
Synthesis, Crystal Structure and Antifungal Activity of (E)-1-(4-Methylbenzylidene)-4-(3-Isopropylphenyl) Thiosemicarbazone: Quantum Chemical and Experimental Studies


Abstract
:1. Introduction
2. Results and Discussion
2.1. Chemical Synthesis
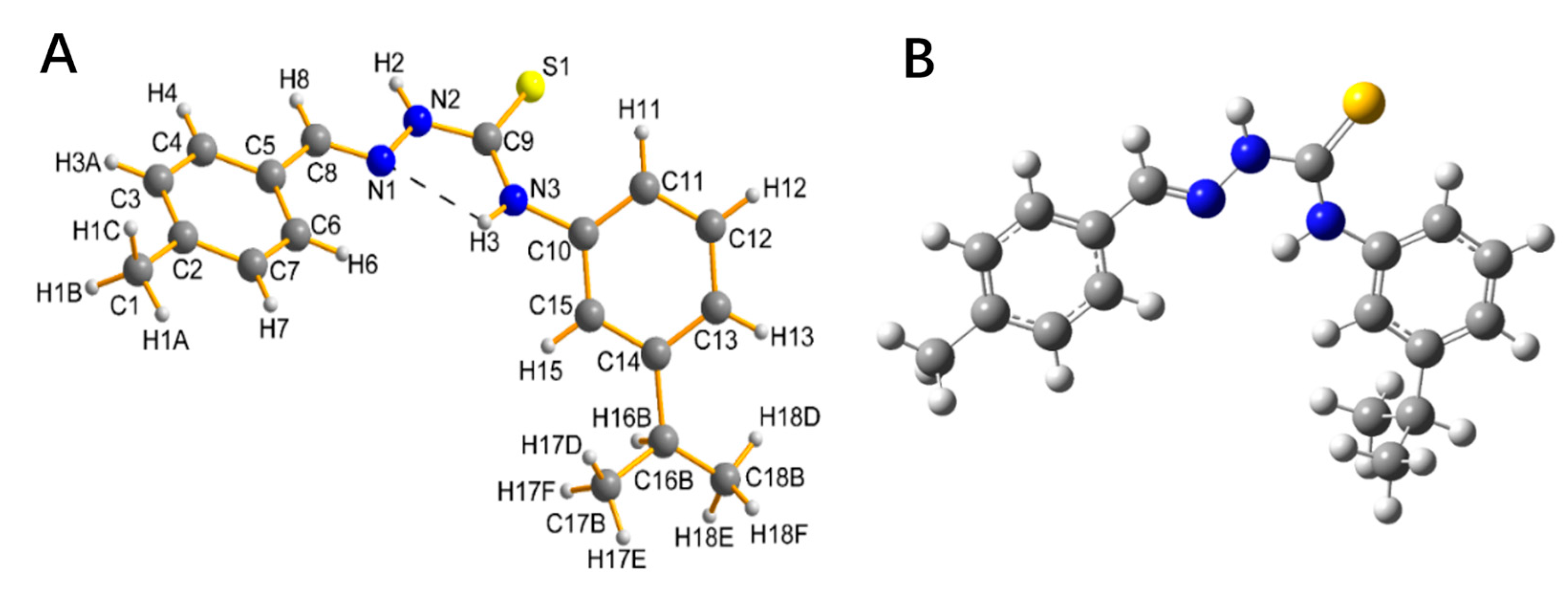
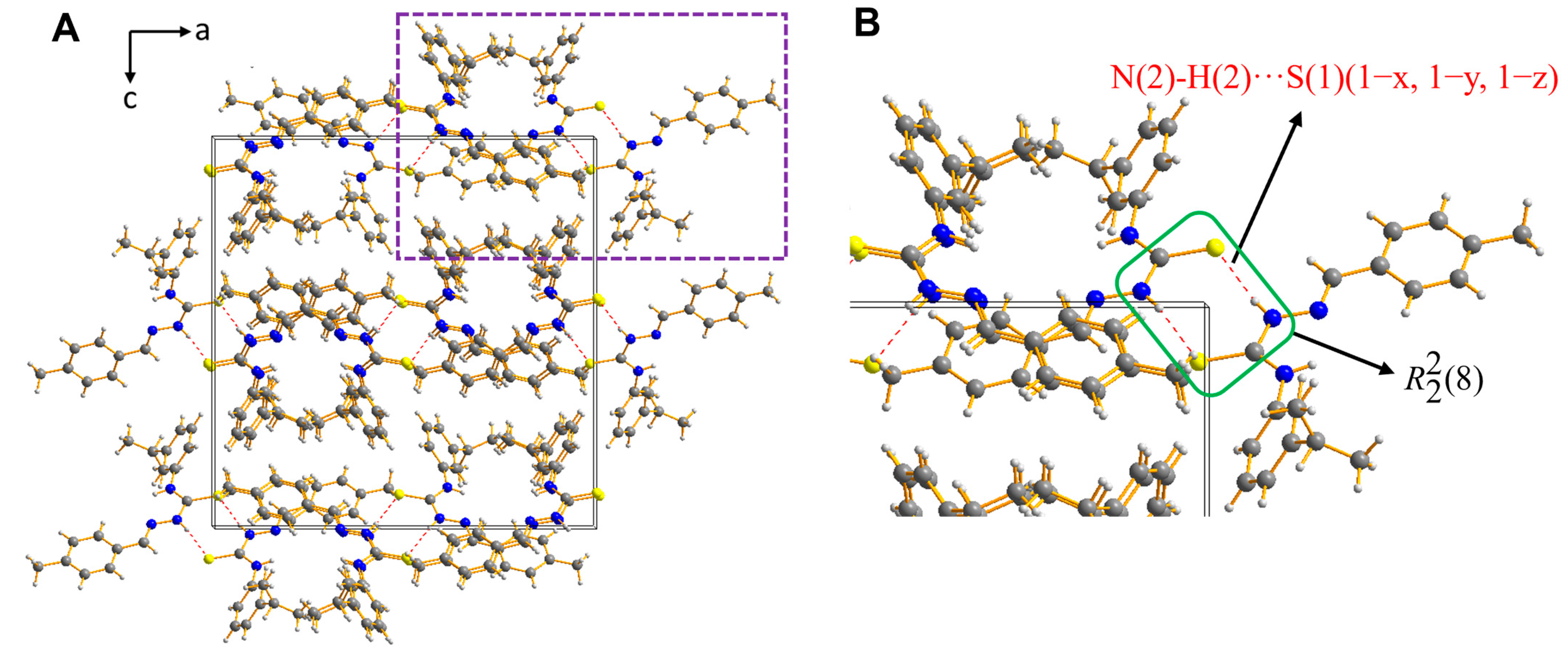
2.2. Description of the Crystal Structure
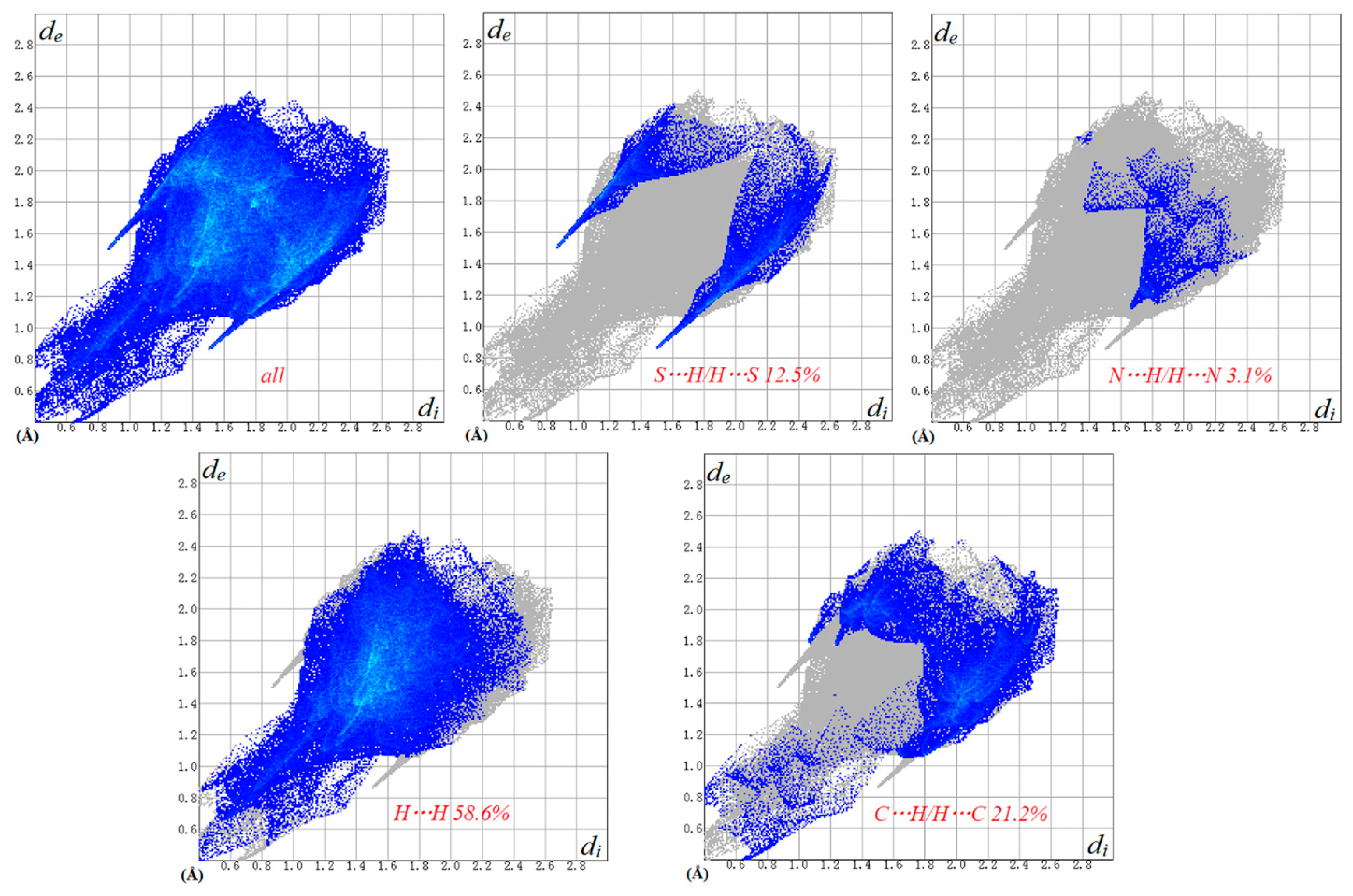
2.3. Hirshfeld Surface Analysis
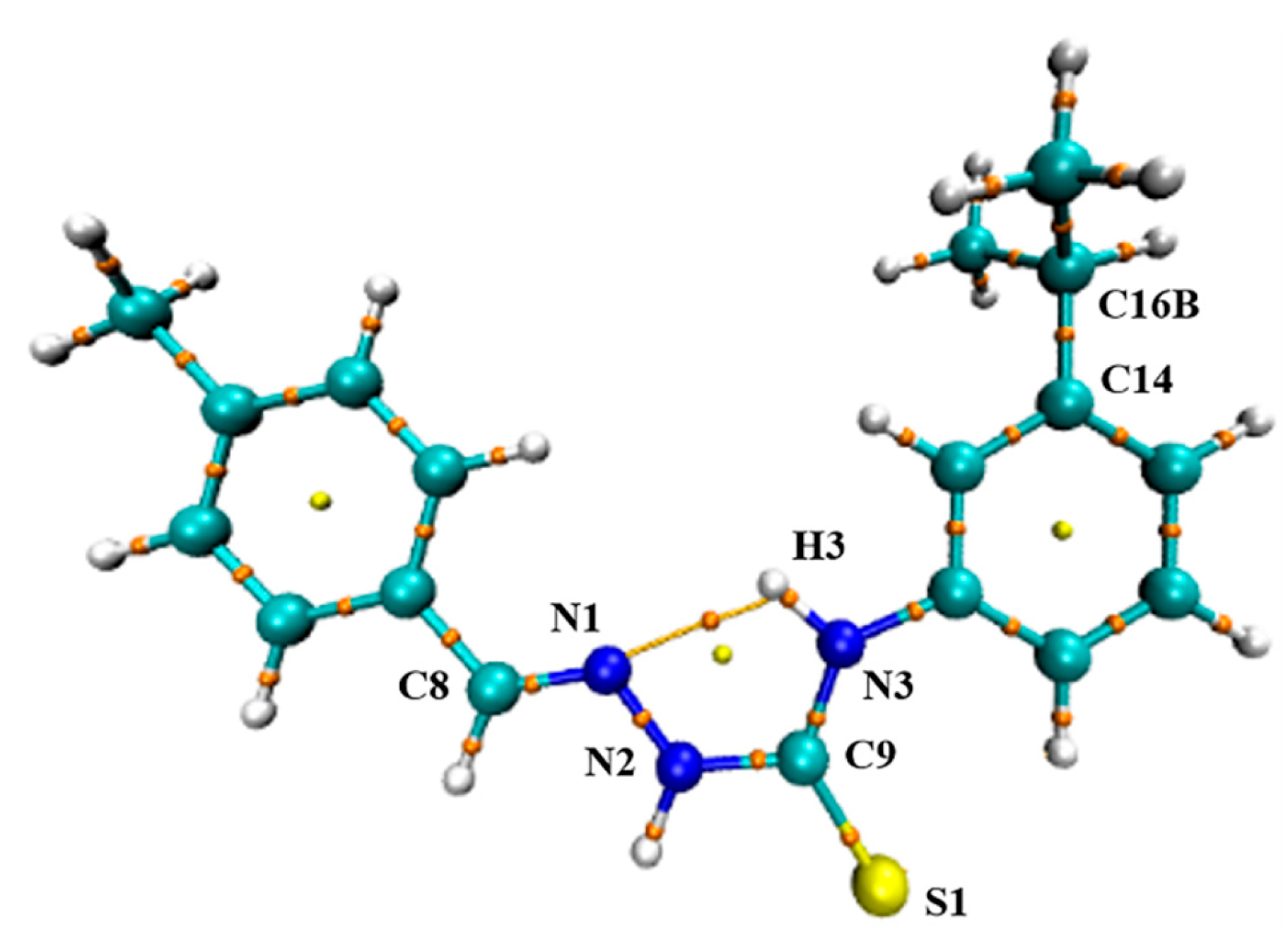
2.4. AIM Theory
2.5. ESP Analysis
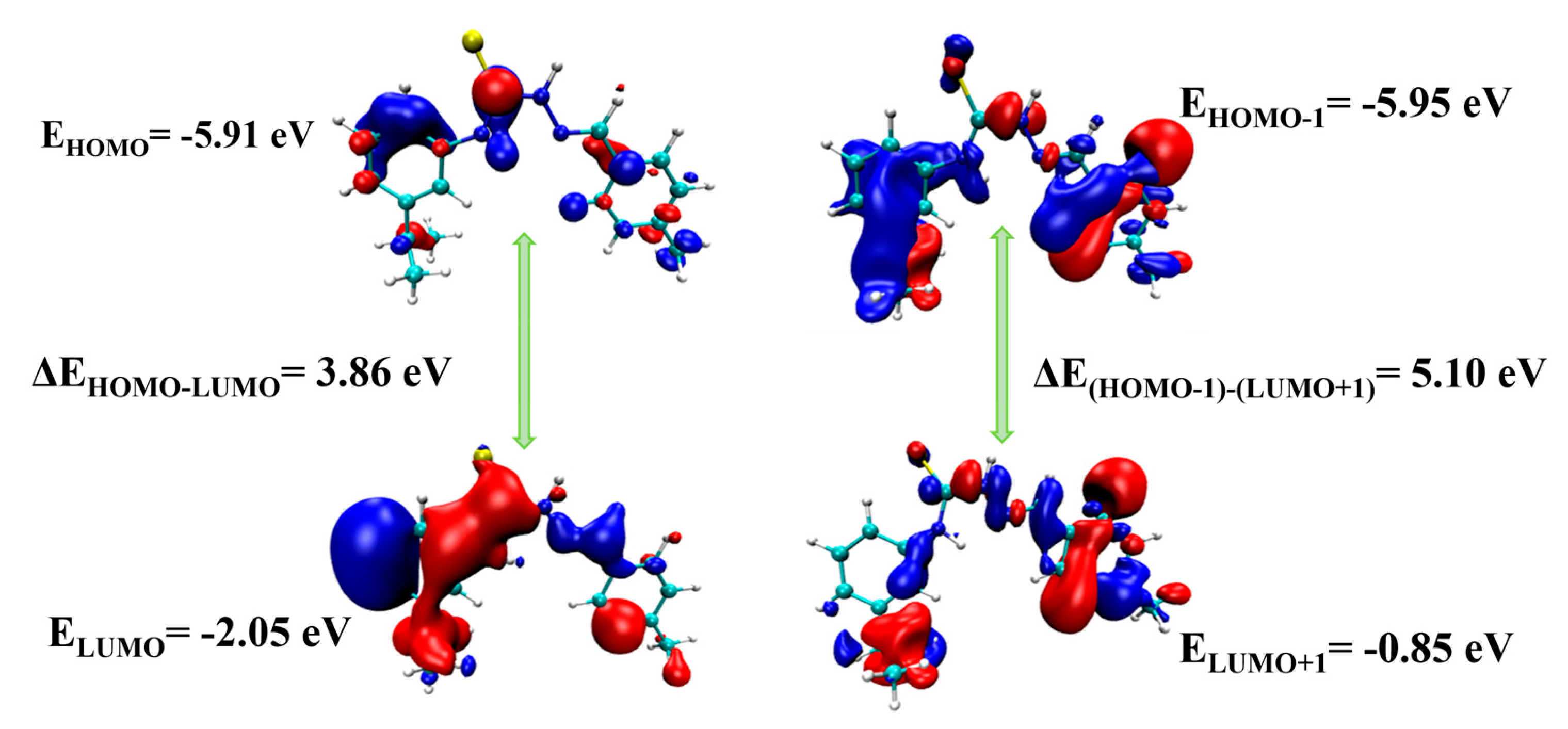
2.6. UV-Visible and HOMO-LUMO Analyses
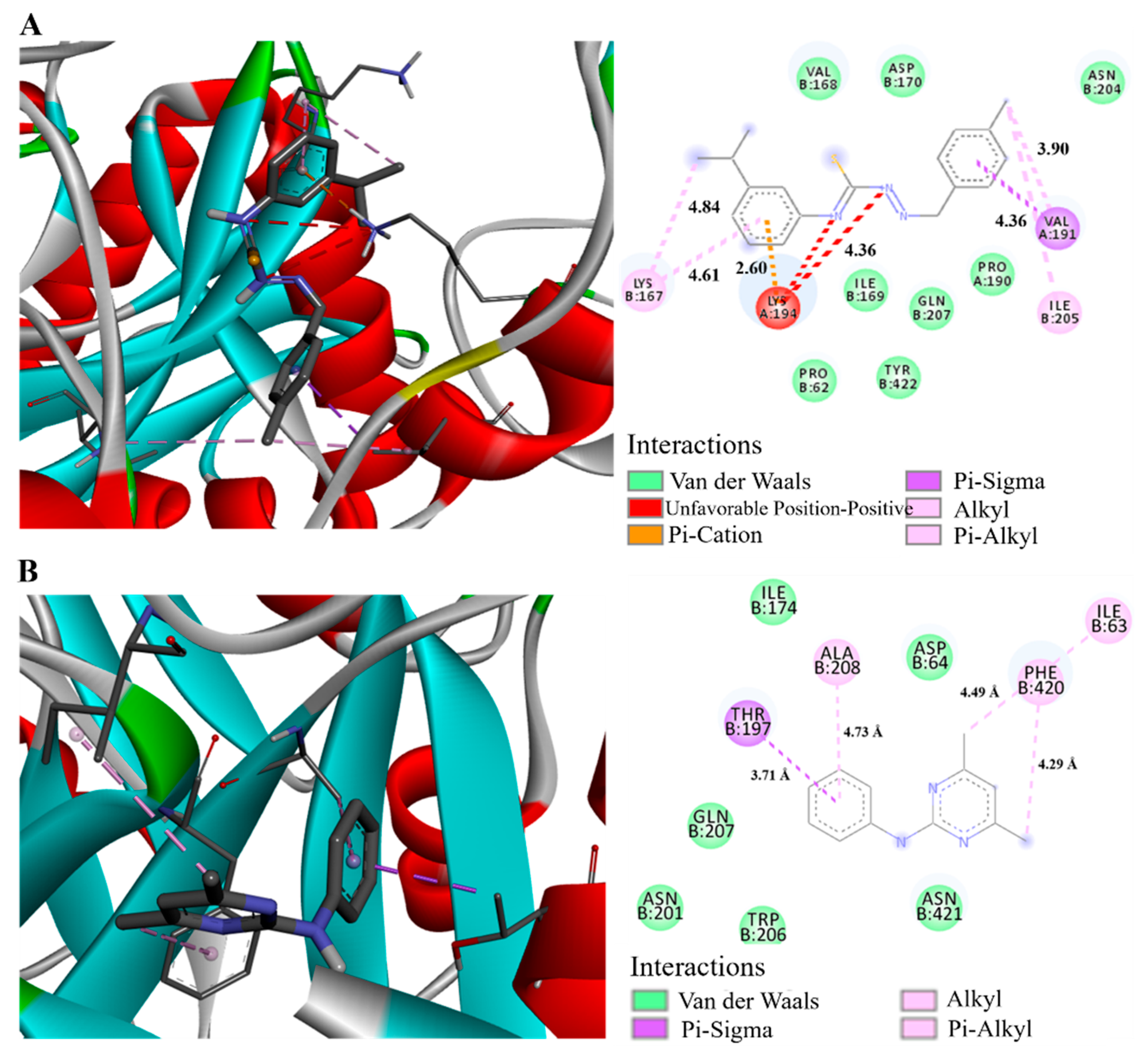
2.7. Molecular Docking
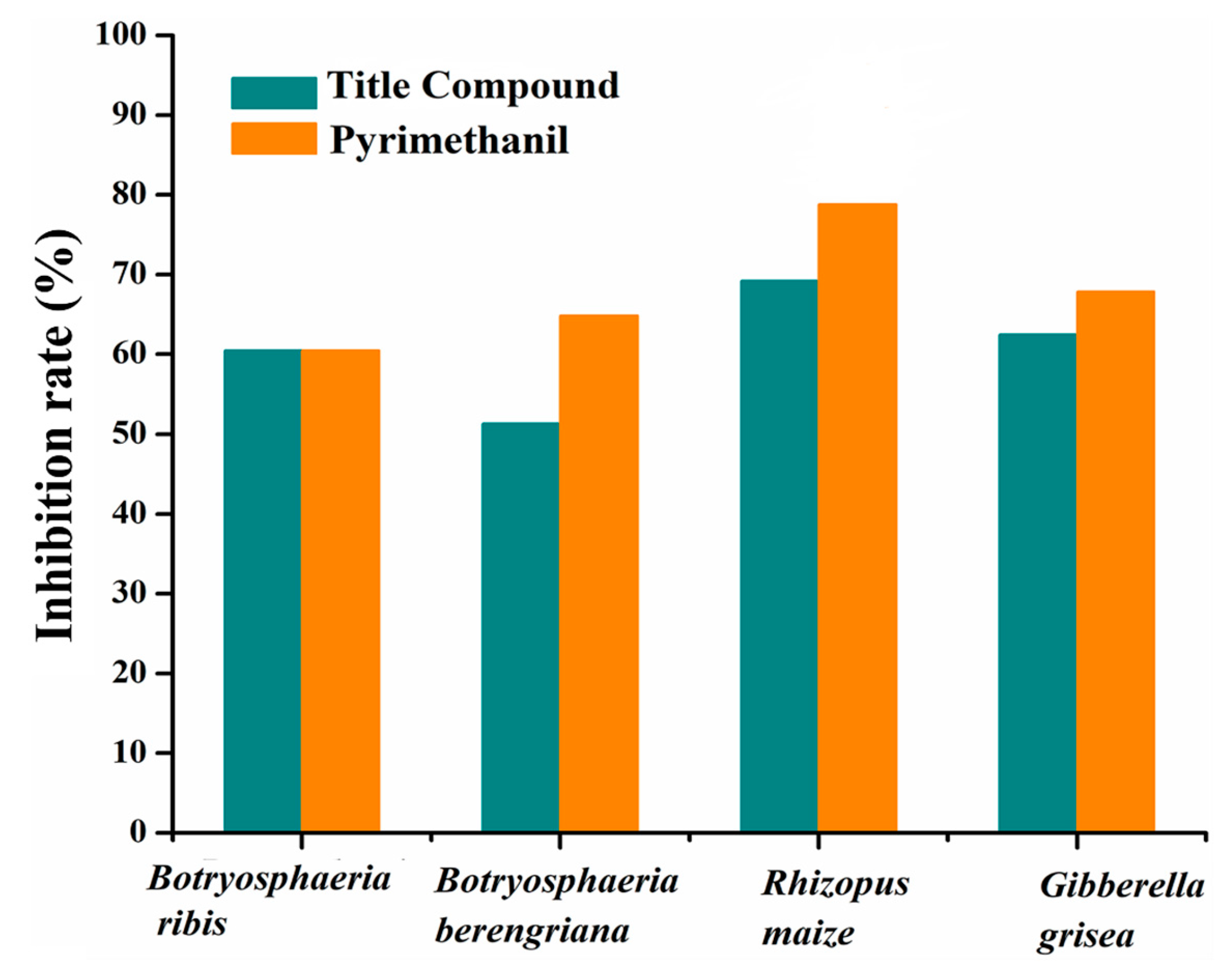
2.8. Antifungal Activity
3. Experimental
3.1. General
3.2. One-Pot Four-Step Synthesis of (E)-1-(4-Methylbenzylidene)-4-(3-Isopropylphenyl) Thiosemi-Carbazone 4 (Scheme 1)
3.3. Analytical Data
3.4. Crystallographic Data Collection and Refinement
3.5. Quantum Chemical Calculation
3.6. Antifungal Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kara, Y.E.; Gülseven Sıdır, Y.; Sıdır, İ. Solvatochromism, optical and electronic properties of thiosemicarbazone derivatives in solution phase. Opt. Quant. Electron. 2024, 56, 658. [Google Scholar] [CrossRef]
- Pandey, S.K.; Pratap, S.; Gozzi, G.; Marverti, G.; Butcher, R.J. Synthesis, molecular structure exploration and in vitro cytotoxicity screening of five novel N, N′-disubstituted thiocarbamide derivatives. Phosphorus Sulfur Silicon Relat. Elem. 2018, 193, 507–514. [Google Scholar] [CrossRef]
- Ashfaq, M.; Khalid, M.; Tahir, M.N.; Ali, A.; Arshad, M.N.; Asiri, A.M. Synthesis of crystalline fluoro-functionalized imines, single crystal investigation, Hirshfeld surface analysis, and theoretical exploration. ACS Omega 2022, 7, 9867–9878. [Google Scholar] [CrossRef] [PubMed]
- Santini, C.; Pellei, M.; Gandin, V.; Porchia, M.; Tisato, F.C.; Marzano, C. Advances in copper complexes as anticancer agents. Chem. Rev. 2014, 114, 815–862. [Google Scholar] [CrossRef] [PubMed]
- Qi, F.; Qi, Q.; Song, J.; Huang, J. Synthesis, Crystal Structure, Biological Evaluation and in Silico Studies on Novel (E)-1-(Substituted Benzylidene)-4-(3-isopropylphenyl) thiosemicarbazone Derivatives. Chem. Biodivers. 2021, 18, e2000804. [Google Scholar] [CrossRef]
- Zhang, X.; Huang, J.; Zhang, Y.; Qi, F.; Wang, S.F.; Song, J.R. Synthesis, crystal structure and non-covalent interactions analysis of novel N-substituted thiosemicarbazone. Chem. Res. Chin. Univ. 2019, 3, 471–477. [Google Scholar] [CrossRef]
- Pandey, S.K.; Pratap, S.; Tiwari, M.K.; Marverti, G.; Jasinski, J.P. Experimental and theoretical exploration of molecular structure and anticancer properties of two N,N′–disubstituted thiocarbamide derivatives. J. Mol. Struct. 2019, 1175, 963–970. [Google Scholar] [CrossRef]
- Larik, F.A.; Saeed, A.; Channar, P.A.; Muqadar, U.; Abbas, Q.; Hassan, M.; Seo, S.Y.; Bolte, M. Design, synthesis, kinetic mechanism and molecular docking studies of novel 1-pentanoyl-3-arylthioureas as inhibitors of mushroom tyrosinase and free radical scavengers. Eur. J. Med. Chem. 2017, 141, 273–281. [Google Scholar] [CrossRef]
- Ferrari, M.B.; Gasparri, F.G.; Leporati, E.; Pelosi, G.; Rossi, R.; Tarasconi, P.; Albertini, R.; Bonati, A.; Lunghi, P.; Pinelli, S. Synthesis, characterisation and biological activity of three copper (II) complexes with a modified nitrogenous base: 5-formyluracil thiosemicarbazone. J. Inorg. Biochem. 1998, 70, 145–154. [Google Scholar] [CrossRef]
- Pandey, S.K.; Singh, D.P.; Marverti, G.; Butcher, R.J.; Pratap, S. Monodentate Coordination of N, N′-Disubstituted Thiocarbamide Ligands: Syntheses, Structural Analyses, In Vitro Cytotoxicity and DNA Damage Studies of Cu(I) Complexes. ChemistrySelect 2018, 3, 3675–3679. [Google Scholar] [CrossRef]
- Hirose, D.; Hobara, S.; Matsuoka, S.; Kato, K.; Tanabe, Y.; Uchida, M.; Kudoh, S.; Osono, T. Diversity and community assembly of moss-associated fungi in ice-free coastal outcrops of continental Antarctica. Fungal Ecol. 2016, 24, 94–101. [Google Scholar] [CrossRef]
- Guo, Z.; Wei, C.; Wang, S.; Qi, F.; He, Z.; Majeed, U.; Huang, J. Crystal structure analysis of three (E)-N-(3-methyl, 5-fluorinephenyl)-2-(4-substituted benzylidene) thiosemicarbazone derivatives: Experimental and theoretical studies. J. Mol. Struct. 2022, 1247, 131383. [Google Scholar] [CrossRef]
- Microbiology, N. Stop neglecting fungi. Nat. Microbiol. 2017, 2, 17120. [Google Scholar]
- Olender, D.; Żwawiak, J.; Lukianchuk, V.; Lesyk, R.; Kropacz, A.; Fojutowski, A.; Zaprutko, L. Synthesis of some N-substituted nitroimidazole derivatives as potential antioxidant and antifungal agents. Eur. J. Med. Chem. 2009, 44, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.L.; Luo, Z.F.; Li, Y.; Liu, X.Y.; Fan, J.D.; Xue, W.; Tang, L.; Li, Y. Synthesis and antifungal activity of imidazo [1,2-b] pyridazine derivatives against phytopathogenic fungi. Bioorg. Med. Chem. Lett. 2020, 30, 127139. [Google Scholar] [CrossRef]
- Rodrigues, M.L.; Nosanchuk, J. Fungal Diseases as Neglected Pathogens: A Wake-up Call to Public Health Officials. Advances in Clinical Immunology, Medical Microbiology, COVID-19, and Big Data; Jenny Stanford Publishing: Singapore, 2021; pp. 399–411. [Google Scholar]
- Bajaj, K.; Buchanan, R.M.; Grapperhaus, C.A. Antifungal activity of thiosemicarbazones, bis (thiosemicarbazones), and their metal complexes. J. Inorg. Biochem. 2021, 225, 111620. [Google Scholar] [CrossRef]
- Kumar, B.; Devi, J.; Dubey, A.; Tufail, N.; Khurana, D. Thiosemicarbazone ligands based transition metal complexes: A multifaceted investigation of antituberculosis, anti-inflammatory, antibacterial, antifungal activities, and molecular docking, density functional theory, molecular electrostatic potential, absorption, distribution, metabolism, excretion, and toxicity studies. Appl. Organomet. Chem. 2024, 38, e7345. [Google Scholar]
- Parr, R.J.; Szentpaly, L.V.; Liu, S. Electrophilicity Index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Ribeiro, M.A.; de-Farias, I.F.; Freire, P.T.C. Synthesis, crystal structure, structural and spectroscopic analysis of (2E)-1-(4-chlorophenyl)-3-(4-methoxyphenyl) prop-2-en-1-one. J. Mol. Struct. 2023, 1294, 136410. [Google Scholar] [CrossRef]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Hirshfeld, F.L. Bonded-atom fragments for describing molecular charge densities. Theor. Chim. Acta 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Gester, R.; Siqueira, M.; Cunha, A.R.; Araújo, R.S.; Provasi, P.F.; Canuto, S. Assessing the dipolar-octupolar NLO behavior of substituted thiosemicarbazone assemblies. Chem. Phys. Lett. 2023, 831, 140807. [Google Scholar] [CrossRef]
- Parkin, A.; Barr, G.; Dong, W.; Gilmore, C.J.; Jayatilaka, D.; McKinnon, J.J.; Spackman, M.A.; Wilson, C.C. Comparing entire crystal structures: Structural genetic fingerprinting. CrystEngComm 2007, 9, 648–652. [Google Scholar] [CrossRef]
- Thamarai, A.; Vadamalar, R.; Raja, M.; Muthu, S.; Narayana, B.; Ramesh, P.; Muhamed, R.R.; Sevvanthi, S.; Aayisha, S. Molecular structure interpretation, spectroscopic (FT-IR, FT-Raman), electronic solvation (UV–Vis, HOMO-LUMO and NLO) properties and biological evaluation of (2E)-3-(biphenyl-4-yl)-1-(4-bromophenyl) prop-2-en-1-one: Experimental and computational modeling approach. Spectrochim. Acta Part A 2020, 226, 117609. [Google Scholar]
- Sriram, D.; Yogeeswari, P.; Dhakla, P.; Senthilkumar, P.; Banerjee, D.; Manjashetty, T.H. 5-Nitrofuran-2-yl derivatives: Synthesis and inhibitory activities against growing and dormant mycobacterium species. Bioorg. Med. Chem. Lett. 2009, 19, 1152–1154. [Google Scholar] [CrossRef]
- Nevagi, R.J.; Dhake, A.S.; Narkhede, H.I.; Kaur, P. Design, synthesis and biological evaluation of novel thiosemicarbazide analogues as potent anticonvulsant agents. Bioorg. Chem. 2014, 54, 68–72. [Google Scholar] [CrossRef]
- Cunha, S.; da Silva, T.L. One-pot and catalyst-free synthesis of thiosemicarbazones via multicomponent coupling reactions. Tetrahedron Lett. 2009, 50, 2090–2093. [Google Scholar] [CrossRef]
- West, D.X.; Castiñeiras, A.; Bermejo, E. Structural and spectral studies of a 1-N(4)-substituted thiosemicarbazone derived from 2-hydroxy-1,4-naphthaquinone. J. Mol. Struct. 2000, 520, 103–106. [Google Scholar] [CrossRef]
- Zhang, J.; Geng, H.; Zhuang, L.; Wang, G. 4-Methylbenzaldehyde thiosemicarbazone. Acta Cryst. 2009, 65, o2244. [Google Scholar] [CrossRef]
- Mahmoudi, G.; Castiñeiras, A.; Garczarek, P.; Bauzá, A.; Rheingold, A.L.; Kinzhybalo, V.; Frontera, A. Synthesis, X-ray characterization, DFT calculations and Hirshfeld surface analysis of thiosemicarbazone complexes of Mn+ ions (n = 2, 3; M = Ni, Cd, Mn, Co and Cu). CrystEngComm 2016, 18, 1009–1023. [Google Scholar] [CrossRef]
- Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Khalaf, M.M.; Abd El-Lateef, H.M.; Gouda, M.; Abdelhamid, A.A.; Abdelbaset, M.; Alsulami, A.H.; Almarri, M.N.; Abdou, A. Designing, DFT, biological, & molecular docking analysis of new Iron (III) & copper (II) complexes incorporating 1-{[-(2-Hydroxyphenyl) methylene] amino}-5,5-diphenylimidazolidine-2,4-dione (PHNS). Comput. Biol. Chem. 2024, 109, 108031. [Google Scholar]
- Koch, W.; Frenking, G.; Gauss, J.; Cremer, D.; Collins, J.R. Helium chemistry: Theoretical predictions and experimental challenge. J. Am. Chem. Soc. 1987, 109, 5917–5934. [Google Scholar] [CrossRef]
- Ganorkar, K.; Mukherjee, S.; Wankar, S.; Joshi, R.; Das, C.; Ghosh, S.K. Exploration of the ESIPT process in a newly designed potential bioactive thiosemicarbazone Schiff base: Spectroscopic analysis accompanied by molecular optimization and crystallographic study. J. Photoch. Photobiol. A 2019, 371, 81–90. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, X.; Qi, F.; Huang, J.; Wei, C.; Guo, Z. Crystal structure analysis of (E)-N-(3,5-dimethylphenyl)-2-(substituted benzylidene) thiosemicarbazone: Experimental and theoretical studies. J. Phys. Org. Chem. 2021, 34, e4138. [Google Scholar] [CrossRef]
- Ayalew, M.E. DFT studies on molecular structure, thermodynamics parameters, HOMO-LUMO and spectral analysis of pharmaceuticals compound quinoline (Benzo [b] Pyridine). J. Biol. Chem. 2022, 13, 29–42. [Google Scholar] [CrossRef]
- Younis, A.M.; El-Gamil, M.M.; Rakha, T.H.; El-Reash, G.M.A. Iron (III), copper (II), cadmium (II), and mercury (II) complexes of isatin carbohydrazone Schiff base ligand (H3L): Synthesis, characterization, X-ray diffraction, cyclic voltammetry, fluorescence, density functional theory, biological activity, and molecular docking studies. Appl. Organomet. Chem. 2021, 35, e6250. [Google Scholar]
- Kumar, V.S.; Potla, K.M.; Srishailam, K.; Kaleeswaran, S.; Javed, S.; Manikandan, A.; Muthu, S. Anti-fibrinolytic activity, electron delocalization, vibrational spectra, drug-likeness properties and bio-activity studies on Anagrelide. Results Chem. 2024, 7, 101379. [Google Scholar] [CrossRef]
- Gil, D.M.; Lestard, M.E.D.; Estévez-Hernández, O.; Duque, J.; Reguera, E. Quantum chemical studies on molecular structure, spectroscopic (IR, Raman, UV-Vis), NBO and Homo-Lumo analysis of 1-benzyl-3-(2-furoyl) thiourea. Spectrochim. Acta Part A 2015, 145, 553–562. [Google Scholar] [CrossRef]
- Baggio, R.; Brovelli, F.; Moreno, Y.; Pinto, M.; Soto-Delgado, J. Structural, electrochemical and theoretical study of a new chalcone derivative containing 3-thiophene rings. J. Mol. Struct. 2016, 1123, 1–7. [Google Scholar] [CrossRef]
- Egorova, S.K.; Gordeev, G.E.; Ananikov, P.V. Biological activity of ionic liquids and their application in pharmaceutics and medicine. Chem. Rev. 2017, 117, 7132–7189. [Google Scholar] [CrossRef] [PubMed]
- Elantabli, F.M.; El-Medani, S.M.; Kozakiewicz-Piekarz, A.; Ramadan, R.M. New transition metal complexes of 1-phenyl-2-((quinolin-2-ylmethylene) amino) ethan-1-ol Schiff base: Spectroscopic, X-ray, DFT, Hirshfeld surface analysis, biological, and molecular docking studies. Appl. Organomet. Chem. 2022, 36, e6779. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112. [Google Scholar] [CrossRef] [PubMed]
- Pettit, G.R.; McNulty, J.; Herald, D.L.; Doubek, D.L.; Chapuis, J.C.; Schmidt, J.M.; Tackett, L.P.; Boyd, M.R. Antineoplastic Agents. 362. Isolation and X-ray Crystal Structure of Dibromophakellstatin from the Indian Ocean Sponge Phakellia mauritiana. J. Nat. Prod. 1997, 60, 180–183. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F.W. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Imran, A.; Shehzad, M.T.; Al Adhami, T.; Rahman, K.M.; Hussain, D.; Alharthy, R.D.; Shafiq, Z.; Iqbal, J. Development of coumarin-thiosemicarbazone hybrids as aldose reductase inhibitors: Biological assays, molecular docking, simulation studies and ADME evaluation. Bioorg. Chem. 2021, 115, 105164. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, X.; Qiao, L.; Ding, Z.-M.; Hang, X.J.; Qin, B.F.; Song, J.R.; Huang, J. Synthesis, structures, drug-likeness, in vitro evaluation and in silico docking on novel N-benzoyl-N′-phenyl thiourea derivatives. J. Mol. Struct. 2019, 1176, 335–345. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical Formula | C18H21N3S |
|---|---|
| Formula weight | 311.44 |
| Temperature (K) | 296 (2) |
| Wavelength (Å) | 0.71073 |
| a (Å) | 20.224 (4) |
| b (Å) | 8.4969 (15) |
| c (Å) | 20.602 (4) |
| α (degree) | 90.00 |
| β (degree) | 90.00 |
| γ (degree) | 90.00 |
| Volume (Å3) | 3540.3 (12) |
| Z | 8 |
| Dc (Mg/m3) | 1.169 |
| Crystal system | Orthorhombic |
| Space group | Pbca |
| Index ranges | −23 < h < 23, −5 < k < 10, −24 < l < 24 |
| F (000) | 1328 |
| Theta range for data collection (°) | 2.2 to 25.0 |
| Reflections collected | 16947 |
| Independent reflections | 3114 |
| Rint | 0.108 |
| Goodness-of-fit on F2 (S) | 0.886 |
| R (all data) | R1 = 0.0670, wR2 = 0.2188 |
| Parameters | Experimental | Calculated |
|---|---|---|
| Bond length (Å) | ||
| S(1)-C(9) | 1.668 | 1.668 |
| N(1)-C(8) | 1.269 | 1.289 |
| N(2)-C(9) | 1.357 | 1.357 |
| N(3)-C(9) | 1.336 | 1.355 |
| C(2)-C(3) | 1.362 | 1.345 |
| C(1)-C(2) | 1.515 | 1.510 |
| N(3)-C(10) | 1.417 | 1.413 |
| Bond angle (°) | ||
| C(17B)-C(16B)-C(18B) | 123.0 | 124.5 |
| C(13)-C(14)-C(16B) | 119.1 | 120.9 |
| C(4)-C(5)-C(8) | 119.9 | 120.0 |
| C(3)-C(2)-C(7) | 117.4 | 117.9 |
| S(1)-C(9)-N(3) | 126.2 | 126.0 |
| Dihedral angle (°) | ||
| C(8)-N(1)-N(2)-C(9) | −179.0 | 0.6 |
| N(1)-N(2)-C(9)-S(1) | −177.3 | −177.3 |
| N(1)-N(2)-C(9)-N(3) | 3.9 | 3.6 |
| C(10)-N(3)-C(9)-S(1) | 0.8 | 1.0 |
| D-H…A | d(D-H) | d(H…A) | d(D…A) | ∠D-H…A |
|---|---|---|---|---|
| N(3)-H(3)···N(1) | 0.8600 Å | 2.1800 Å | 2.618(5) Å | 117.000° |
| N(2)-H(2)···S(1) a | 0.8600 Å | 2.5200 Å | 3.372(3) Å | 171.000° |
| Bond (X-Y) | ρ | ▽2ρ(BCP) | HBCP | Position (x, y, z) |
|---|---|---|---|---|
| N1=C8 | 0.00366 | −0.00505 | −0.00625 | (−4.17751, −3.09275, 0.00007) |
| N1-N2 | 0.00361 | −0.00695 | −0.00359 | (−1.84763, −3.81886, 0.00008) |
| C9-S1 | 0.00210 | −0.03291 | −0.00247 | (2.57242, −6.32864, 0.00013) |
| N3-H3 | 0.00341 | −0.01834 | −0.00510 | (1.28564, −1.27543, −0.00007) |
| C14-C16B | 0.00250 | −0.00583 | −0.00203 | (7.15390, 4.32850, −0.00006) |
| Interaction N1···H3 | 0.00232 | 0.08696 | 0.00198 | (−0.39692, −1.75043, −0.00003) |
| Excited State | Wavelength (nm) | Excitation Energy (eV) | Oscillator Strength (f) | Assignment | |
|---|---|---|---|---|---|
| Theoretical | Experimental | ||||
| S1 | 346 | 335 | 3.58 | 0.95 | HOMO→LUMO (67%) |
| S2 | 259 | 220 | 4.78 | 0.31 | HOMO-1→LUMO+1 (65%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ren, H.; Qi, F.; Zhao, Y.; Labidi, A.; Miao, Z. Synthesis, Crystal Structure and Antifungal Activity of (E)-1-(4-Methylbenzylidene)-4-(3-Isopropylphenyl) Thiosemicarbazone: Quantum Chemical and Experimental Studies. Molecules 2024, 29, 4702. https://doi.org/10.3390/molecules29194702
Ren H, Qi F, Zhao Y, Labidi A, Miao Z. Synthesis, Crystal Structure and Antifungal Activity of (E)-1-(4-Methylbenzylidene)-4-(3-Isopropylphenyl) Thiosemicarbazone: Quantum Chemical and Experimental Studies. Molecules. 2024; 29(19):4702. https://doi.org/10.3390/molecules29194702
Chicago/Turabian StyleRen, Haitao, Fan Qi, Yuzhen Zhao, Abdelkader Labidi, and Zongcheng Miao. 2024. "Synthesis, Crystal Structure and Antifungal Activity of (E)-1-(4-Methylbenzylidene)-4-(3-Isopropylphenyl) Thiosemicarbazone: Quantum Chemical and Experimental Studies" Molecules 29, no. 19: 4702. https://doi.org/10.3390/molecules29194702