzo[h]chromeno[2,3-d]pyrimidine on Human Breast Cancer Cell Line MCF-7

 , , , , and
, , , , and
Abstract
1. Introduction
2. Results and Discussion
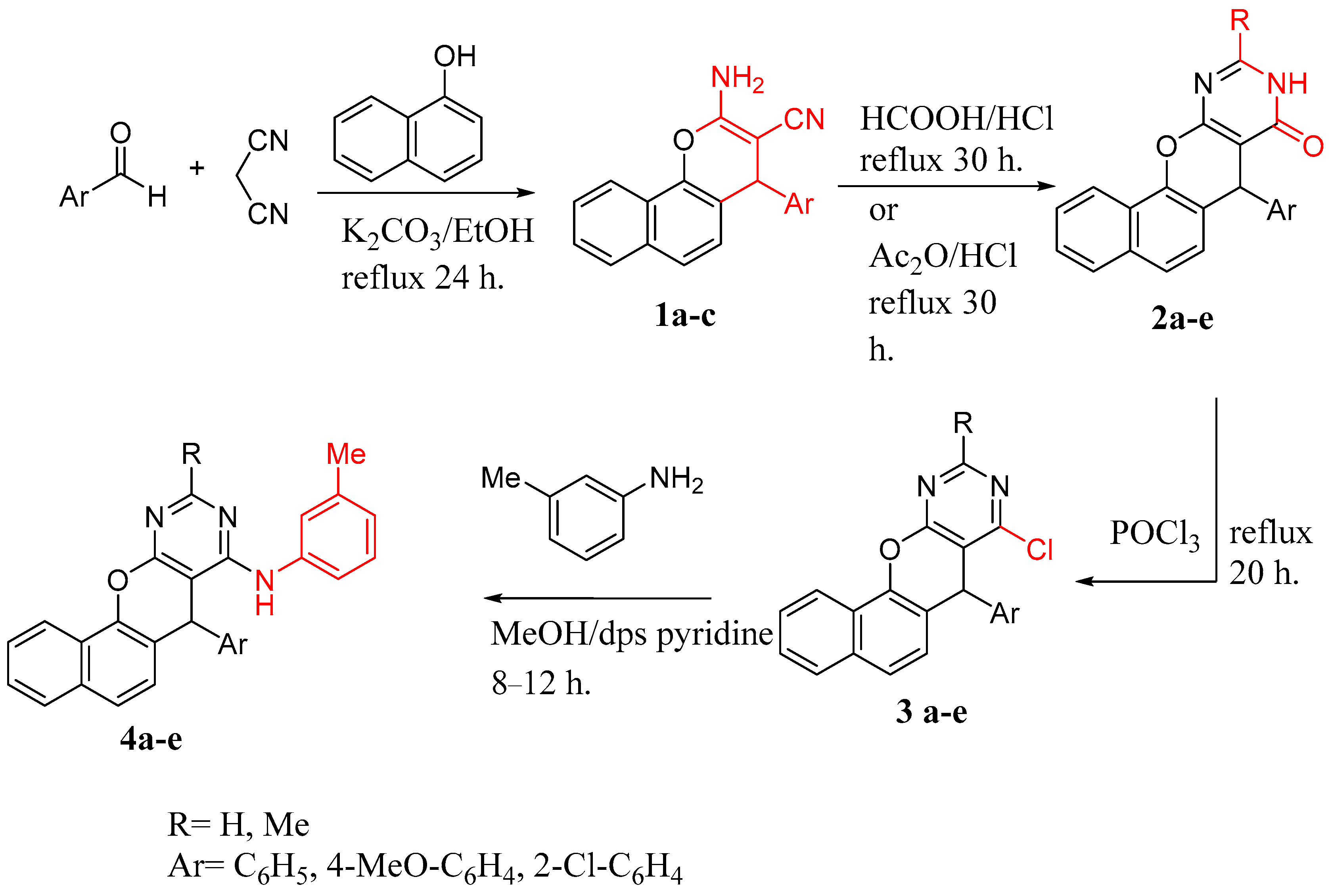
2.1. Chemistry
2.2. Anti-Cancer Result and Discussion
2.2.1. Single-Dose Testing
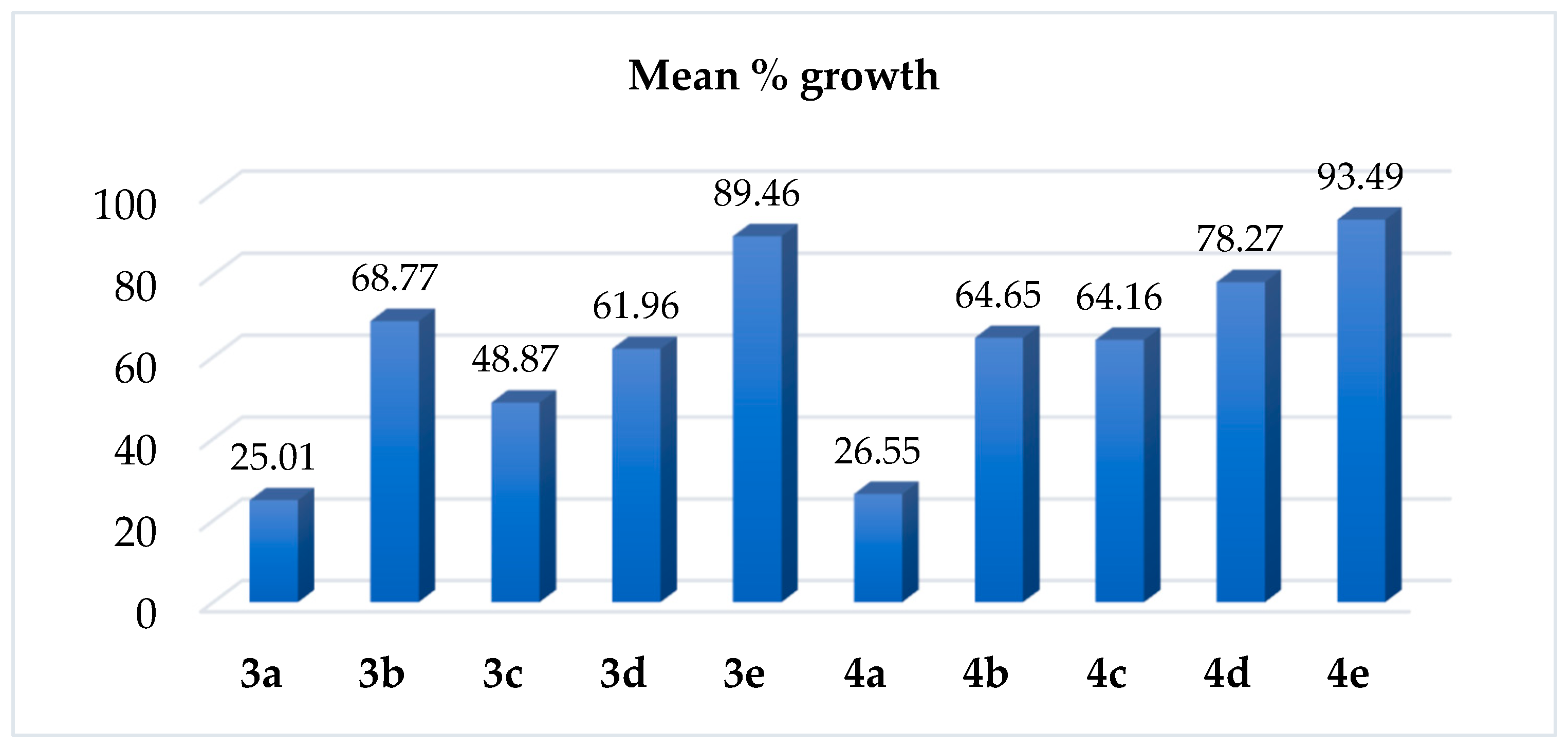
2.2.2. Five-Dose Assay and Selectivity Estimation
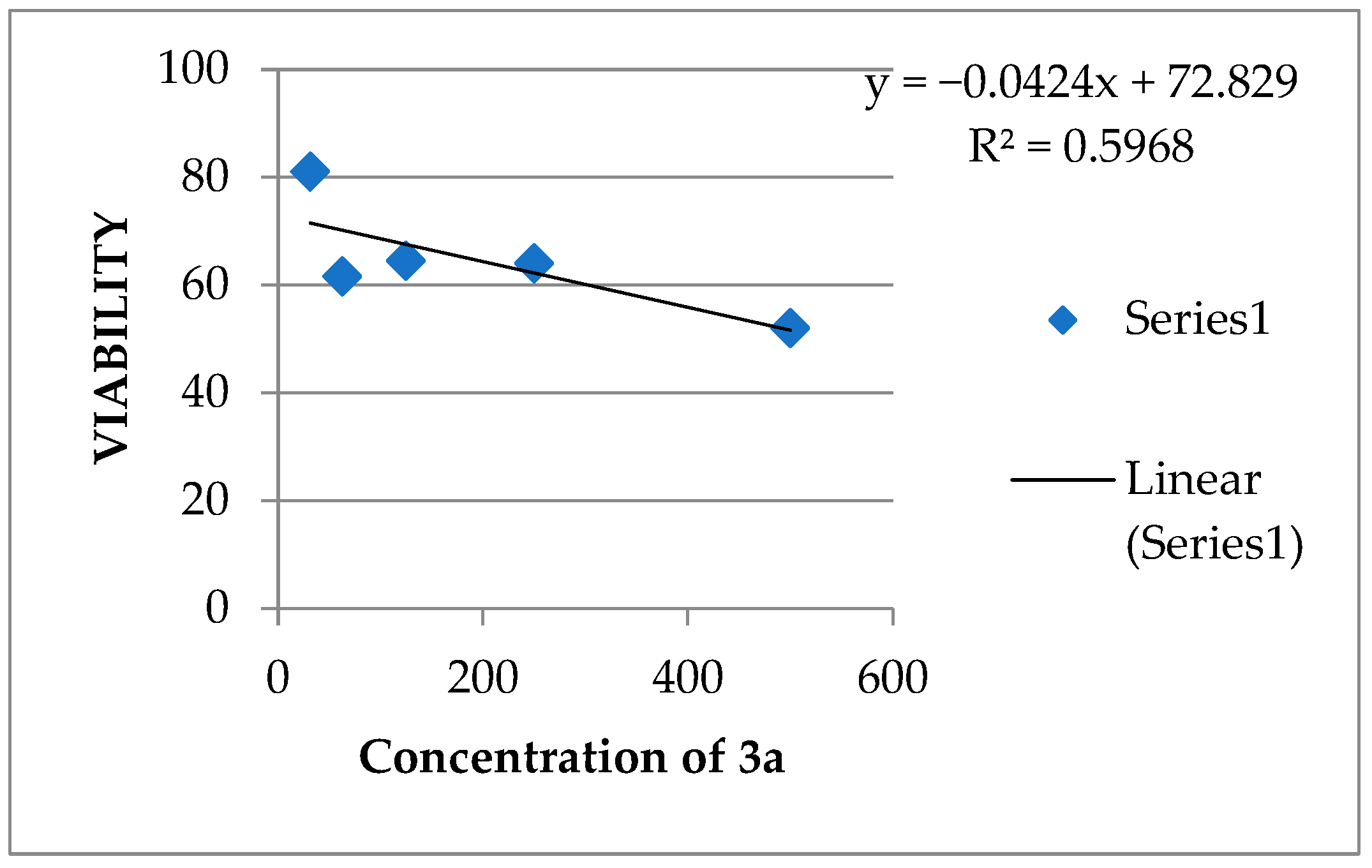
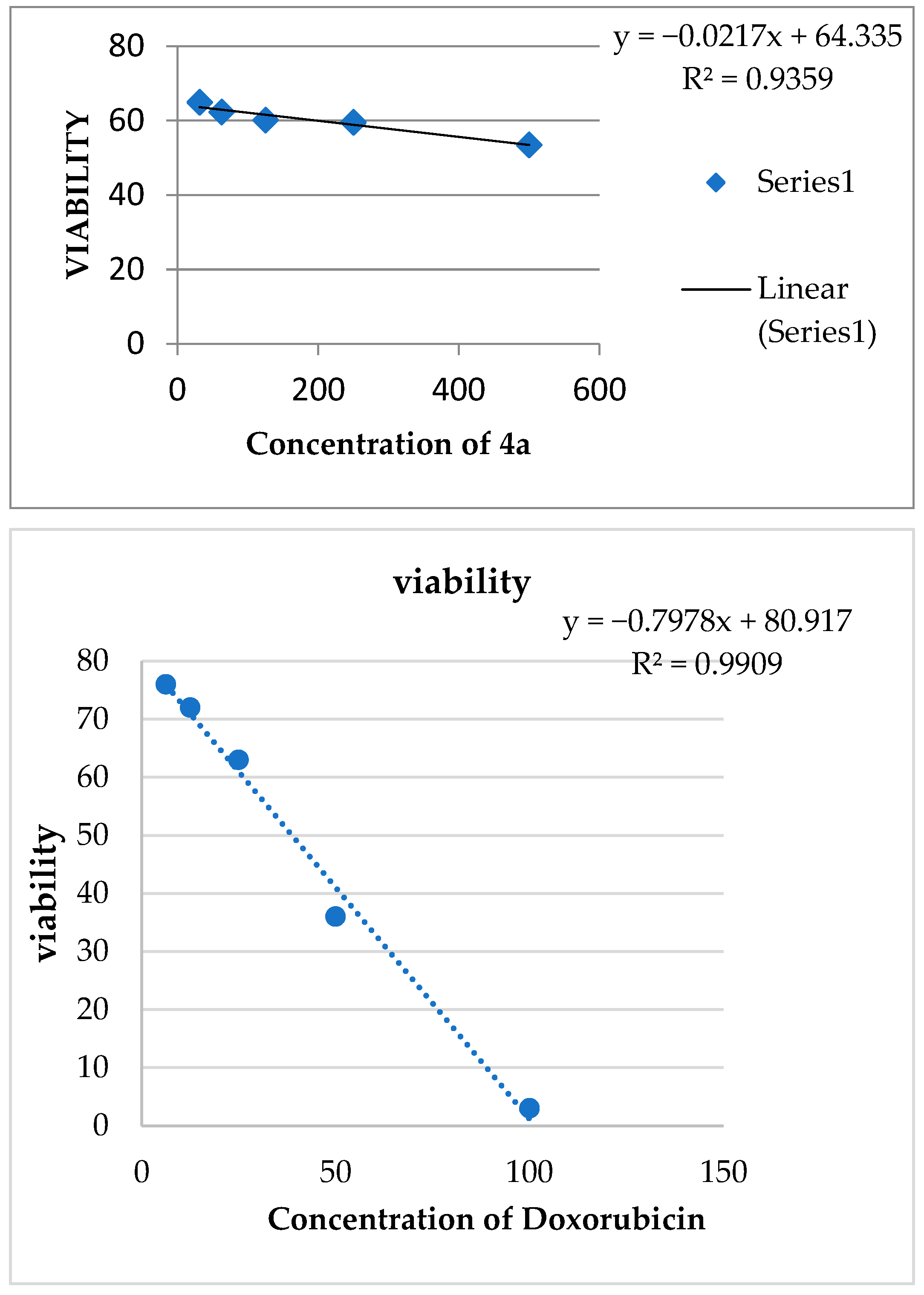
2.2.3. Cytotoxicity/Viability Assay for the Most Active Compounds (3a and 4a) versus Positive Control against MCF7 Cell Line
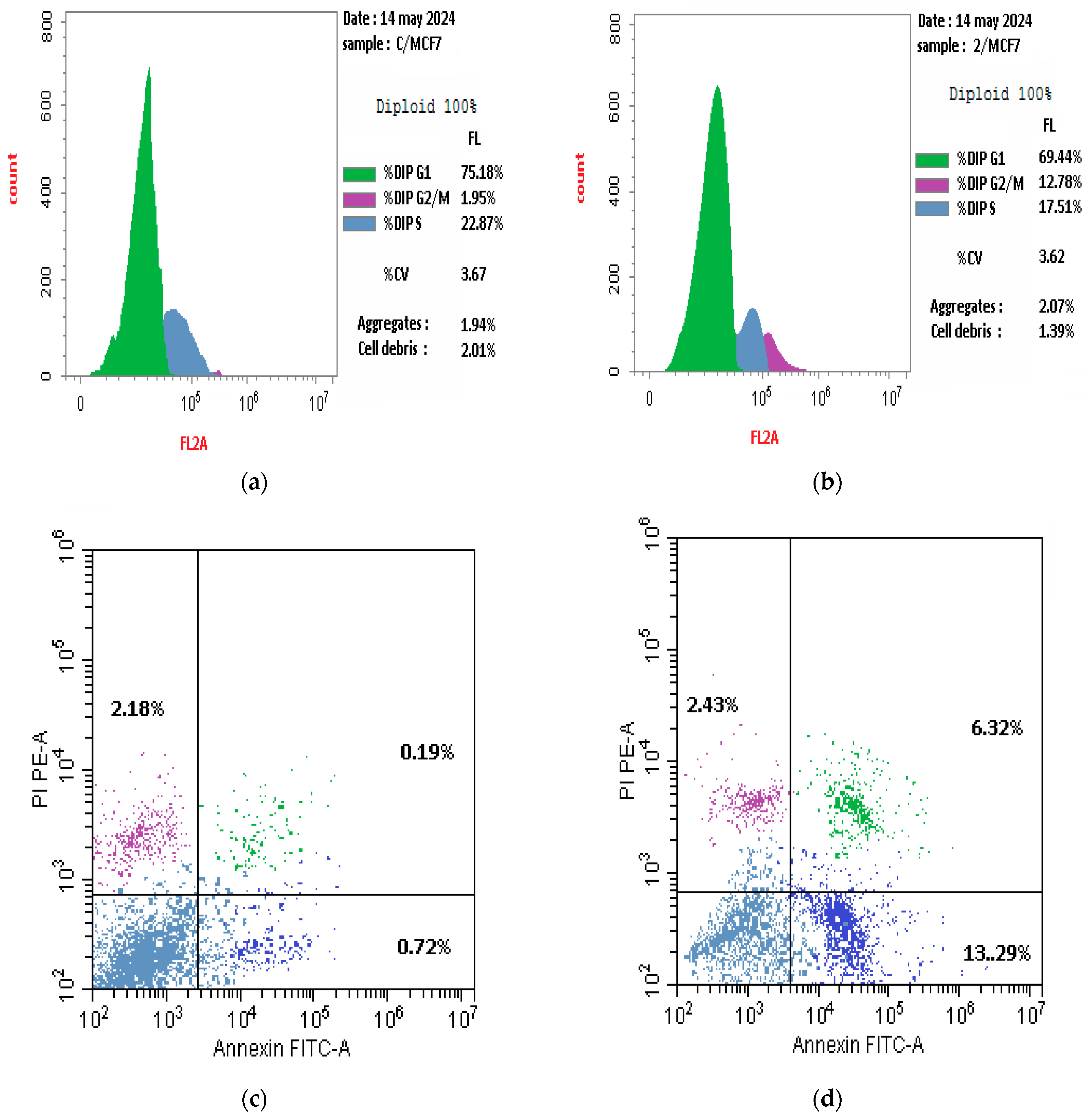
2.2.4. Cell Cycle Arrest and Apoptotic Cells Formation
2.2.5. RT-PCR
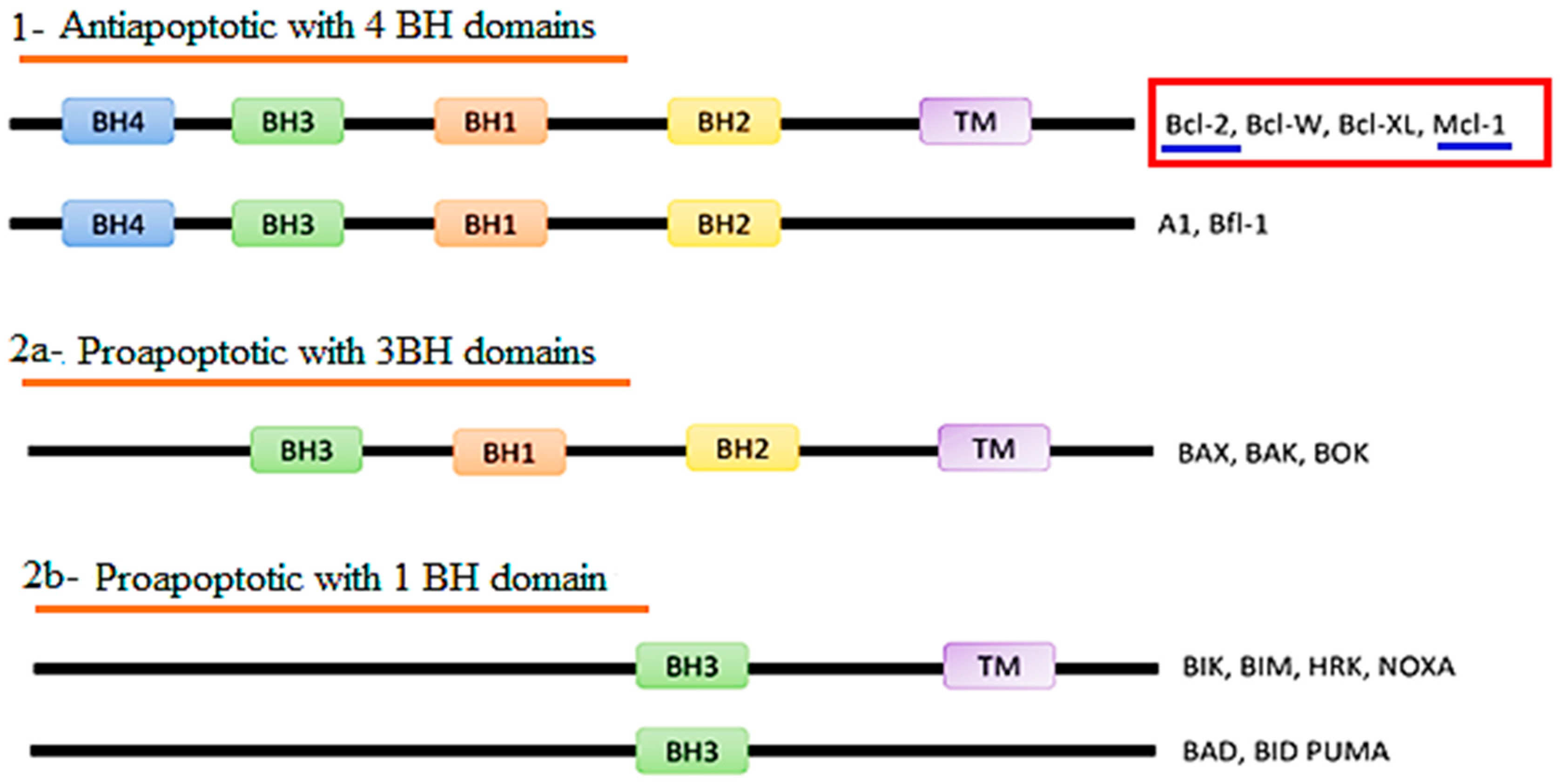
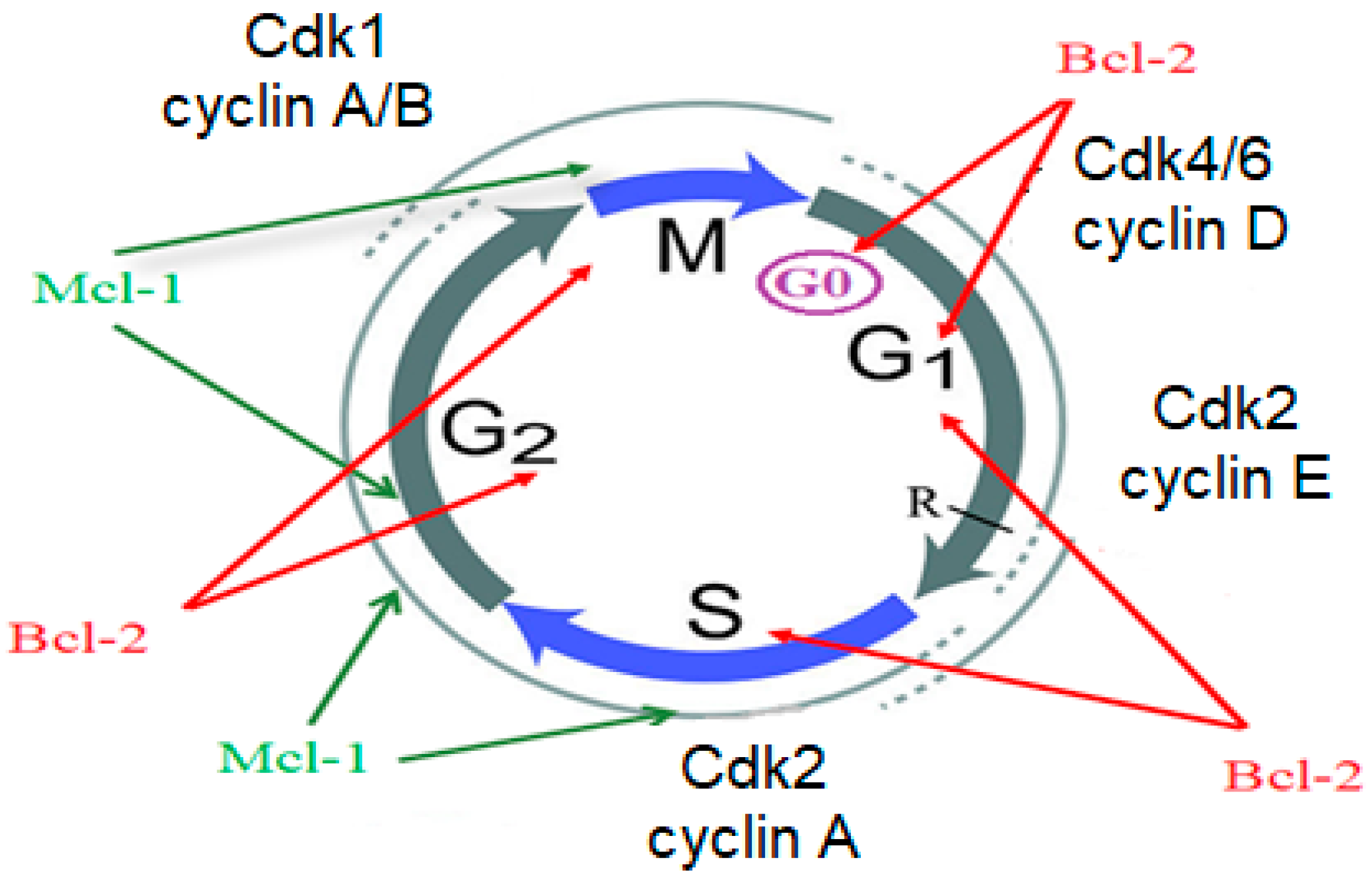
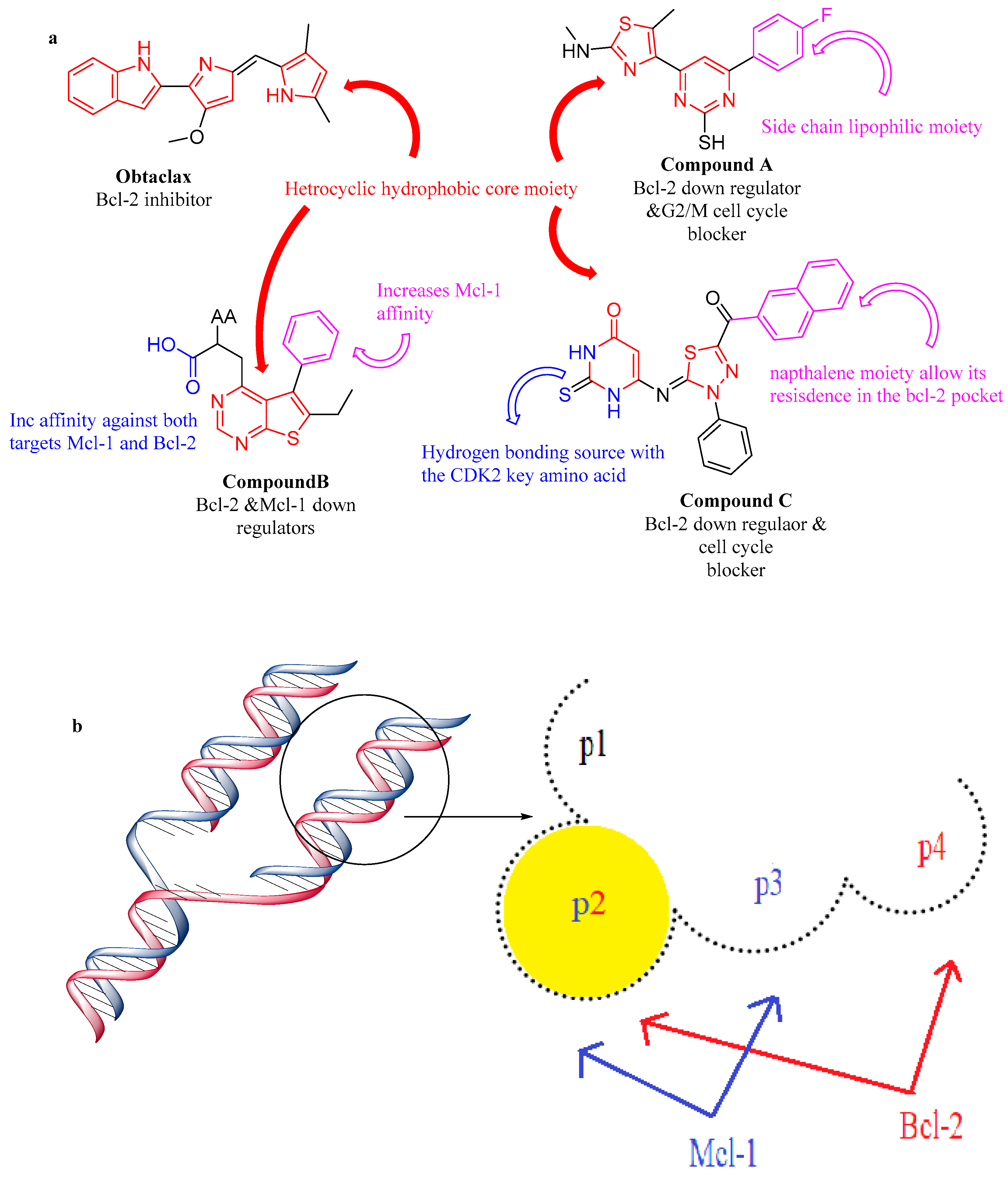
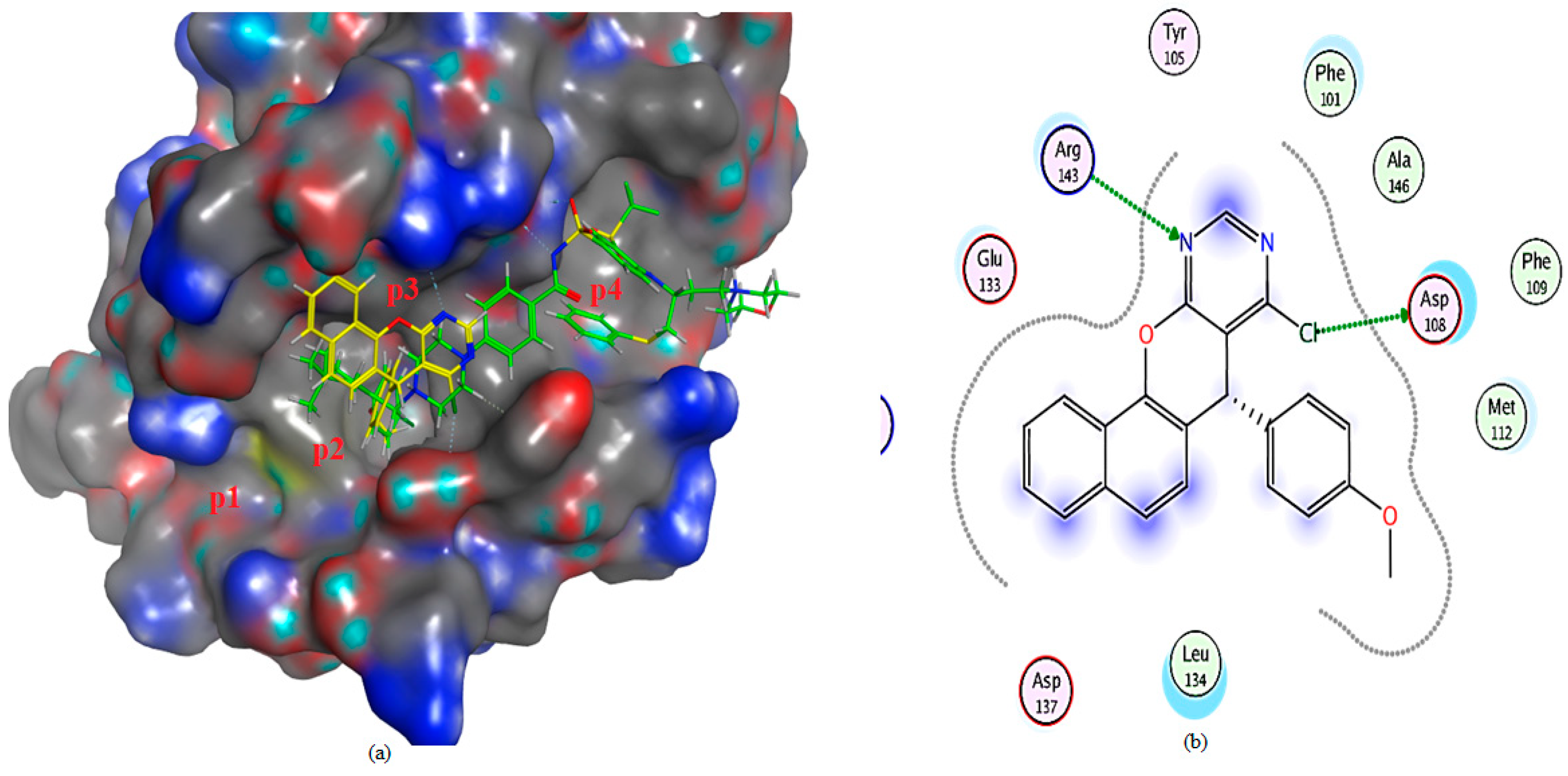
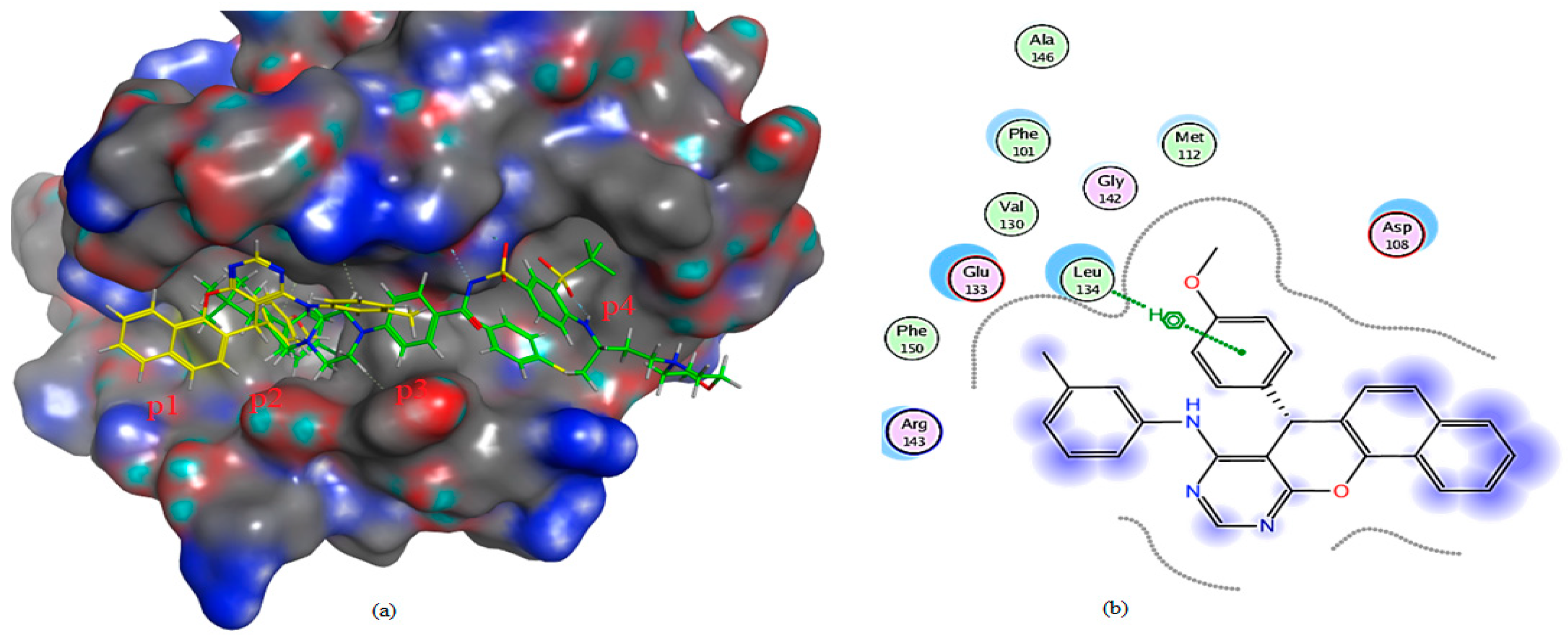
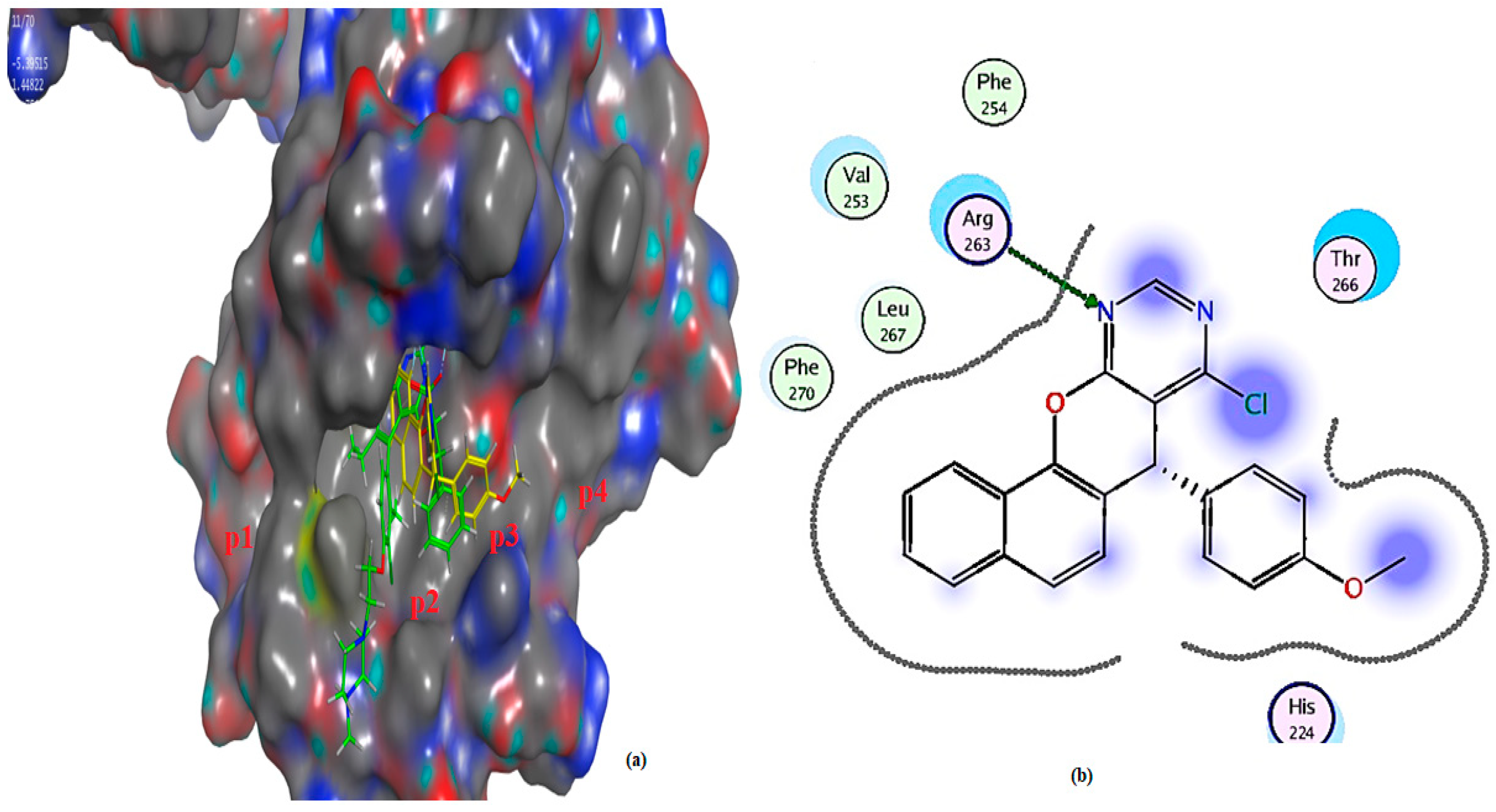
2.3. Molecular Docking Analysis
2.3.1. Structure Activity Correlation
2.3.2. Molecular Docking Investigation
3. Materials and Methods
3.1. Synthesis of Lead Compounds
3.1.1. General Procedure for the Synthesis of Compounds 3a–e
3.1.2. General Procedure for the Synthesis of Compounds 4a–e
3.2. Assessment of Anti-Cancer Activity
3.2.1. The NCI-60 Human Tumor Cell Lines Screen
Selection Guidelines in NCI
Interpretation of One-Dose Data
NCI-60 Cell Five-Dose Screen
3.2.2. Cell Viability and Cytotoxicity against MCF-7 Cell Line
3.2.3. Cell Cycle and Apoptosis Detection by Flow Cytometry
3.2.4. Gene Expression Analysis of Bcl2 and Mcl1 by Reverse Transcription-Quantitative Polymerase Chain Reaction (RT-qPCR)
3.3. Docking Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gomes, J.P.M.; Cardoso, C.R.P.; Varanda, E.A.; Molina, J.M.; Fernandez, M.F.; Olea, N.; Carlos, I.Z.; Vilegas, W. Antitumoral, Mutagenic and (Anti)Estrogenic Activities of Tingenone and Pristimerin. Braz. J. Pharmacogn. 2011, 21, 963–971. [Google Scholar] [CrossRef]
- Nagai, H.; Kim, Y.H. Cancer Prevention from the Perspective of Global Cancer Burden Patterns. J. Thorac. Dis. 2017, 9, 448–451. [Google Scholar] [CrossRef]
- WHO Web Cite. Available online: https://www.who.int/news-room/fact-sheets/detail/breast-cancer (accessed on 5 March 2021).
- Raha, P.; Thomas, S.; Thurn, K.T.; Park, J.; Munster, P.N. Combined Histone Deacetylase Inhibition and Tamoxifen Induces Apoptosis in Tamoxifen-Resistant Breast Cancer Models, by Reversing Bcl-2 Overexpression. Breast Cancer Res. 2015, 17, 26. [Google Scholar] [CrossRef] [PubMed]
- Doshi, J.M.; Tian, D.; Xing, C. Structure-Activity Relationship Studies of Ethyl 2-Amino-6-Bromo-4-(1- Cyano-2-Ethoxy-2-Oxoethyl)-4H-Chromene-3-Carboxylate (HA 14-1), an Antagonist for Antiapoptotic Bcl-2 Proteins to Overcome Drug Resistance in Cancer. J. Med. Chem. 2006, 49, 7731–7739. [Google Scholar] [CrossRef]
- Merino, D.; Lok, S.W.; Visvader, J.E.; Lindeman, G.J. Targeting BCL-2 to Enhance Vulnerability to Therapy in Estrogen Receptor-Positive Breast Cancer. Oncogene 2016, 35, 1877–1887. [Google Scholar] [CrossRef] [PubMed]
- Farghaly, T.A.; Masaret, G.S.; Muhammad, Z.A.; Harras, M.F. Discovery of Thiazole-Based-Chalcones and 4-Hetarylthiazoles as Potent Anticancer Agents: Synthesis, Docking Study and Anticancer Activity. Bioorg. Chem. 2020, 98, 103761. [Google Scholar] [CrossRef]
- Albratty, M.; Alhazmi, H.A. Novel Pyridine and Pyrimidine Derivatives as Promising Anticancer Agents: A Review. Arab. J. Chem. 2022, 15, 103846. [Google Scholar] [CrossRef]
- Meier, P.; Finch, A.; Evan, G. Apoptosis in Development. Nature 2000, 407, 796–801. [Google Scholar] [CrossRef] [PubMed]
- Hengartner, M.O. The Biochemistry of Apoptosis. Nature 2000, 407, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Arnett, E.; Pahari, S.; Leopold Wager, C.M.; Hernandez, E.; Bonifacio, J.R.; Lumbreras, M.; Renshaw, C.; Montoya, M.J.; Opferman, J.T.; Schlesinger, L.S. Combination of MCL-1 and BCL-2 Inhibitors Is a Promising Approach for a Host-Directed Therapy for Tuberculosis. Biomed. Pharmacother. 2023, 168, 115738. [Google Scholar] [CrossRef]
- Sancho, M.; Leiva, D.; Lucendo, E.; Orzáez, M. Understanding MCL1: From Cellular Function and Regulation to Pharmacological Inhibition. FEBS J. 2022, 289, 6209–6234. [Google Scholar] [CrossRef] [PubMed]
- Widden, H.; Placzek, W.J. The Multiple Mechanisms of MCL1 in the Regulation of Cell Fate. Commun. Biol. 2021, 4, 1029. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Shi, Y. Apoptosis Signaling Pathways and Lymphocyte Homeostasis. Cell Res. 2007, 17, 759–771. [Google Scholar] [CrossRef] [PubMed]
- Kvansakul, M.; Hinds, M.G. The Bcl-2 Family: Structures, Interactions and Targets for Drug Discovery. Apoptosis 2015, 20, 136–150. [Google Scholar] [CrossRef] [PubMed]
- Ploumaki, I.; Triantafyllou, E.; Koumprentziotis, I.A.; Karampinos, K.; Drougkas, K.; Karavolias, I.; Trontzas, I.; Kotteas, E.A. Bcl-2 Pathway Inhibition in Solid Tumors: A Review of Clinical Trials. Clin. Transl. Oncol. 2023, 25, 1554–1578. [Google Scholar] [CrossRef] [PubMed]
- Bonnefoy-Berard, N.; Aouacheria, A.; Verschelde, C.; Quemeneur, L.; Marçais, A.; Marvel, J. Control of Proliferation by Bcl-2 Family Members. Biochim. Biophys. Acta Mol. Cell Res. 2004, 1644, 159–168. [Google Scholar] [CrossRef]
- Jamil, S.; Sobouti, R.; Hojabrpour, P.; Raj, M.; Kast, J.; Duronio, V. A Proteolytic Fragment of Mcl-1 Exhibits Nuclear Localization and Regulates Cell Growth by Interaction with Cdk1. Biochem. J. 2005, 387, 659–667. [Google Scholar] [CrossRef]
- Zinkel, S.; Gross, A.; Yang, E. BCL2 Family in DNA Damage and Cell Cycle Control. Cell Death Differ. 2006, 13, 1351–1359. [Google Scholar] [CrossRef]
- Qian, S.; Wei, Z.; Yang, W.; Huang, J.; Yang, Y.; Wang, J. The Role of BCL-2 Family Proteins in Regulating Apoptosis and Cancer Therapy. Front. Oncol. 2022, 12, 985363. [Google Scholar] [CrossRef]
- Vogler, M.; Dinsdale, D.; Dyer, M.J.S.; Cohen, G.M. Bcl-2 Inhibitors: Small Molecules with a Big Impact on Cancer Therapy. Cell Death Differ. 2009, 16, 360–367. [Google Scholar] [CrossRef]
- El-Saidi, M.M.T.; El-Sayed, A.A.; Pedersen, E.B.; Tantawy, M.A.; Mohamed, N.R.; Gad, W.A. Synthesis, Characterization and Docking Study of Novel Pyrimidine Derivatives as Anticancer Agents. Indones. J. Chem. 2020, 20, 1163–1177. [Google Scholar] [CrossRef]
- Bruncko, M.; Wang, L.; Sheppard, G.S.; Phillips, D.C.; Tahir, S.K.; Xue, J.; Erickson, S.; Fidanze, S.; Fry, E.; Hasvold, L.; et al. Structure-Guided Design of a Series of MCL-1 Inhibitors with High Affinity and Selectivity. J. Med. Chem. 2015, 58, 2180–2194. [Google Scholar] [CrossRef] [PubMed]
- Leverson, J.D.; Zhang, H.; Chen, J.; Tahir, S.K.; Phillips, D.C.; Xue, J.; Nimmer, P.; Jin, S.; Smith, M.; Xiao, Y.; et al. Potent and Selective Small-Molecule MCL-1 Inhibitors Demonstrate on-Target Cancer Cell Killing Activity as Single Agents and in Combination with ABT-263 (Navitoclax). Cell Death Dis. 2015, 6, e1590. [Google Scholar] [CrossRef] [PubMed]
- Tron, A.E.; Belmonte, M.A.; Adam, A.; Aquila, B.M.; Boise, L.H.; Chiarparin, E.; Cidado, J.; Embrey, K.J.; Gangl, E.; Gibbons, F.D.; et al. Discovery of Mcl-1-Specific Inhibitor AZD5991 and Preclinical Activity in Multiple Myeloma and Acute Myeloid Leukemia. Nat. Commun. 2018, 9, 5341. [Google Scholar] [CrossRef]
- Yuda, J.; Will, C.; Phillips, D.C.; Abraham, L.; Alvey, C.; Avigdor, A.; Buck, W.; Besenhofer, L.; Boghaert, E.; Cheng, D.; et al. Selective MCL-1 Inhibitor ABBV-467 Is Efficacious in Tumor Models but Is Associated with Cardiac Troponin Increases in Patients. Commun. Med. 2023, 3, 154. [Google Scholar] [CrossRef]
- Zhu, J.; Wang, Z.; Guo, Z.; Zhang, X.; Song, T.; Guo, Y.; Ji, T.; Zhang, Z. Structure-Based Design, Synthesis, and Evaluation of Bcl-2/Mcl-1 Dual Inhibitors. Arch. Pharm. 2020, 353, e2000005. [Google Scholar] [CrossRef]
- Wendt, M.D.; Shen, W.; Kunzer, A.; McClellan, W.J.; Bruncko, M.; Oost, T.K.; Ding, H.; Joseph, M.K.; Zhang, H.; Nimmer, P.M.; et al. Discovery and Structure-Activity Relationship of Antagonists of B-Cell Lymphoma 2 Family Proteins with Chemopotentiation Activity in Vitro and in Vivo. J. Med. Chem. 2006, 49, 1165–1181. [Google Scholar] [CrossRef]
- Lu, X.; Liu, Y.C.; Orvig, C.; Liang, H.; Chen, Z.F. Discovery of a Copper-Based Mcl-1 Inhibitor as an Effective Antitumor Agent. J. Med. Chem. 2020, 63, 9154–9167. [Google Scholar] [CrossRef]
- Zhang, Z.; Hou, L.; Bai, L.; Pei, J.; Zhao, S.; Luan, S.; Liu, D.; Huang, M.; Zhao, L. Discovery and Structure-Activity Relationship Studies of Novel Bcl-2/Mcl-1 Dual Inhibitors with Indole Scaffold. Bioorg. Chem. 2022, 125, 105845. [Google Scholar] [CrossRef]
- Badrey, M.G.; Gomha, S.M. 3-Amino-8-Hydroxy-4-Imino-6-Methyl-5-Phenyl-4,5-Dihydro-3h-Chromeno [2,3-d]Pyrimidine: An Effecient Key Precursor for Novel Synthesis of Some Interesting Triazines and Triazepines as Potential Anti-Tumor Agents. Molecules 2012, 17, 11538–11553. [Google Scholar] [CrossRef]
- Pordeli, M.; Nakhjiri, M.; Safavi, M.; Ardestani, S.K.; Foroumadi, A. Anticancer Effects of Synthetic Hexahydrobenzo [g]Chromen-4-One Derivatives on Human Breast Cancer Cell Lines. Breast Cancer 2017, 24, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.S.; Moustafa, G.O.; Awad, H.M. Synthesis and in Vitro Anticancer Activity of Pyrazolo[1,5-a]Pyrimidines and Pyrazolo[3,4-d][1,2,3]Triazines. Synth. Commun. 2017, 47, 1963–1972. [Google Scholar] [CrossRef]
- Mohamed, M.M.; Khalil, A.K.; Abbass, E.M.; El-Naggar, A.M. Design, Synthesis of New Pyrimidine Derivatives as Anticancer and Antimicrobial Agents. Synth. Commun. 2017, 47, 1441–1457. [Google Scholar] [CrossRef]
- El-Bakhshawangy, N.M.; El-Nassan, H.B.; Kassab, A.E.; Taher, A.T. Design, Synthesis and Biological Evaluation of Chromenopyrimidines as Potential Cytotoxic Agents. Future Med. Chem. 2018, 10, 1465–1481. [Google Scholar] [CrossRef]
- Bhosle, M.R.; Wahul, D.B.; Bondle, G.M.; Sarkate, A.; Tiwari, S.V. An Efficient Multicomponent Synthesis and in Vitro Anticancer Activity of Dihydropyranochromene and Chromenopyrimidine-2,5-Diones. Synth. Commun. 2018, 48, 2046–2060. [Google Scholar] [CrossRef]
- Alblewi, F.F.; Okasha, R.M.; Hritani, Z.M.; Mohamed, H.M.; El-Nassag, M.A.A.; Halawa, A.H.; Mora, A.; Fouda, A.M.; Assiri, M.A.; Al-Dies, A.A.M.; et al. Antiproliferative Effect, Cell Cycle Arrest and Apoptosis Generation of Novel Synthesized Anticancer Heterocyclic Derivatives Based 4H-Benzo[h]Chromene. Bioorg. Chem. 2019, 87, 560–571. [Google Scholar] [CrossRef]
- Eghtedari, M.; Azimzadeh Arani, M.; Sarrafi, Y.; Shafiei, M.; Alimohammadi, K.; Safari, F.; Foroumadi, A. Synthesis and Antitumor Activity Evaluation of Novel Pyrimidoquinoline Derivatives. Polycycl. Aromat. Compd. 2022, 42, 4359–4373. [Google Scholar] [CrossRef]
- Bhale, P.S.; Chavan, H.V.; Shringare, S.N.; Khedkar, V.M.; Tigote, R.M.; Mali, N.N.; Jadhav, T.D.; Kamble, N.B.; Kolat, S.P.; Bandgar, B.P.; et al. Design, Synthesis of Anticancer and Anti-Inflammatory 4-(1-Methyl-1 H -Indol-3-Yl)-6-(Methylthio) Pyrimidine-5-Carbonitriles. Synth. Commun. 2022, 52, 733–744. [Google Scholar] [CrossRef]
- Boddiboyena, R.; Reddy, G.N.; Seelam, N.; Sarma, M.; Gudisela, M. reddy. Design, Synthesis, Anticancer Evaluation, Molecular Docking, and in Silico ADME Analysis of Novel Chalcones Incorporated Indole-Pyrimidine Derivatives as Promising Anticancer Agents. Chem. Data Collect. 2022, 39, 100852. [Google Scholar] [CrossRef]
- Sigmond, J.; Peters, G.J. Pyrimidine and Purine Analogues, Effects on Cell Cycle Regulation and the Role of Cell Cycle Inhibitors to Enhance Their Cytotoxicity. Nucleosides Nucleotides Nucleic Acids 2005, 24, 1997–2022. [Google Scholar] [CrossRef]
- Kassab, A.E.; Gedawy, E.M.; El-Malah, A.A.; Abdelghany, T.M.; Abdel-Bakky, M.S. Synthesis, Anticancer Activity, Effect on Cell Cycle Profile, and Apoptosis-Inducing Ability of Novel Hexahydrocyclooctathieno[2,3-d]-Pyrimidine Derivatives. Chem. Pharm. Bull. 2016, 64, 490–496. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, M.F.; Abdelmoniem, A.M.; Elwahy, A.H.M.; Abdelhamid, I.A. DNA Fragmentation, Cell Cycle Arrest, and Docking Study of Novel Bis Spiro-Cyclic 2-Oxindole of Pyrimido[4,5-b]Quinoline-4,6-Dione Derivatives against Breast Carcinoma. Curr. Cancer Drug Targets 2018, 18, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Ragab, F.A.; Nissan, Y.M.; Seif, E.M.; Maher, A.; Arafa, R.K. Synthesis and in Vitro Investigation of Novel Cytotoxic Pyrimidine and Pyrazolopyrimidne Derivatives Showing Apoptotic Effect. Bioorg. Chem. 2020, 96, 103621. [Google Scholar] [CrossRef] [PubMed]
- Nallajennugari, V.; Pajaniradje, S.; Subramanian, S.; Bhat, S.A.; Parthasarathi, D.; Bhaskaran, S.; Syed Ali Padusha, M.; Rajagopalan, R. A Novel Anticancer Chromeno-Pyrimidine Analogue Inhibits Epithelial-Mesenchymal Transition in Lung Adenocarcinoma Cells. Toxicol. Mech. Methods 2021, 31, 401–412. [Google Scholar] [CrossRef]
- Elgaafary, M.; Fouda, A.M.; Mohamed, H.M.; Hamed, A.; El-Mawgoud, H.K.A.; Jin, L.; Ulrich, J.; Simmet, T.; Syrovets, T.; El-Agrody, A.M. Synthesis of β-Enaminonitrile-Linked 8-Methoxy-1H-Benzo[f]Chromene Moieties and Analysis of Their Antitumor Mechanisms. Front. Chem. 2021, 9, 759148. [Google Scholar] [CrossRef]
- Albalawi, F.F.; El-Nassag, M.A.A.; El-Eisawy, R.A.; Mohamed, M.B.I.; Fouda, A.M.; Afifi, T.H.; Elhenawy, A.A.; Mora, A.; El-Agrody, A.M.; El-Mawgoud, H.K.A. Synthesis of 9-Hydroxy-1H-Benzo[f]Chromene Derivatives with Effective Cytotoxic Activity on MCF7/ADR, P-Glycoprotein Inhibitors, Cell Cycle Arrest and Apoptosis Effects. Int. J. Mol. Sci. 2023, 24, 49. [Google Scholar] [CrossRef]
- Karoui, S.; Dhiabi, M.; Fakhfakh, M.; Abid, S.; Limanton, E.; Le Guével, R.; Charlier, T.D.; Mainguy, A.; Mignen, O.; Paquin, L.; et al. Design and Synthesis of Novel N-Benzylidene Derivatives of 3-Amino-4-Imino-3,5-Dihydro-4H-Chromeno[2,3-d]Pyrimidine under Microwave, In Silico ADME Predictions, In Vitro Antitumoral Activities and In Vivo Toxicity. Pharmaceuticals 2024, 17, 458. [Google Scholar] [CrossRef]
- Haiba, M.E.; Al-Abdullah, E.S.; Ghabbour, H.A.; Riyadh, S.M.; Abdel-Kader, R.M. Inhibitory Activity of Benzo[h]Quinoline and Benzo[h]Chromene in Human Glioblastoma Cells. Trop. J. Pharm. Res. 2016, 15, 2337–2343. [Google Scholar] [CrossRef]
- Abdelatef, S.A.; El-Saadi, M.T.; Amin, N.H.; Abdelazeem, A.H.; Omar, H.A.; Abdellatif, K.R.A. Design, Synthesis and Anticancer Evaluation of Novel Spirobenzo[h]Chromene and Spirochromane Derivatives with Dual EGFR and B-RAF Inhibitory Activities. Eur. J. Med. Chem. 2018, 150, 567–578. [Google Scholar] [CrossRef]
- Schmitt, F.; Kasparkova, J.; Brabec, V.; Begemann, G.; Schobert, R.; Biersack, B. New (Arene)Ruthenium(II) Complexes of 4-aryl-4H-naphthopyrans with Anticancer and Anti-Vascular Activities. J. Inorg. Biochem. 2018, 184, 69–78. [Google Scholar] [CrossRef]
- Schmitt, F.; Gold, M.; Rothemund, M.; Andronache, I.; Biersack, B.; Schobert, R.; Mueller, T. New Naphthopyran Analogues of LY290181 as Potential Tumor Vascular-Disrupting Agents. Eur. J. Med. Chem. 2019, 163, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.S.; Jarhad, D.B.; Kim, G.; Nayak, A.; Zhao, L.X.; Yu, J.; Kim, H.R.; Lee, J.Y.; Mulamoottil, V.A.; Chandra, G.; et al. Design, Synthesis and Anticancer Activity of Fluorocyclopentenyl-Purines and—Pyrimidines. Eur. J. Med. Chem. 2018, 155, 406–417. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, N.F.H.; Ghareeb, E.A. Synthesis of Novel Substituted Tetrahydropyrimidine Derivatives and Evaluation of Their Pharmacological and Antimicrobial Activities. J. Heterocycl. Chem. 2019, 56, 81–91. [Google Scholar] [CrossRef]
- Li, L.; Liu, J.; Yang, Z.; Zhao, H.; Deng, B.; Ren, Y.; Mai, R.; Huang, J.; Chen, J. Discovery of Thieno[2,3-d]Pyrimidine-Based KRAS G12D Inhibitors as Potential Anticancer Agents via Combinatorial Virtual Screening. Eur. J. Med. Chem. 2022, 233, 114243. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.V.; Sarkate, A.P.; Lokwani, D.K.; Pansare, D.N.; Gattani, S.G.; Sheaikh, S.S.; Jain, S.P.; Bhandari, S.V. Bioorganic & Medicinal Chemistry Letters Explorations of Novel Pyridine-Pyrimidine Hybrid Phosphonate Derivatives as Aurora Kinase Inhibitors. Bioorg. Med. Chem. Lett. 2022, 67, 128747. [Google Scholar]
- Mohamed, M.S.; Sayed, A.I.; Khedr, M.A.; Nofal, S.; Soror, S.H. Evaluation of Novel Pyrrolopyrimidine Derivatives as Antiviral against Gastroenteric Viral Infections. Eur. J. Pharm. Sci. 2019, 127, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Eweas, A.F.; Abdallah, Q.M.A.; Hassan, E.S.I. Design, Synthesis, Molecular Docking of New Thiopyrimidine-5- Carbonitrile Derivatives and Their Cytotoxic Activity against HepG2 Cell Line. J. Appl. Pharm. Sci. 2014, 4, 102–111. [Google Scholar]
- Gordon, J.L.; Brown, M.A.; Reynolds, M.M. Cell-Based Methods for Determination of Efficacy for Candidate Therapeutics in the Clinical Management of Cancer. Diseases 2018, 6, 85. [Google Scholar] [CrossRef]
- Adan, A.; Kiraz, Y.; Baran, Y. Cell Proliferation and Cytotoxicity Assays. Curr. Pharm. Biotechnol. 2016, 17, 1213–1221. [Google Scholar] [CrossRef]
- Fang, X.J.; Jiang, H.; Zhu, Y.Q.; Zhang, L.Y.; Fan, Q.H.; Tian, Y. Doxorubicin Induces Drug Resistance and Expression of the Novel CD44st via NF-ΚB in Human Breast Cancer MCF-7 Cells. Oncol. Rep. 2014, 31, 2735–2742. [Google Scholar] [CrossRef] [PubMed]
- Hird, A.W.; Tron, A.E. Recent Advances in the Development of Mcl-1 Inhibitors for Cancer Therapy. Pharmacol. Ther. 2019, 198, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Wei, A.H.; Roberts, A.W.; Spencer, A.; Rosenberg, A.S.; Siegel, D.; Walter, R.B.; Caenepeel, S.; Hughes, P.; McIver, Z.; Mezzi, K.; et al. Targeting MCL-1 in Hematologic Malignancies: Rationale and Progress. Blood Rev. 2020, 44, 100672. [Google Scholar] [CrossRef] [PubMed]
- Awad, S.; Mohamed, M.; Khodair, M.; Hameed, R. Synthesis and Evaluation of Cytotoxic Activity of Certain Benzo[h]Chromene Derivatives. Anticancer. Agents Med. Chem. 2021, 21, 963–986. [Google Scholar] [CrossRef] [PubMed]
- El-hameed, R.H.A.; Mohamed, M.S.; Awad, S.M.; Bardes, B.; Khodair, M.A.E.; Mansour, Y.E.; Abd, R.H.; Mohamed, M.S.; Awad, S.M. Novel Benzo Chromene Derivatives: Design, Synthesis, Molecular Docking, Cell Cycle Arrest, and Apoptosis Induction in Human Acute Myeloid Leukemia HL-60 Cells. J. Enzym. Inhib. Med. Chem. 2023, 38, 405–422. [Google Scholar] [CrossRef]
- Evans, B.E.; Rittle, K.E.; Bock, M.G.; DiPardo, R.M.; Freidinger, R.M.; Whitter, W.L.; Lundell, G.F.; Veber, D.F.; Anderson, P.S.; Chang, R.S.L.; et al. Methods for Drug Discovery: Development of Potent, Selective, Orally Effective Cholecystokinin Antagoniststs. J. Med. Chem. 1988, 31, 2235–2246. [Google Scholar] [CrossRef] [PubMed]
- NCI Web Site. Available online: https://dtp.cancer.gov/ (accessed on 11 August 2024).
- NCI Methodology. Available online: https://dtp.cancer.gov/discovery_development/nci-60/methodology.htm (accessed on 11 August 2024).
- Boyd, M.R.; Paull, K.D. Some Practical Considerations and Applications of the National Cancer Institute in Vitro Anticancer Drug Discovery Screen. Drug Dev. Res. 1995, 34, 91–109. [Google Scholar] [CrossRef]
- Kalalbandi, V.K.A.; Seetharamappa, J. 1-[(2E)-3-Phenylprop-2-Enoyl]-1H-Benzimidazoles as Anticancer Agents: Synthesis, Crystal Structure Analysis and Binding Studies of the Most Potent Anticancer Molecule with Serum Albumin. MedChemComm 2015, 6, 1942–1953. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Thabrew, M.I.; Hughes, R.D.; McFarlane, I.G. Screening of Hepatoprotective Plant Components Using a HepG2 Cell Cytotoxicity Assay. J. Pharm. Pharmacol. 1997, 49, 1132–1135. [Google Scholar] [CrossRef] [PubMed]
- El-Menshawi, B.S.; Fayad, W.; Mahmoud, K.; El-Hallouty, S.M.; El-Manawaty, M.; Olofsson, M.H.; Linder, S. Screening of Natural Products for Therapeutic Activity against Solid Tumors. Indian J. Exp. Biol. 2010, 48, 258–264. [Google Scholar]
- Kojima, K.; Nakamura, H.; Komeya, M.; Yamanaka, H.; Makino, Y.; Okada, Y.; Akiyama, H.; Torikai, N.; Sato, T.; Fujii, T.; et al. Neonatal Testis Growth Recreated in Vitro by Two-Dimensional Organ Spreading. Biotechnol. Bioeng. 2018, 115, 3030–3041. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cytotoxicity of Most Active Derivatives (5-Dose Active) against Standard ** | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Tested Compound | 3a (4-Chloro) | 4a (4-Tolylamino) | ||||||||
| Different Dilution (µg/mL) | Different Dilution (µg/mL) | |||||||||
| 500 | 250 | 125 | 62.5 | 31.25 | 500 | 250 | 125 | 62.5 | 31.25 | |
| Viability % | 52 ± 1.22 | 64 ± 1.16 | 64.5 ± 1.22 | 61.5 ± 1.35 | 81 ± 1.70 | 53.5 ± 0.44 | 59.5 ± 0.79 | 60.5 ± 0.80 | 62.37 ± 0.59 | 65 ± 0.43 |
| Cytotoxicity % | 48 ± 1.22 | 36 ± 1.16 | 35.5 ± 1.22 | 38.4 ± 1.35 | 19 ± 1.70 | 46.5 ± 0.44 | 40.4 ± 0.79 | 39.4 ± 0.80 | 37.63 ± 0.59 | 35 ± 0.43 |
| * IC50 µg/ml | 399.6891 | 631.2369 | ||||||||
| Groups | Bcl-2 | Mcl-1 |
|---|---|---|
| Control | 1 ± 0.00 | 1 ± 0.00 |
| Compound 3a | 0.77 ± 0.05 * | 0.71 ± 0.04 * |
| Compound | Docking Score (S) Kcal/mol | RMSD | E-Score 1 (London dG) Kcal/mol | E-Score 2 (London dG) Kcal/mol | Binding Interaction (Ligand Receptor) |
|---|---|---|---|---|---|
| 3a | −5.5000 | 2.0588 | −8.1277 | −5.5000 | (N-Arg143) (H-b) (Cl-Asp108) (H-b) |
| 4a | −5.67806 | 1.65981 | −8.56915 | −5.67806 | (benzene H-Leu 134) (pi-H) |
| Receptor ligand, (1XJ) | −8.7886 | 2.0588 | −8.8739 | −8.7886 | (benzene H-Asn 140) (pi-H) (S-Asp 100) (H-b) (Gly 142-O) (H-b) |
| Compound | Docking Score (S) Kcal/mol | RMSD | E-Score 1 (London dG) Kcal/mol | E-Score 2 (London dG) Kcal/mol | Binding Interaction (Ligand Receptor) |
|---|---|---|---|---|---|
| 3a | −5.3952 | 1.4482 | −8.7543 | −5.3952 | (N-Arg 263) (H-b) |
| 4a | −5.9229 | 1.7320 | −9.7526 | −5.9229 | (benzene + Arg363) (Pi- +) |
| Receptor ligand, (JLH) | −6.9102 | 1.4881 | −9.9729 | −8.7886 | (Arg 263-O) (H-b) (Arg 263-O) (H-b) |
| Target | Sequence |
|---|---|
| Bcl-2 | F 5′-ATCGCCCTGTGGATGACTGAGT-3′ R 5′-GCCAGGAGAAATCAAACAGAGGC-3′ |
| Mcl-1 | F 5′-CCAAGAAAGCTGCATCGAACCAT-3′ R 5′-CAGCACATTCCTGATGCCACCT-3′ |
| GAPDH | F 5′-GTCTCCTCTGACTTCAACAGCG-3′ R 5′-ACCACCCTGTTGCTGTAGCCAA-3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khoder, Z.M.; Mohamed, M.S.; Awad, S.M.; Gharib, A.F.; Aly, O.; Khodair, M.A.E.-F.; Fatahala, S.S.; El-Hameed, R.H.A. Synthesis, Anti-Cancer Activity, Cell Cycle Arrest, Apoptosis Induction, and Docking Study of Fused Benzo[h]chromeno[2,3-d]pyrimidine on Human Breast Cancer Cell Line MCF-7. Molecules 2024, 29, 4697. https://doi.org/10.3390/molecules29194697
Khoder ZM, Mohamed MS, Awad SM, Gharib AF, Aly O, Khodair MAE-F, Fatahala SS, El-Hameed RHA. Synthesis, Anti-Cancer Activity, Cell Cycle Arrest, Apoptosis Induction, and Docking Study of Fused Benzo[h]chromeno[2,3-d]pyrimidine on Human Breast Cancer Cell Line MCF-7. Molecules. 2024; 29(19):4697. https://doi.org/10.3390/molecules29194697
Chicago/Turabian StyleKhoder, Zainab M., Mosaad S. Mohamed, Samir M. Awad, Amal F. Gharib, Omnia Aly, Marwa Abd El-Fattah Khodair, Samar S. Fatahala, and Rania H. Abd El-Hameed. 2024. "Synthesis, Anti-Cancer Activity, Cell Cycle Arrest, Apoptosis Induction, and Docking Study of Fused Benzo[h]chromeno[2,3-d]pyrimidine on Human Breast Cancer Cell Line MCF-7" Molecules 29, no. 19: 4697. https://doi.org/10.3390/molecules29194697
APA StyleKhoder, Z. M., Mohamed, M. S., Awad, S. M., Gharib, A. F., Aly, O., Khodair, M. A. E.-F., Fatahala, S. S., & El-Hameed, R. H. A. (2024). Synthesis, Anti-Cancer Activity, Cell Cycle Arrest, Apoptosis Induction, and Docking Study of Fused Benzo[h]chromeno[2,3-d]pyrimidine on Human Breast Cancer Cell Line MCF-7. Molecules, 29(19), 4697. https://doi.org/10.3390/molecules29194697