Abstract
Herein, the pharmacokinetic profiles, binding interactions, and molecular properties of fluoroquinolone derivatives as prospective antiviral drugs are examined using a combination of docking, ADME, and DFT simulations. The effectiveness of the ligands is compared with the clinically tested and FDA-authorized medicine remdesivir. The findings demonstrated encouraging binding energies, indicating possible inhibitory effectiveness against SARS-CoV-2 Mpro. The fluoroquinolone derivatives also exhibit promising ADME characteristics, although compounds 5, 6, 9, 12–20 possess poor values, suggesting that oral administration may be possible. The potential of the selected compounds as SARS-CoV-2 Mpro inhibitors is thoroughly understood because of the integrated analysis of DFT, with compound 11 demonstrating the highest energy gap of 0.2604 eV of, docking with viral targets with docking scores of −7.9 to −5.9 kcal/mol, with compound 18 demonstrating the highest docking score, which is at the 13th position in energy difference in the DFT data. Their favorable electrical properties, robust binding interactions with viral targets, and attractive pharmacokinetic profiles boost their potential as prospective study subjects. These substances have the potential to be transformed into cutting-edge antiviral therapies that specifically target SARS-CoV-2 Mpro and related coronaviruses.
Keywords:
fluoroquinolones; SARS-CoV-2 Mpro; theoretical studies; DFT; molecular docking; ADME; cardiotoxicity 1. Introduction
After SARS-CoV-2 broke out in December 2019, the WHO labeled it an infectious disease on 11 March 2020. This led to a global health emergency [1,2,3]. As of 11 June 2020, COVID-19 has spread all over the world; there have been more than 7 million reported cases and over 400 thousand confirmed deaths (World Meter, 11 June 2020). In reaction to the pandemic, numerous countries imposed strict lockdowns or nationwide isolations to slow the virus’s spread. As an outcome, more than one-third of the world’s population was subjected to various sorts of limitations (Business Insider, the seventeenth of April 2020) [4].
In the early studies conducted on COVID-19, it was discovered that the spike protein receptor-binding domain alongside the host receptor ACE2 was responsible for controlling the movement of the virus across species and its spread from one person to another, similar to what happened during the SARS-CoV-2 outbreak of 2020 [5,6]. At present, no particular vaccine or drug is suitable for COVID-19 treatment; however, numerous FDA-approved antimalarial and antiviral drugs have been anticipated, again as supportive care. As a result, the rapid creation of novel compounds for possible COVID-19 treatment has become a vital goal [7,8].
A recent study on SARS-CoV-2 has highlighted the importance of the Mpro hydrolase (chymotrypsin-like protease), referred to as the new coronavirus’s primary protease; 3-chymotrypsin-like protease (Mpro or 3CLpro) is the primary protease. This kind of protease is important in the coronavirus life cycle, and blocking the virus’s replication could be a promising treatment for COVID-19 [9]. Traditional Chinese medicine has gained popularity among Chinese people for the treatment of COVID-19. Because of their efficiency and cost-effectiveness, in silico methods such as molecular docking and molecular dynamics have become popular for identifying possible inhibitors for specific diseases [10]. Numerous scientists are conducting virtual screening with various potent libraries, including antiviral agents, antimalarial medications, plant sources, fungi, and synthetic substances, so that they can find suitable blockers for COVID-19 protease; one instance of this is a major protease (Mpro) of diseases like constant corona [11,12,13]. Computer-aided drug design, especially molecular docking and molecular dynamic simulations, has garnered much interest in drug development for specific disease treatments. For this purpose, we used the protein from the database with PDB-ID: 6LU7.
Fluoroquinolone antibiotics, based on their wide-ranging systemic effects, are useful in combating urinary tract and respiratory infections [14,15,16]. Gram-positive and Gram-negative aerobic bacteria, representing a wide range of species, can be affected by fluoroquinolones. These are used to alleviate infections caused by bacteria in humans, animals, and animal farming, notably chicken farming. Mostly all quinolone drugs are fluoroquinolones that contain a fluoro element in their chemical structure and are effective against bacteria [17,18,19]. Ciprofloxacin, one of the most extensively used medicines in the world, belongs to this class. Many fluoroquinolone analogs have been produced and licensed by the FDA in past years as wide-ranging defensive medicines used to cure respiratory and urinary bladder diseases [20,21].
Several widely viable fluoroquinolones are designed for treating infectious diseases and are effective in non-bacterial occurrences. Fluoroquinolones are also used to treat antiviral, antibacterial, and antimalarial-related diseases [22]. The derivatives of quinolones are also effective against biological action, such as displaying kinase, RAS, cardiotonic-sensitive PDE III inhibitory, anticancer, anti-inflammatory, antiallergic, antifungal, and antiparasitic effects [23]. Recently, ciprofloxacin and moxifloxacin have shown strong binding energy with SARS-CoV-2 Mpro [23,24,25]. As a conclusion, pulmonary fluoroquinolones could be utilized as an adjunct treatment in COVID-19 patients. The 20 fluoroquinolones were chosen based on the variety of their structural makeup and previous studies that suggested they would be effective antiviral drugs. Through the docking experiment, we hope to uncover new structural requirements for the effective binding and suppression of the viral protein [26,27,28].
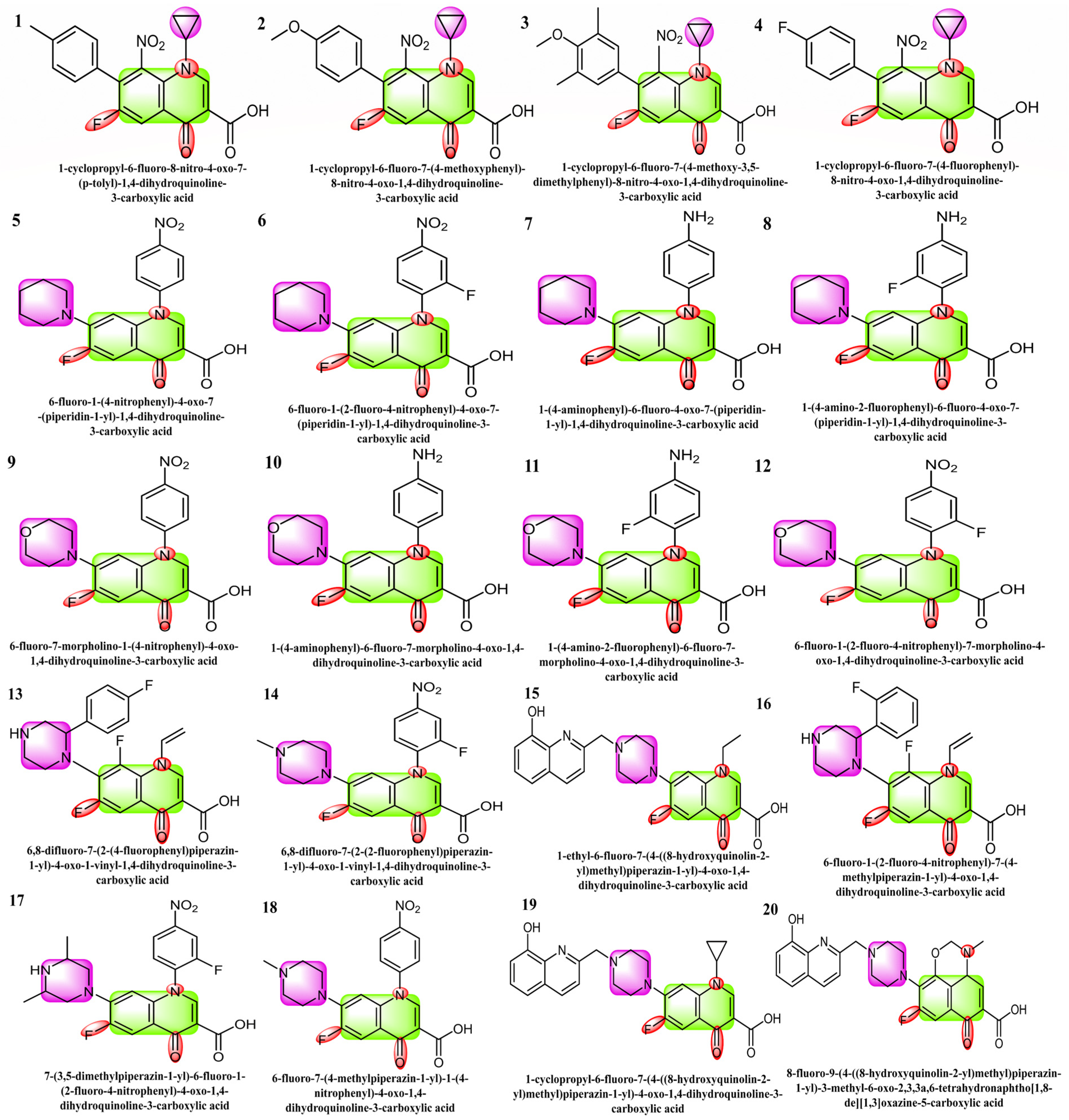
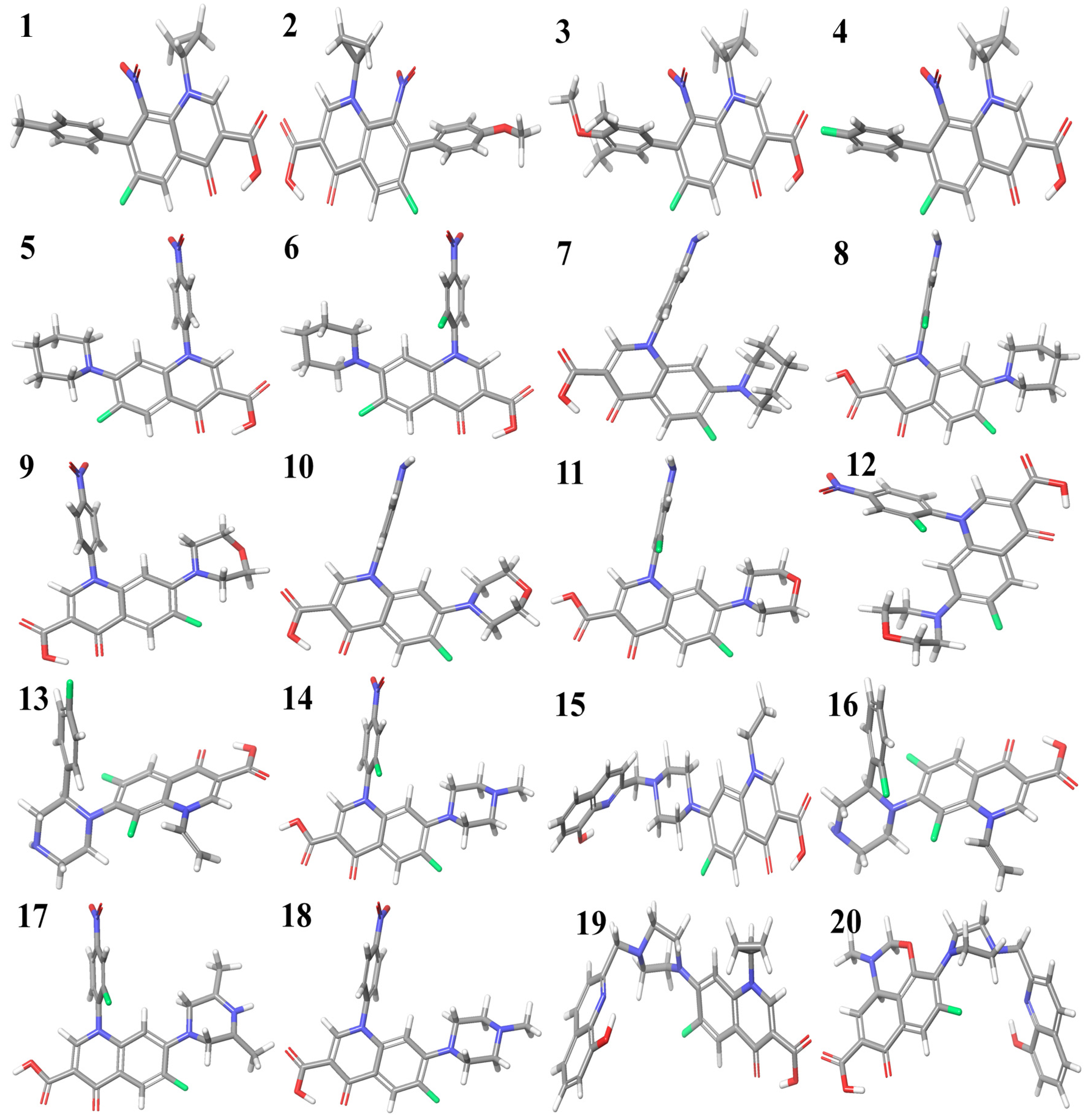
The selected 20 fluoroquinolone-based compounds including carboxylic acid with cyclopropyl, piperidine-substituted fluoroquinolones, morpholine-containing fluoroquinolones, and piperazine-containing fluoroquinolones as shown in Figure 1, were investigated against SARS-CoV-2’s protease (Mpro) [29,30,31]. The structural, electronic, in silico, ADME, and cardiotoxicity analyses/properties of these molecules have not been explored before by any research group. The present research mainly focused on the DFT studies of selected fluoroquinolone-based drugs and the binding of SARS-CoV-2 main protease with selected ligands and their toxicity studies. SARS-CoV-2 Mpro (PDB ID: 6LU7) was employed, which plays a vital role in viral replication [32,33,34]. The 96% sequence homology of SARS-CoV-2 Mpro is comparable to that of the prior SARS-CoV and resembles a dimer when complexed with N3, a peptide-like inhibitor [35,36]. Therefore, the selected 20 inhibitors were screened against SARS-CoV-2 Mpro by structure-based drug design.
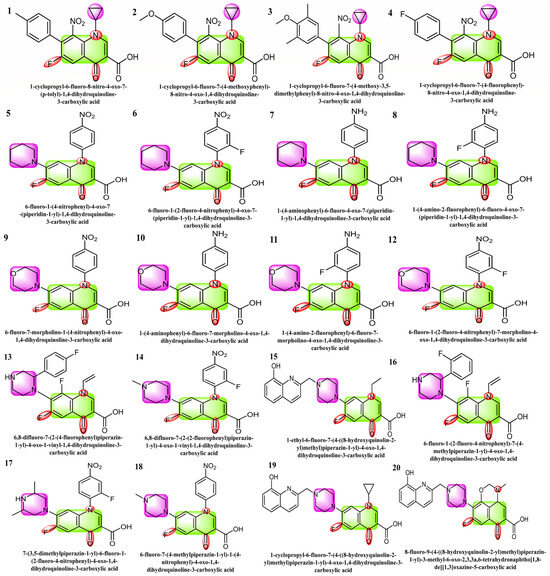
Figure 1.
Structures 1 to 4 are the derivatives of fluoroquinolone with cyclopropyl substitutions, 5 to 8 are piperidine-substituted, 9 to 12 are morpholine-substituted, and 13 to 20 are piperazine-substituted fluoroquinolones.
Chemical reactivity, natural bond orbital analysis, equilibrium geometry, frontier molecular orbital analysis, and molecular electrostatic potential are some of the theoretical predictions that were investigated and computed using RCAM-B3LYP/6-311++G (2d, p) basis sets of DFT. The description of absorption, distribution, metabolism, and excretion (ADME), pharmacokinetics drug-likeness, and the cardiotoxicity of compounds were studied to explore the safety considerations for a new medicine, based on which risk-based evaluations can be made.
2. Results and Discussions
2.1. DFT Study
The optimized structures of the selected compounds are shown in Figure 2. The geometries of the RCAM-B3LYP/6-311++G (2d, p) optimized clusters in the Cartesian coordinates of compounds 1–20 have been presented in the Supplementary Materials for researchers to reuse for future research purposes.
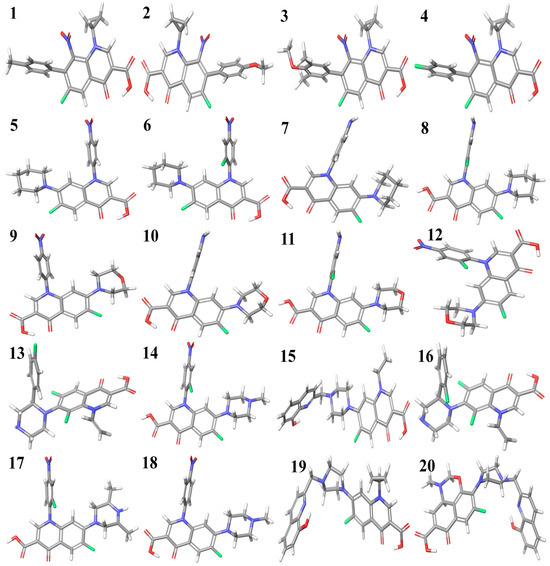
Figure 2.
Optimized structures of fluoroquinolone derivatives with cyclopropyl (1 to 4), piperidine (5 to 8), morpholine (9 to 12), and piperazine substitutions (13 to 20).
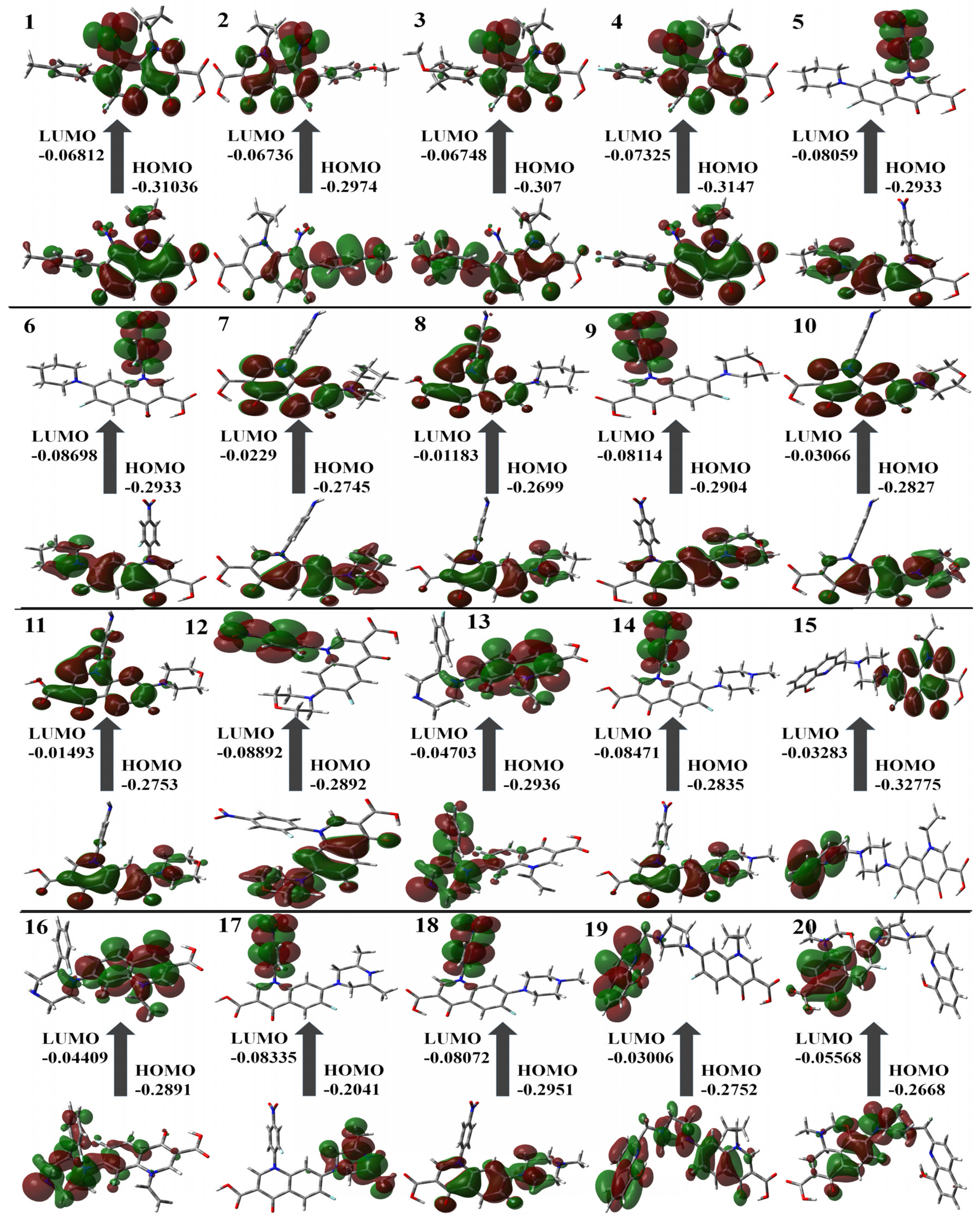
Table 1 contains the computed results for ionization potential, energy gap, electron affinity, and other descriptors. The stability of the structure is indicated by the energy gap; therefore, a ligand with a larger energy gap would be more stable compared to a molecule with a smaller gap. The order of the energy gaps for the selected compounds is 11 > 8 > 10 > 7 > 13 > 19 > 16 > 15 > 1 > 4 > 3 and so on. The largest energy gap of compound 11, according to the results, makes it more stable than the rest of the compounds due to the amine group shown in Figure 3. Compounds 4 and 11 possess the highest ionization potential, compound 12 shows the highest electron affinity, 11 possesses the highest hardness, 5, 6, 12, and 20 are chemically softer as compared to others, 6 is highly electronegative, 16 has the highest chemical potential, 17 has highest electrophilicity index (all are in the mild range, N < 2) [37].

Table 1.
The basis set, RCAM-B3LYP/6-311++G (2d, p), was used to compute the DFT level descriptors for selected compounds for the HOMO-LUMO energy differences.
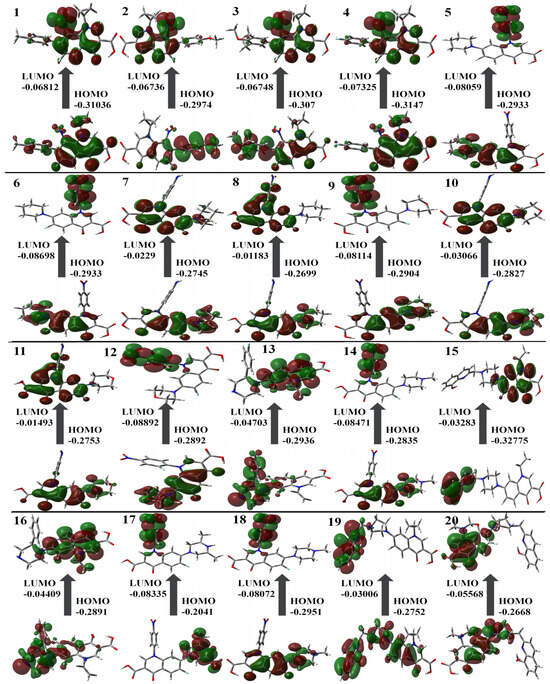
Figure 3.
Molecular orbital surfaces and energy levels for the HOMO and LUMO of the compounds computed at RCAM-B3LYP/6-311++G (2d, p) basis set.
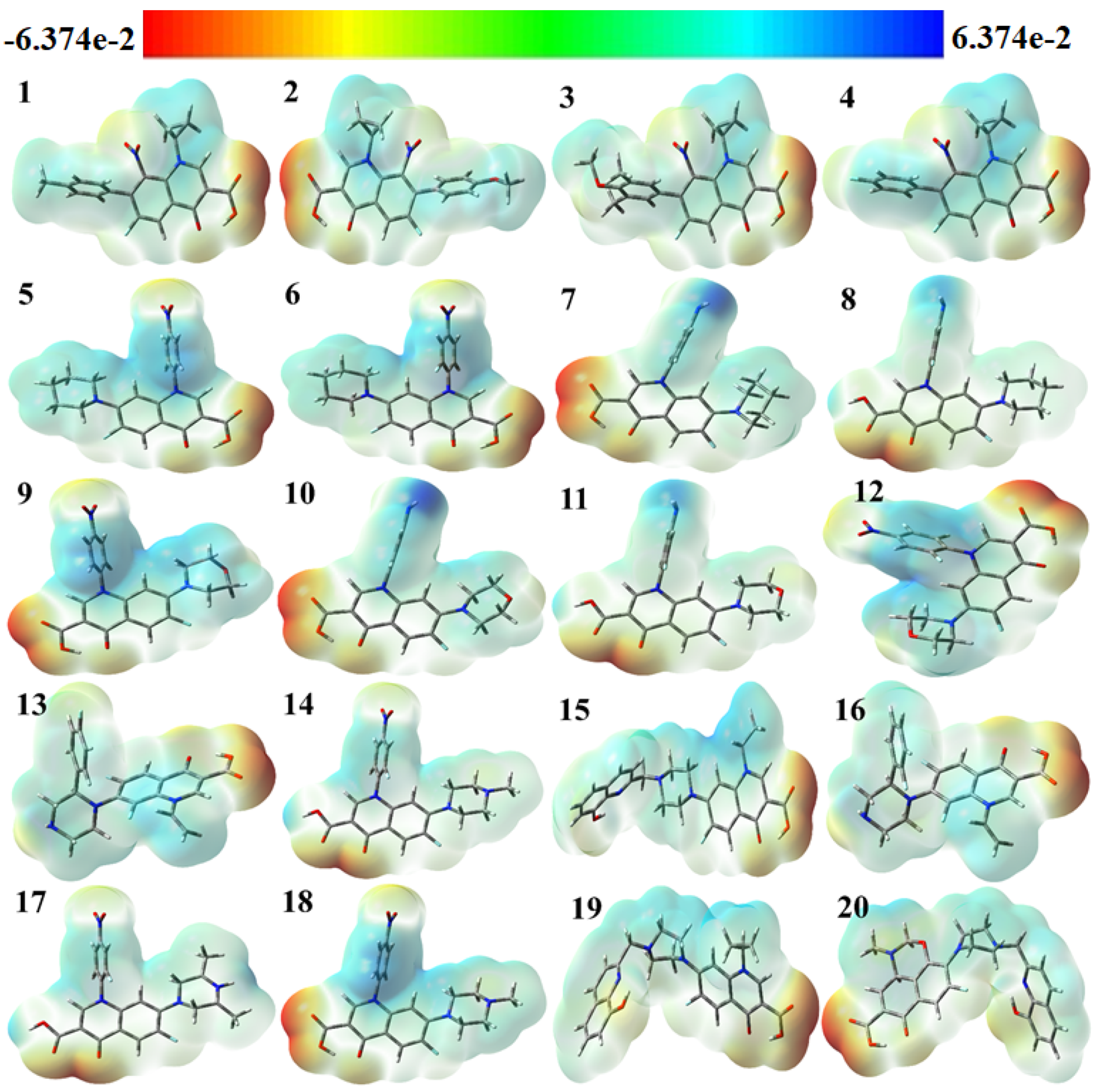
The use of color grading in MEP helps to indicate a molecule’s size, shape, and different possible charges, as shown in Figure 4 The colors are red, orange, yellow, green, and blue in descending order of potential. The blue color indicates the most electrostatically powerful area, meaning there are no electrons, while low electrostatic potential areas are indicated by the red hue, which has more electrons, hence making it an ideal place for an electrophilic attack [38]. With DFT calculations from the optimized structure and basis set as depicted in Figure 4, the surface analysis determined the MEP of the compounds. It is worth noting that gas-phase MEP surfaces have values ranging from −6.374 e−2 eV and +6.374 e−2 eV.
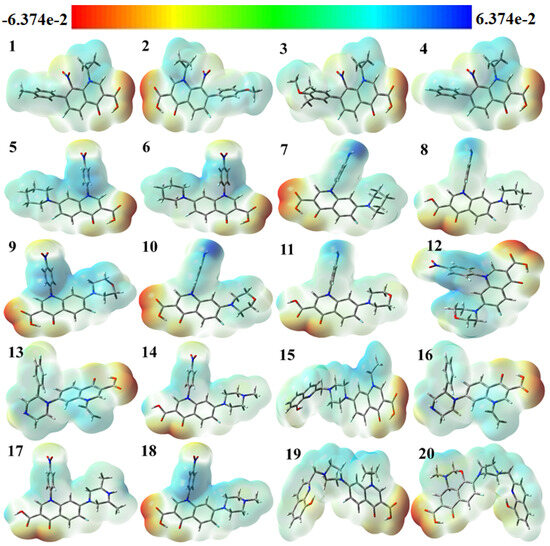
Figure 4.
Molecular electrostatic potential (MEP) surfaces of selected compounds.
The hydroxyl and carbonyl groups of the selected fluoroquinolones have their oxygen atoms located in the most electrophilic area, as shown by the yellow region in Figure 4. From the MEP map, it can be inferred that the title molecules can work as antiviral molecules by entering a reaction system mainly through N atoms, because N atoms contain the nucleophilic part of the molecules, making them more suitable for reaction with negatively charged anions or radicals [39]. It was theoretically proved in 2005 by Hay et al. that, in the presence of an electron-withdrawing substituent, aryl C–H moieties become strong hydrogen bond donors and form stronger receptor–anion complexes than those of traditional hydrogen-bonding donor groups like N–H O–H, since nitro substitution can induce the receptors towards the binding of anions [40].
2.2. Molecular Docking Study
Molecular docking of selected compounds has been carried out through the MOE 2015 to find out their interaction with SARS-CoV-2 Mpro, which is responsible for virus activity. Remdesivir has been used as a standard in this study. All except four ligands showed very efficient results compared to the standard and predicted results, indicating that these are very potent inhibitors of SARS-CoV-2 Mpro. Their docking values demonstrated that these compounds can bind very efficiently with the main protease of SARS-CoV-2 Mpro. Receptor–ligand interaction plays an important role in biological processes like the multiplication of microorganisms. The resultant data of the given compounds show that they can be used in the drugs and these are excellent inhibitors of SARS-CoV-2 Mpro. Table 2 provides a summary of the outcomes from the effective docking of the investigated ligands (1–20).
Table 2.
Docking score (kcal/mol) of selected compounds with SARS-CoV-2 (6LU7).
2.3. Validation of MOE Software through Inhibitory Mechanism of N3
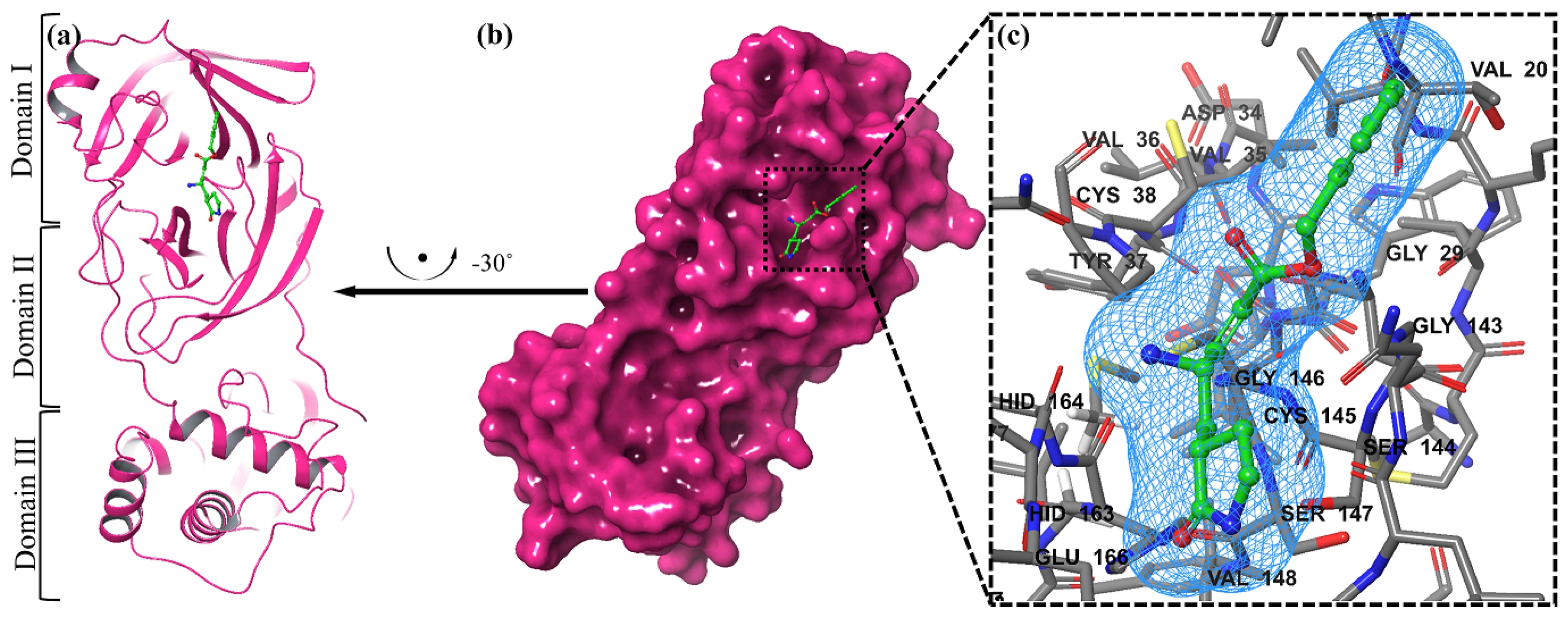
We examined a crystal structure at 2.1 Å resolution that included SARS-CoV-2 Mpro together with an N3 compound, which is a Michael acceptor inhibitor developed using computer-aided drug design [7]. The crystal’s asymmetric unit contains a single polypeptide chain. Nonetheless, these polypeptide chains are designated as protomers through a crystallographic fold symmetry axis (Figure 5b). Notably, electron density maps confirm the visibility of all residues. The protomer is composed of three distinct domains (Figure 5a). The beta-barrel structures are formed by domains I (residues 1–123) and II (residues 123–194) in an antiparallel arrangement. The third domain (residues 195–306) has five alpha helices that usually appear to be arranged in an anti-parallel globular cluster, whereas its connection point to domain II is through a stretched loop region (residues 195–286). The catalytic dyad of Cys145-His41 can be found in SARS-CoV-2 Mpro, and the hydrolysis catalyzing center is located inside a gap between both domains I and II. These structural attributes bear a resemblance to previously reported SARS structures in other coronaviruses [7].
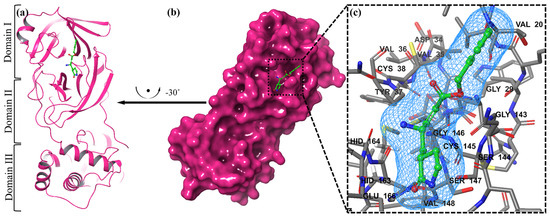
Figure 5.
The representation of a single protomer from the SARS-inhibitor complex is depicted in a cartoon format (a). The homodimeric structure of SARS is shown in a surface representation. The protomer is shown by the pink colors, while the N3 inhibitor is depicted as green sticks (b). The substrate-binding pocket is highlighted in an enlarged view. Important binding pocket residues are represented as sticks. The density map (2Fo-Fc) contoured at 1.2σ is presented around the N3 molecule (blue mesh) (c).
The electron density map clearly illustrates the binding of N3 within the substrate-binding pocket, adopting an extended conformation (Figure 5c). The inhibitor’s backbone atoms establish an antiparallel sheet formation, with the interconnecting loop between domain I on one side and domain II on the other.
Specific interactions between N3 and SARS-CoV-2 Mpro are elucidated in detail (Figure 5c). The electron density highlights the formation of a covalent bond between the Sγ atom of C145 in the protomer and the Cβ atom of the vinyl group, thus confirming the occurrence of Michael addition. This establishes a hydrogen bond with LEU 4 with ammonia in the protomer.
Previous studies have suggested a conserved substrate-recognition pocket within SARS-CoV-2 Mpro, indicating its potential as a drug target for designing broad-spectrum inhibitors [7,41,42]. With the emergence of new types of coronaviruses and the accumulation of structural data for SARS-CoV-2 Mpro across diverse species, this hypothesis gains further support. Superimposing the crystal structures of SARS-CoV-2 Mpro with compound (N3) underscores the variability of helical domain I and surface loops while confirming the high conservation of the substrate-binding pocket located between domain I and domain II across all coronaviruses. This suggests that antiviral inhibitors targeting this pocket have the potential for broad-spectrum efficacy against coronaviruses (Figure 5) [43].
2.4. Visualization of Docking Results
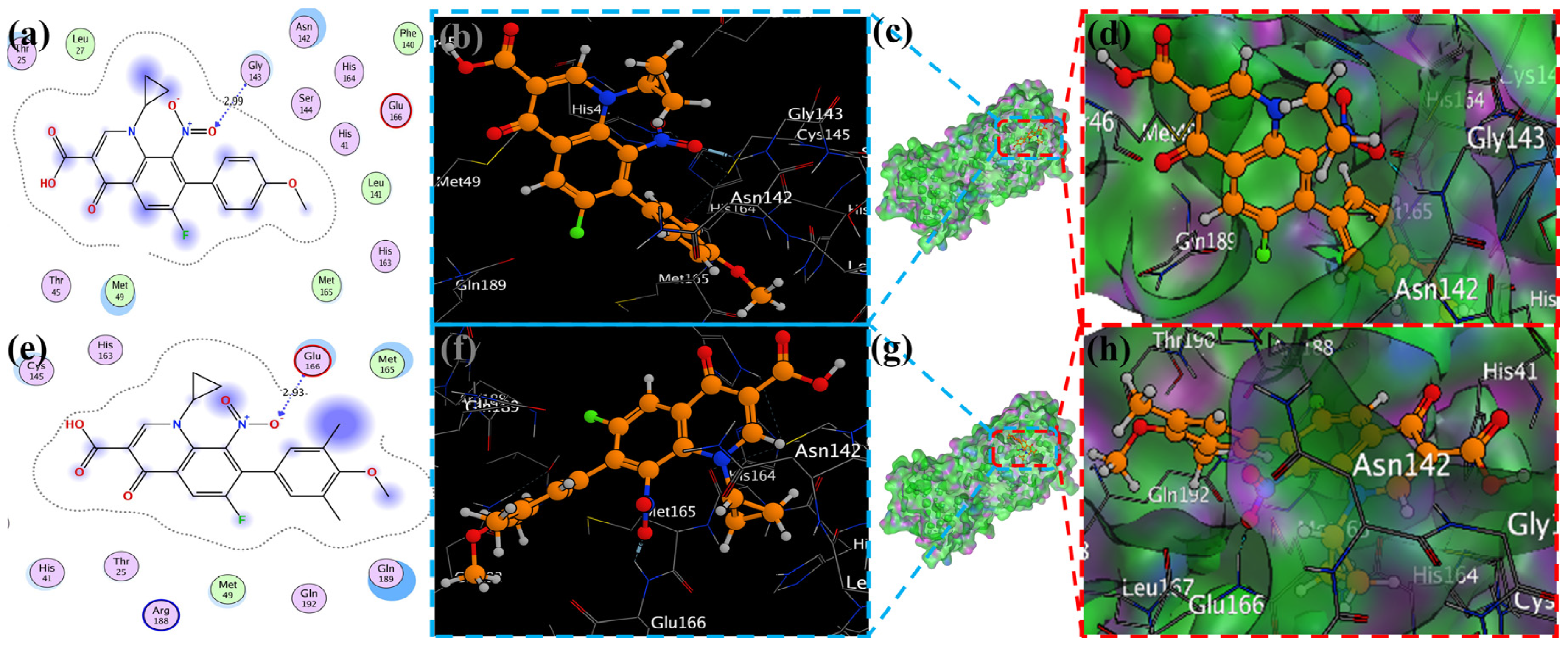
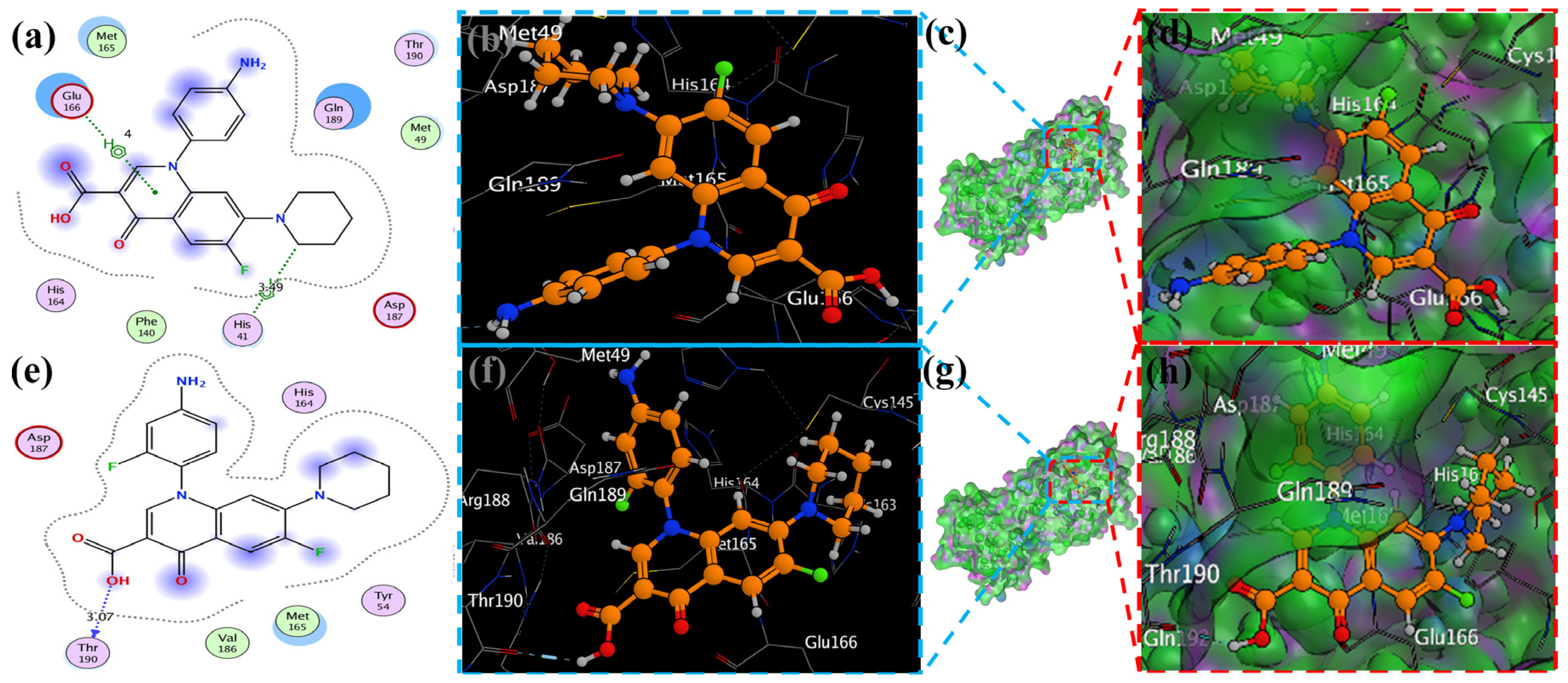
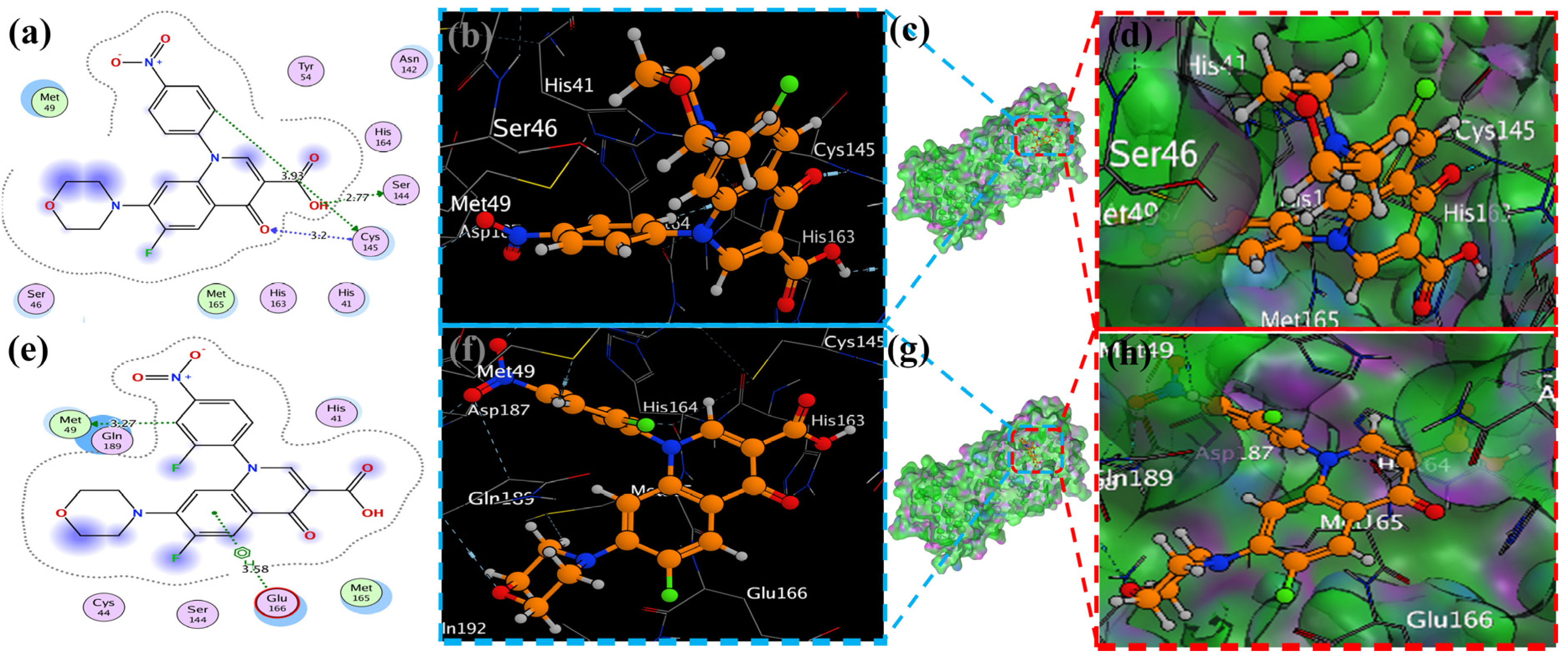
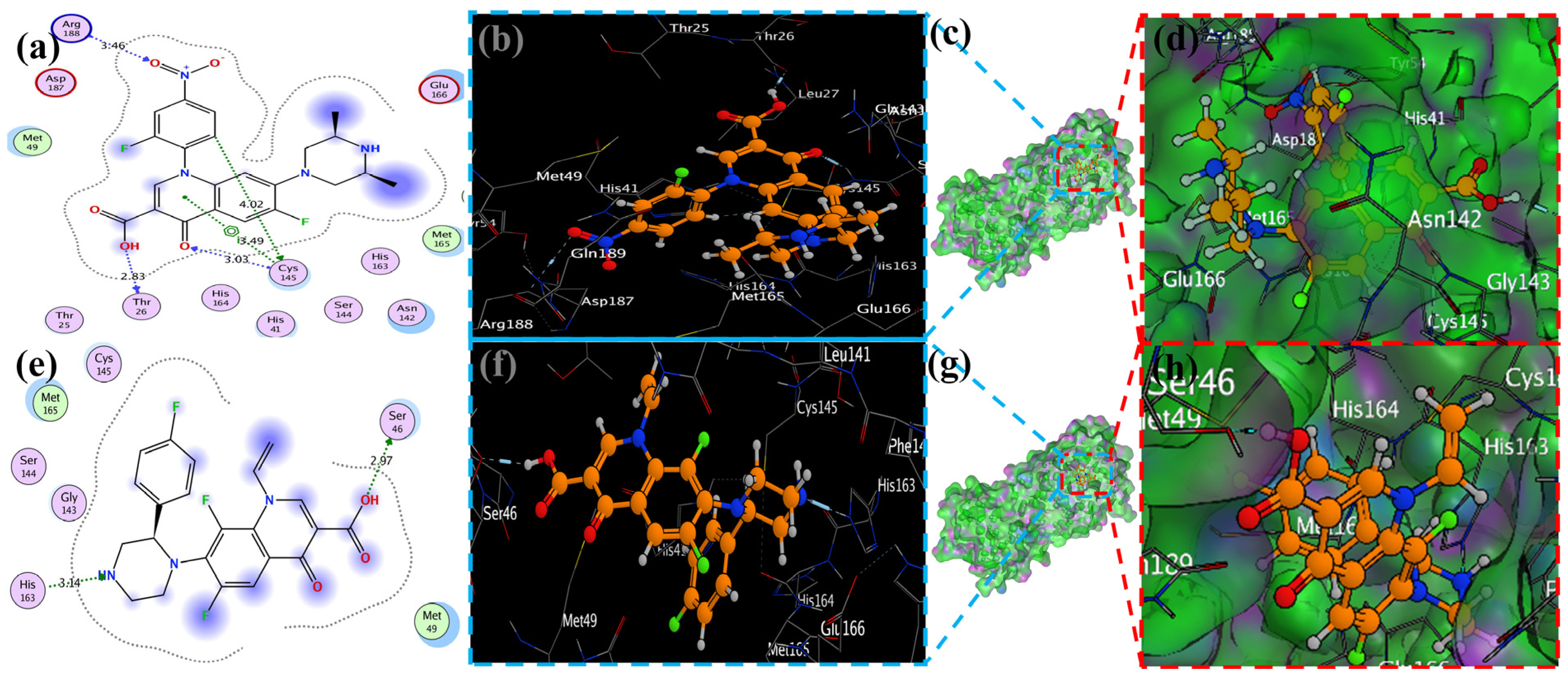
MOE has proven to be effective in creating 2D interactive ball-and-stick diagrams, as well as surface diagrams for selected compounds; a comparison was made with a standard FDA-approved drug (remdesivir). The docked result for the standard was −6.5, whereas these compounds demonstrated a potential docked score when compared with the standard. Compounds 2, 3, 8, 11, 14, 15, 19, 20, and 18 showed only one hydrogen bond at the Gly 143, Glu 166, Thr 190, Gly 143, Thr 190, Thr 25, Met 49, Thr 26, and His 163 residues with 2.99 Å, 2.93 Å, 3.07 Å, 3.04 Å, 2.84 Å, 3.8 Å, 4.23 Å, 3.22 Å and 3.48 Å in length, respectively (Figure 6, Figure 7 and Figures S1–S3). Compounds 13 and 9 formed two hydrogen bonds at Ser 46, His 163, Ser 144, and Cys 145 with a bond length of 2.97 Å, 3.14 Å, 2.77 Å, and 3.2 Å, respectively (Figure 8 and Figure 9).
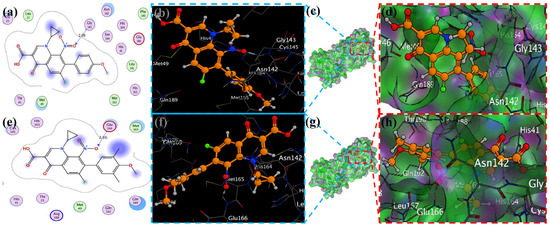
Figure 6.
Docking with the 2 and 3 cyclopropyl ligands in the SARS-CoV-2 Mpro (6LU7). (a,e) The best binding mode of a protein (ligands 2 and 3, depicted as brown and red sticks), (b,d,f,h) The amino acid residues involved in bonding interactions (blue and green dashed line represents the ligand 2 and 3 interaction (c,g) (2D)).
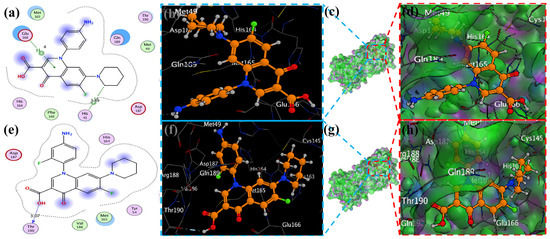
Figure 7.
The piperidine ligands 7 and 8’s docking visualization in SARS-CoV-2 Mpro (6LU7); (a,e) show the optimal binding mode of the protein (ligands 7 and 8, depicted as brown and red sticks) while (b,d,f,h) highlight the amino acid residues engaged in bonding interaction (the blue and green dashed line represents the binding interaction (2D) of ligands 7 and 8 (c,g).
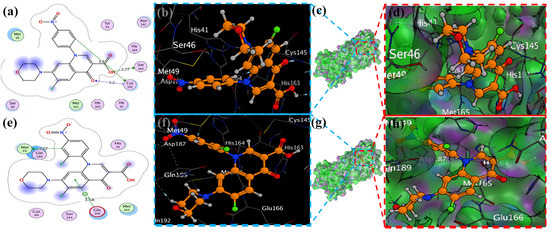
Figure 8.
Docking representation of morpholine ligands 9 and 12 into SARS-CoV-2 Mpro (6LU7) (a,e): the most preferred binding configuration of the protein (ligands 9 and 12, shown as brown and red sticks, respectively) (b,d,f,h); interactions between amino acid residues participating in the bonding process (2D) ((c,g) show blue and green dashed lines, representing the binding interaction of ligands 9 and 12).
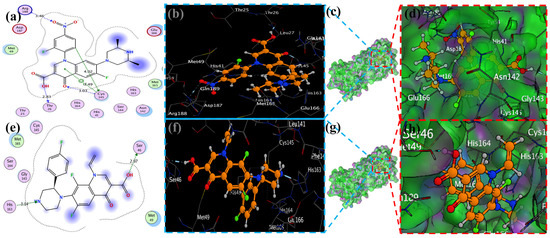
Figure 9.
The docking of piperazine ligands 13 and 17 into SARS-CoV-2 Mpro (6LU7) was visualized as follows: (a,e) best binding mode of the protein (ligands 13 and 17, depicted as brown and red stick), (b,d,f,h). The amino acid residues involved in the bonding interaction (blue and green dashed lines represent the binding interaction (2D) of ligands 13 and 17 (c,g).
Compound 17 formed three hydrogen bonds at the Thr 26, Cys 145, and Arg 188 residues with 2.83 Å, 3.03 Å, and 3.46 Å respectively (Figure 9). Compounds 1, 4, 9, and 11 formed double bonds at the Glu 166, Glu 166, Cys 145, and His 41 residues with 4.42 Å, 4.3 Å, 3.93 Å and 4.44 Å, respectively (Figure 8, Figures S3 and S4). Similarly, compounds 14, 17, 7, and 12 all showed two double bonds with Glu 166, Glu 166, Cys 145, Cys 145, His 41, Glu 166, Met 49, and Glu 166 residues, which were 4.39 Å, 3.73 Å, 3.49 Å, 4.02 Å, 3.49 Å, 4.0 Å, 3.27 Å and 3.58 Å in length, respectively (Figure 7, Figure 8 and Figure 9 and Figure S4). The remaining 5, 6, 16, and 10 are compounds that could not show good results (Figures S5 and S6). When the compounds were docked with the protease (6LU7), it was discovered that the ligands fit perfectly in the core pocket region of the SARS-CoV-2 Mpro at the boundary between domain I and domain II, as shown in Figure 5 [42].
Table 3 presents the data of some of the best ligands against the docking results of FDA-approved and clinically tested antiviral and antimalarial drugs regarding the main protease of COVID-19 (6LU7). The binding energy data of remdesivir have been reported by researchers and are comparable with ligands 1, 3, 15, 17, and 18. The energy difference, ionization potential, electron affinity, chemical hardness, chemical softness, and electrophilicity index values are also comparatively better than other ligands, making them more suitable for binding with protein residues and also in protein pockets [44].
Table 3.
A comparison of some of the best ligands with FDA-approved and clinically tested drugs to treat the main protease of COVID-19.
2.5. ADME Parameters, Pharmacokinetics, and Drug Likeness
The ADME (absorption, distribution, metabolism, and excretion) properties of twenty fluoroquinolone molecules as potential SARS-CoV inhibitors were analyzed in this study. The molecular weight (MW), hydrogen bond donors (HBD), hydrogen bond acceptors (HBA), QPlog Po/w, QPlogs, QPPCaco, metabolic stability, QPlog Khsa, and % age of human oral absorption were calculated using the QikProp command of the Schrodinger software, Maestro 11.2.
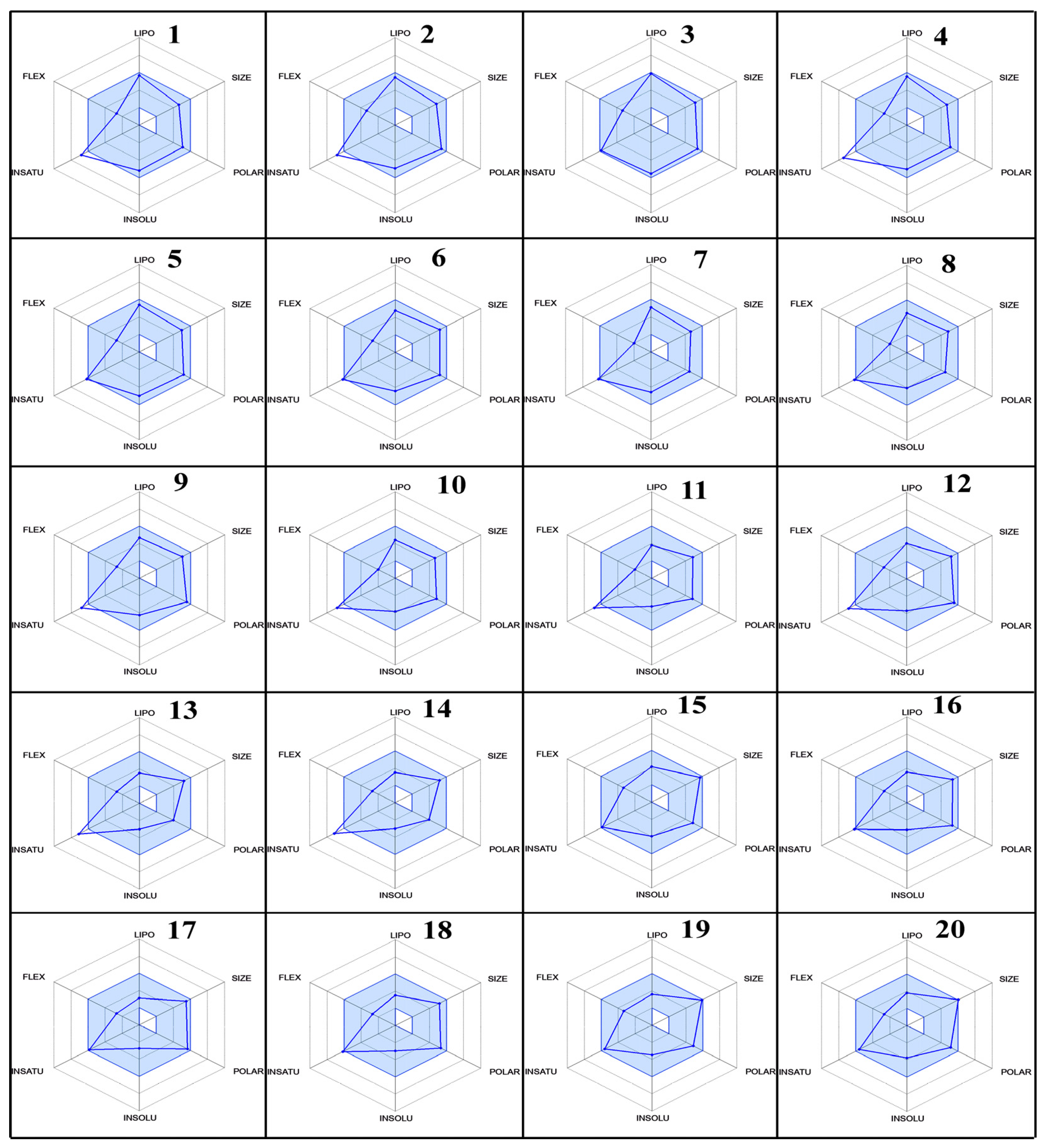
The results of the ADME analysis (Table 4) revealed significant variations among the selected fluoroquinolones. The molecular weight range should be from 130.0 to 725.0, and all selected molecules have values with the permitted range. Regarding the total number of hydrogen bonds donated by solute to water molecules in an aqueous solution (donor HB), the total number of hydrogen bonds accepted by solute from water molecules in an aqueous solution (accept HB), the predicted octanol/water partition coefficient (QP logPo/w), the predicted aqueous solubility (QP logS), the number of likely metabolic reactions (Metab), the prediction of binding to human serum albumin (Qplog Khsa), the predicted qualitative human oral absorption, and the predicted human oral absorption on a 0 to 100% scale, all values were within the permissible range, as mentioned in footnote of Table 4. In contrast, with the predicted apparent Caco-2 cell permeability in nm/sec, Caco-2 cells are a model for the gut–blood barrier; Caco-2 monolayers in the prediction of intestinal drug absorption range is <25 poor, >500 great, and the compounds 5, 6, 9, 12–20, showed poor intestinal absorption.
Table 4.
In silico absorption, distribution, metabolism, and excretion (ADME) study of manufactured molecules.
Furthermore, the % age of human oral absorption predictions indicated that all compounds had relatively high values, ranging from 25.64% to 79.41%. Compound 20 had the lowest % age of human oral absorption, while compound 3 showed the highest value. Lipinski’s rule of five showed that all compounds had a logP value less than 5, which is good for oral and intestinal absorption. These results provide valuable insights into the ADME properties of fluoroquinolones as SARS-CoV inhibitors with their radar (Figure 10), which was predicted by Swiss ADME online services. The variations in molecular weight, lipophilicity, metabolic stability, blood–brain barrier penetration, and oral absorption can aid in the selection and optimization of lead compounds for further development as effective antiviral agents.
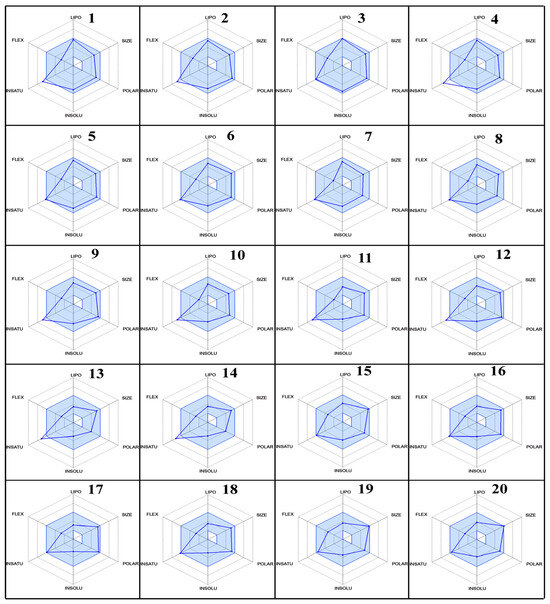
Figure 10.
The radar plots of bioavailability data (lipophilicity, size, polarity, insolubility, instauration, and flexibility) of compounds 1–20.
2.6. Cardiotoxicity of Compounds
Pred-hERG 5.0 may provide information regarding the interaction of various compounds with hERG (the gene for human ether-a-go-go-related cardiac potassium channel), which is crucial in controlling heart rhythm fluctuations. The majority of compounds are classified as “non-blockers,” implying an expectation of minimal impact on the hERG channel. Confidence percentages accompanying these predictions vary from 67.29% to 99.83%, signifying diverse levels of certainty. The concept of “applicability domain” is introduced, with most compounds falling “Outside” this domain, suggesting potential limitations in prediction reliability. Categorical assessments of “Categorical Potency” further characterize the compounds, primarily as “Moderate blockers”, despite the overarching non-blocking prediction. Numerical “Potency” values add a quantitative dimension, showcasing the strength of potential interactions. Notably, the dataset highlights the model’s reliance on categorical assessments, with a tendency to predict non-blocking behavior even when categorical potency suggests otherwise. These findings underscore the need for the careful consideration of confidence levels, applicability domain, and categorical potency when interpreting predictions from Pred-hERG 5.0, enriching our understanding of the nuanced landscape of hERG channel interactions shown in Table 5.
Table 5.
Predicted cardiotoxicity of all substances.
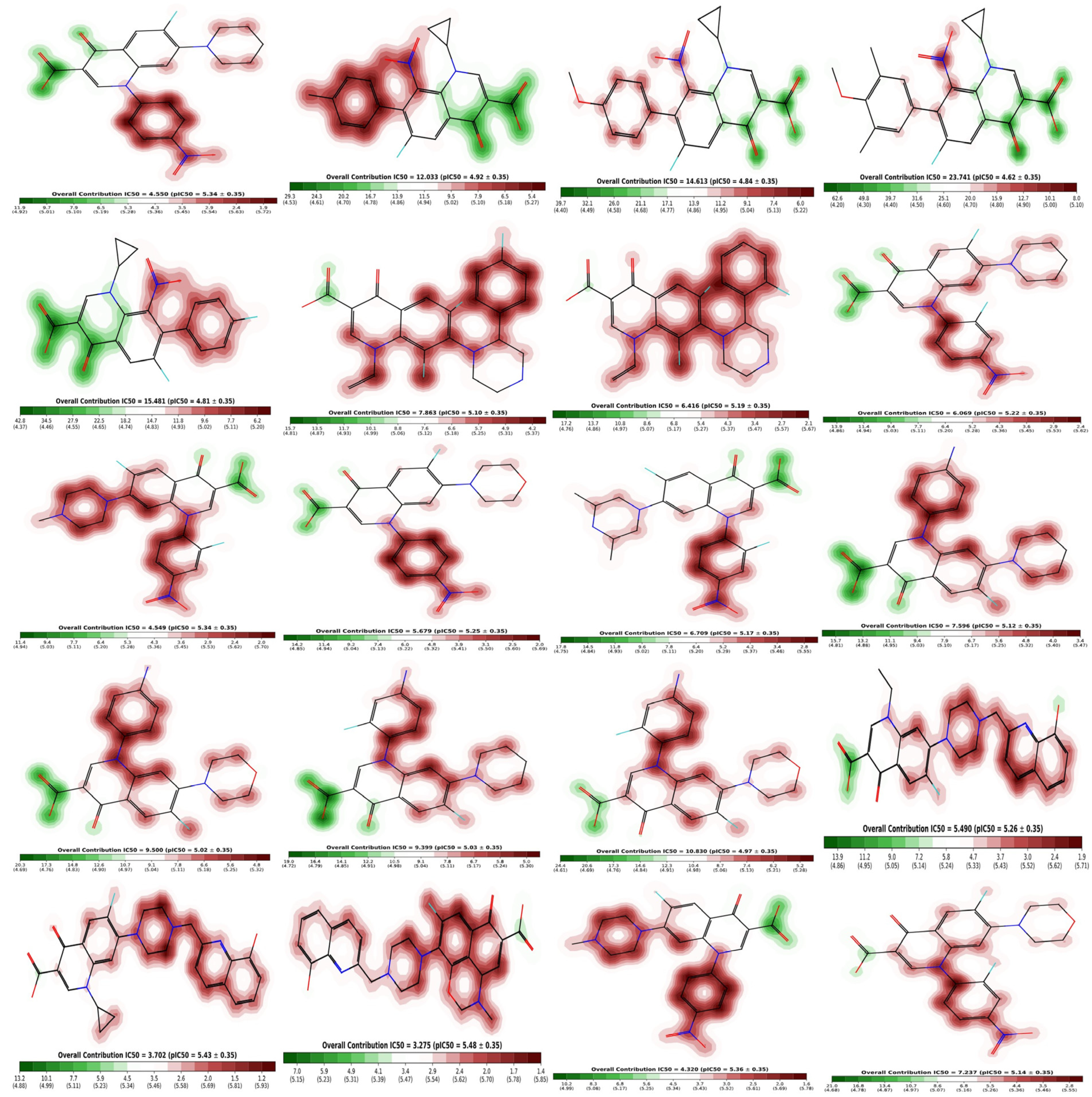
The fragment contribution map produced by Pred-hERG for the most and least strong compounds is shown in Figure 11. Each atom is given a specific color based on how much it contributes to the activity. The colors red, red-orange, and orange are associated with the negative gradient contribution, whereas the colors yellow, yellow-green, and green are associated with the positive atomic contribution. In this study, we used an in silico technique to predict the cardiotoxicity of all substances. All compounds showed potential non-cardiotoxicity, except compounds 6 and 18, which are potentially cardiotoxic and they are predicted as blockers.
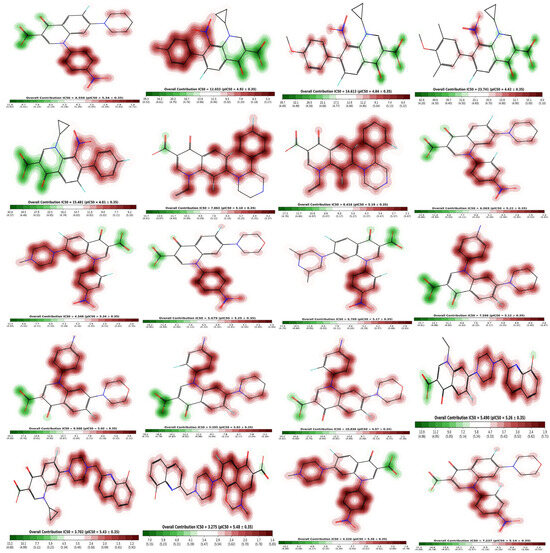
Figure 11.
Fragment contribution maps for the regression model with IC50 and PIC50 values of compounds 1–20.
3. Materials and Methods
The energy gap of analogs of fluoroquinolones was demonstrated by considering their HOMO-LUMO energies and the role they play in chemical interactions. The work also utilized molecular electrostatic potential to predict sites of nucleophilic and electrophilic attack on molecules, while several computational tools were applied to investigate their ability to inhibit SARS-CoV-2. The DFT calculations were performed using Gaussian 09 W Revision D.01, while binding affinity calculations were performed using MOE 2015, Pred-hERG was used for cardiotoxicity prediction, and Maestro 13.5 was utilized for calculating drug properties. This study explains the mechanism of action for the selected fluoroquinolones and their respective potentials of being therapeutic agents against SARS-CoV-2 via understanding their activities, ADME profiles, and cardio-toxicity risks [38,51,52,53,54,55].
3.1. Density Functional Theory (DFT) Study
We used DFT to determine the chemical hardness, ionization potential and softness, electrophilicity, and total energy of a process. The terms LUMO and HOMO refer to the lowest and highest occupied molecular orbitals, respectively. Molecules’ ionization energy and electron affinity are correlated with their HOMO and LUMO energies (Table 1) [56].
Koopmans’ theorem equation: A = −ELUMO and I = −EHOMO
The energy difference reveals details about the molecules’ reactivity. While a lower energy difference suggests that a molecule is the least stable and most reactive, a bigger gap suggests that a molecule is extremely steady and less reactive. The electronic chemical potential is the name given to a structure’s negative electronegativity and is represented by the symbol µ.
µ = (EHOMO − ELUMO)/2
This quantifies an electron’s ability to leave a system. A molecule is said to be more reactive and unstable when its value is greater. The ability of a system to draw an electron is gauged by the electrophilicity index [57]. This formula is used to compute it:
Ꞷ = µ2/2η
The solidity and reactivity of a compound are connected to chemical hardness. It calculates the degree of resistance to changes in the electron distribution [58].
η = (I − A)/2
The electronegativity χ of a molecule indicates its capacity to attract electrons.
χ = (IP + EA)/2
Tetracyanoethylene, the most electrophilic neutral molecule, has an index called nucleophilicity N that is correlated with its HOMO energy (TCE).
N = EHOMO (nucleophile) − EHOMO(TCE)
With Gaussian 09 W Revision D.01, all the structures of compounds 1–20 have their energy minimized by DFT using the RCAMB3LYP functional and 6-31++G(2d, p) basis set in the DMSO solvent. A similar approach has been used for Gauss view version 5 for MEP computations. Using the principles of quantum mechanics, Gaussian makes predictions about molecular structure and spectroscopic data. The Gauss view and Gaussian09 were used to calculate the different energies of the selected substances. The Hartree–Fock approach, simulation methods, molecular mechanics, and post-Hartree–Fock investigations utilizing density functional theory (DFT) are all used to investigate various structural and electrical properties of organic molecules [32,33]. The well-known HOMO-LUMO energies of quantum mechanics influence numerous chemical interactions. One of the more beneficial discoveries for explaining a molecule’s chemical stability is the FMOs theory (Frontier molecular orbitals), which uses HOMO and LUMO. The MEP of the selected structures has been predicted using the Gauss view software 09 [34,59,60].
3.2. Docking Study Using MOE (Molecular Operating Environment)
Initially, the MPro: N3 complex’s crystal structure (PDB ID: 6LU7; Version 2, 2.16 Å resolution, 4) was downloaded. Although Mpro typically operates as a homodimer, as mentioned in the Introduction, we only computed the monomeric unit found in the PDB file, because the dimer interface does not directly interact with the N3 ligand. Using the MOE tool, we further processed the molecular structure. The 84 crystal water molecules were all saved, and the FMO calculation was then performed on them. The peptide-like N3 inhibitor is shown in Figure 5a as the protein’s ligand. Take note that the Michael addition has given this ligand a covalent connection with Cys145. Standard modeling activities, such as pKa adaption (pH 7) and hydrogen attachment, were carried out. Ser1 and Gln306 were given the charged N- and C-termini, respectively. Within all attached hydrogen atoms, the ligand, and the amino acid residues within 4.5 Å of the ligand, molecular mechanics-based energy minimization (using the AMBER10:EHT force field in MOE) was performed; a tether mask of 1.0 Å was employed within the important pharmacophore region. Figure 5b displays the whole structure of the processed protein–ligand complex along with a close-up of the pharmacophore. Based on the geometrical relationships determined by MOE, Figure 5c is a schematic representation of the interactions between ligands and residues. [61,62].
3.3. ADME Studies
The Maestro Schrodinger and Swiss ADME online services have been used to forecast the substances’ assessment of absorption, distribution, metabolism, and excretion (ADME). Using a large database, the server may make high-precision assumptions about physicochemical attributes, pharmacokinetics, drug similarity, and different pharmacological effects. For the ADME study, the selected compounds, after optimization by DFT, were used in the Maestro Schrodinger and Swiss ADME services, which gives the ADME values in the form of table and radar form [63].
3.4. Cardiotoxicity Studies of Compounds
Pred-hERG, a web-enabled computer framework, was utilized to calculate the prediction, confidence, and probability map for all the drugs’ potential cardiac toxicity. The Pred-hERG server aids users in identifying hERG blockers and non-blockers by providing a quick, user-friendly interface. We used an optimized compound from DFT in CHK file for cardiotoxicity [64].
4. Conclusions
In conclusion, this study used computational chemistry methods to assess the potential of several fluoroquinolone compounds as SARS-CoV-2 Mpro inhibitors. The molecular characteristics of the chosen compounds, such as their HOMO and LUMO energies, ionization potential, energy gap, electron affinity, and other descriptors were well understood by DFT analysis. The more stable properties of the compounds with bigger energy gaps point to their potential as inhibitors. The selected compounds’ electron distribution and reactivity were further shown by the HOMO and LUMO plots. An anti-aromatic molecule was suggested by a tiny HOMO-LUMO gap, whereas a hard, stable, and less reactive molecule was suggested by a broad gap. These results help us understand the stability and chemical behavior of fluoroquinolone derivatives. The molecular electrostatic potential (MEP) analysis also made it possible to locate the molecules’ nucleophilic and electrophilic attack sites. The visual representation of the positive, negative, and neutral regions offered by the MEP surfaces allowed for the evaluation of possible interactions and inhibitory capacities. Furthermore, the molecular docking analyses of all compounds except six, utilizing MOE, showed the chosen compounds’ propensities for attaching to the SARS-CoV-2 Mpro with the docking score between −7.9 kcal/mol and −5.9 kcal/mol. The effectiveness of the compounds in attaching to the main protease of SARS-CoV-2 Mpro was shown by the docking scores. Numerous substances outperformed remdesivir in terms of docking scores, indicating that they could be effective SARS-CoV-2 Mpro inhibitors. The chosen fluoroquinolone compounds appear to have potential as SARS-CoV-2 Mpro inhibitors, according to the combined findings of the DFT analysis, HOMO-LUMO plots, MEP analysis, and molecular docking investigations. These results offer useful information for future research and the creation of possible antiviral medications to treat COVID-19 infections. The obtained result of the ADME study showed the pharmacokinetics and drug-likeness of these compounds, where all the ligands showed good physicochemical properties, lipophilicity, water solubility, pharmacokinetics, drug-likeness, and medicinal chemistry features. The ligands 1, 3, 15, 17, and 18 showed better electronic, structural, in silico, ADME and cardiotoxicity properties, so these fluoroquinolone compounds have a good scope for further drug development.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules29194721/s1, Figure S1 Visualization of docking of Ligand 15, 19 docked in protein (6LU7) (a and e). Best binding mode of protein (Ligand 15, 19 as brown and red stick), (b, d, f and h) Amino acid residues involved in bonding interaction (Blue and green dash line represents binding interaction (2D) of ligand 15, 19). Protein with ligand with their best pose (c and g). Figure S2 Visualization of docking of Ligand 20, 18 docked in protein (6LU7) (a and e). Best binding mode of protein (Ligand 20, 18 as brown and red stick), (b, d, f and h) Amino acid residues involved in bonding interaction (Blue and green dash line represents binding interaction (2D) of ligand 20, 18).Protein with ligand with their best pose(c and g). Figure S3 Visualization of docking of Ligand 1, 4 docked in protein (6LU7) (a and e). Best binding mode of protein (Ligand 1, 4 as brown and red stick), (b, d, f and h) Amino acid residues involved in bonding interaction (Blue and green dash line represents binding interaction (2D) of ligand 1, 4).Protein with ligand with their best pose(c and g). Figure S4 Visualization of docking of cyclopropyl Ligand 11, 14 docked in protein (6LU7) (a and e). Best binding mode of protein (Ligand 11, 14 as brown and red stick), (b, d, f and h) Amino acid residues involved in bonding interaction (Blue and green dash line represents binding interaction (2D) of ligand 11, 14).Protein with ligand with their best pose(c and g). Figure S5 Visualization of docking of cyclopropyl Ligand 5, 6 docked in protein (6LU7) (a and e). Best binding mode of protein (Ligand 5, 6 as brown and red stick), (b, d, f and h) Amino acid residues involved in bonding interaction (Blue and green dash line represents binding interaction (2D) of ligand 5, 6).Protein with ligand with their best pose(c and g). Figure S6 Visualization of docking of Ligand 16, 10 docked in protein (6LU7) (a and e). Best binding mode of protein (Ligand 16, 10 as brown and red stick), (b, d, f and h) Amino acid residues involved in bonding interaction (Blue and green dash line represents binding interaction (2D) of ligand 16, 10).Protein with ligand with their best pose(c and g).
Author Contributions
Conceptualization, M.A.K. and S.M.; methodology, M.A.T.; software, validation, M.A.K. and A.A.A.; formal analysis, A.A.A.; investigation, S.M.; resources, S.M. and M.A.K.; writing—original draft preparation, S.M. and M.A.K.; writing—review and editing, M.A.T.; visualization, S.M.; supervision, S.M. and M.A.K.; project administration, S.M. and S.M. All authors have read and agreed to the published version of the manuscript.
Funding
Linyi University High-level Talent (Ph.D.) Scientific Research Start-up Fund (Natural Sciences) No. (Z6124019). This research was funded by King Saud University, Riyadh, Saudi Arabia, through the Researchers Supporting Project No. (RSPD2024R852).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in the study are included in the article and Supplementary Materials, further inquiries can be directed to the corresponding authors.
Acknowledgments
Sadaf Mutahir and Muhammad Asim Khan acknowledge Linyi University for High-level Talent (Ph.D.) Research Start-up Fund (Natural Sciences) No. (Z6124019). The authors are grateful to King Saud University, Riyadh, Saudi Arabia, for funding the work through the Researchers Supporting Project No. (RSPD2024R852).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Jebril, N.M.T. World Health Organization declared a pandemic public health menace: A systematic review of the coronavirus disease 2019 “COVID-19”. SSRN Electron. J. 3566298 2020. [Google Scholar] [CrossRef]
- Cucinotta, D.; Vanelli, M. WHO declares COVID-19 a pandemic. Acta. Biomed. 2020, 91, 157. [Google Scholar]
- Tsergouli, K.; Karampatakis, T.; Haidich, A.; Metallidis, S.; Papa, A. Nosocomial infections caused by Crimean–Congo haemorrhagic fever virus. J. Hosp. Infect. 2020, 105, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef]
- Sun, M.; Yang, J.; Sun, Y.; Su, G. Inhibitors of RAS might be a good choice for the therapy of COVID-19 pneumonia. Chin. J. Tuberc. Respir. Dis. 2020, 43, E014. [Google Scholar]
- Batiha, G.E.-S.; Moubarak, M.; Shaheen, H.M.; Zakariya, A.M.; Usman, I.M.; Rauf, A.; Adhikari, A.; Dey, A.; Alexiou, A.; Hetta, H.F. Favipiravir in SARS-CoV-2 Infection: Is it Worth it? Comb. Chem. High Throughput Screen. 2022, 25, 2413–2428. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef]
- Yang, Y.; Islam, M.S.; Wang, J.; Li, Y.; Chen, X. Traditional Chinese medicine in the treatment of patients infected with 2019-new coronavirus (SARS-CoV-2): A review and perspective. Int. J. Biol. Sci. 2020, 16, 1708. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, Y. Traditional Chinese medicine treatment of COVID-19. Complement. Ther. Clin. Pract. 2020, 39, 101165. [Google Scholar] [CrossRef]
- Sampaio, F.; Sequeira, C.; Teixeira, L. Impact of COVID-19 outbreak on nurses’ mental health: A prospective cohort study. Environ. Res. 2021, 194, 110620. [Google Scholar] [CrossRef] [PubMed]
- Ling, C.-q. Traditional Chinese medicine is a resource for drug discovery against 2019 novel coronavirus (SARS-CoV-2). J. Integr. Med. 2020, 18, 87. [Google Scholar] [CrossRef] [PubMed]
- Hong-Zhi, D.; Xiao-Ying, H.; Yu-Huan, M.; Huang, B.-S.; Da-Hui, L. Traditional Chinese Medicine: An effective treatment for 2019 novel coronavirus pneumonia (NCP). Chin. J. Nat. Med. 2020, 18, 206–210. [Google Scholar]
- Feng, M.; Wang, X.; Chen, J.; Qu, R.; Sui, Y.; Cizmas, L.; Wang, Z.; Sharma, V.K. Degradation of fluoroquinolone antibiotics by ferrate (VI): Effects of water constituents and oxidized products. Water Res. 2016, 103, 48–57. [Google Scholar] [CrossRef]
- De Briyne, N.; Atkinson, J.; Borriello, S.; Pokludová, L. Antibiotics used most commonly to treat animals in Europe. Vet. Rec. 2014, 175, 325. [Google Scholar] [CrossRef]
- Daneman, N.; Chateau, D.; Dahl, M.; Zhang, J.; Fisher, A.; Sketris, I.S.; Quail, J.; Marra, F.; Ernst, P.; Bugden, S. Fluoroquinolone use for uncomplicated urinary tract infections in women: A retrospective cohort study. Clin. Microbiol. Infect. 2020, 26, 613–618. [Google Scholar] [CrossRef]
- Zhao, X.; Zhao, Y.; Ren, Z.; Li, Y. Combined QSAR/QSPR and molecular docking study on fluoroquinolones to reduce biological enrichment. Comput. Biol. Chem. 2019, 79, 177–184. [Google Scholar] [CrossRef]
- Kawczak, P.; Bober, L.; Bączek, T. Application of QSAR analysis and different quantum chemical calculation methods in activity evaluation of selected fluoroquinolones. Comb. Chem. High Throughput Screen. 2018, 21, 468–475. [Google Scholar] [CrossRef]
- Buglak, A.A.; Shanin, I.A.; Eremin, S.A.; Lei, H.-T.; Li, X.; Zherdev, A.V.; Dzantiev, B.B. Ciprofloxacin and clinafloxacin antibodies for an immunoassay of quinolones: Quantitative structure–activity analysis of cross-reactivities. Int. J. Mol. Sci. 2019, 20, 265. [Google Scholar] [CrossRef]
- Scientifico-Disciplinare, S.; Falabella, P.; Luca, F.; Scrano, L. Advanced Oxidation Processes (AOPs): Solutions for the degradation of emerging contaminants in liquid phases. Water 2023, 15, 1615. [Google Scholar]
- Appelbaum, P.; Hunter, P. The fluoroquinolone antibacterials: Past, present and future perspectives. Int. J. Antimicrob. Agents 2000, 16, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Dalhoff, A. Immunomodulatory activities of fluoroquinolones. Infection 2005, 33 (Suppl. 2), 55–70. [Google Scholar] [CrossRef]
- Kula, K.; Nagatsky, R.; Sadowski, M.; Siumka, Y.; Demchuk, O.M. Arylcyanomethylenequinone Oximes: An Overview of Synthesis, Chemical Transformations, and Biological Activity. Molecules 2023, 28, 5229. [Google Scholar] [CrossRef] [PubMed]
- Marciniec, K.; Beberok, A.; Pęcak, P.; Boryczka, S.; Wrześniok, D. Ciprofloxacin and moxifloxacin could interact with SARS-CoV-2 protease: Preliminary in silico analysis. Pharmacol. Rep. 2020, 72, 1553–1561. [Google Scholar] [CrossRef] [PubMed]
- Scroggs, S.L.; Offerdahl, D.K.; Flather, D.P.; Morris, C.N.; Kendall, B.L.; Broeckel, R.M.; Beare, P.A.; Bloom, M.E. Fluoroquinolone antibiotics exhibit low antiviral activity against SARS-CoV-2 and MERS-CoV. Viruses 2020, 13, 8. [Google Scholar] [CrossRef]
- Chaudhary, K.K.; Mishra, N.J.D. A review on molecular docking: Novel tool for drug discovery. JSM Chem. 2016, 3, 1029. [Google Scholar]
- Santos, E.S.; Silva, P.C.; Sousa, P.S.A.; Aquino, C.C.; Pacheco, G.; Teixeira, L.F.L.S.; Araujo, A.R.; Sousa, F.B.M.; Barros, R.O.; Ramos, R.M.; et al. Antiviral potential of diminazene aceturate against SARS-CoV-2 proteases using computational and in vitro approaches. Chem. Biol. Interact. 2022, 367, 110161. [Google Scholar] [CrossRef]
- Barciszewska, A.-M. Elucidating of oxidative distress in COVID-19 and methods of its prevention. Chem. Biol. Interact. 2021, 344, 109501. [Google Scholar] [CrossRef]
- Zhao, Y.-L.; Chen, Y.-L.; Sheu, J.-Y.; Chen, I.-L.; Wang, T.-C.; Tzeng, C.-C.; Chemistry, M. Synthesis and antimycobacterial evaluation of certain fluoroquinolone derivatives. Bioorg. Med. Chem. 2005, 13, 3921–3926. [Google Scholar] [CrossRef]
- Srinivasan, S.; Shafreen, R.M.B.; Nithyanand, P.; Manisankar, P.; Pandian, S.K. Synthesis and in vitro antimicrobial evaluation of novel fluoroquinolone derivatives. Eur. J. Med. Chem. 2010, 45, 6101–6105. [Google Scholar] [CrossRef]
- Al-Trawneh, S.A.; El-Abadelah, M.M.; Al-Abadleh, M.M.; Zani, F.; Incerti, M.; Vicini, P. A new efficient route to 7-aryl-6-fluoro-8-nitroquinolones as potent antibacterial agents. Eur. J. Med. Chem. 2014, 86, 364–367. [Google Scholar] [CrossRef] [PubMed]
- Chhetri, A.; Chettri, S.; Rai, P.; Sinha, B.; Brahman, D. Exploration of inhibitory action of Azo imidazole derivatives against COVID-19 main protease (Mpro): A computational study. J. Mol. Struct. 2021, 1224, 129178. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Jing, X.; Wang, J.; Zheng, Y.; Qiu, Y.; Ji, H.; Peng, L.; Jiang, S.; Wu, W.; Guo, D. Safety considerations of chloroquine in the treatment of patients with diabetes and COVID-19. Chem. Biol. Interact. 2022, 361, 109954. [Google Scholar] [CrossRef]
- Ye, N.; Yang, Z.; Liu, Y. Applications of density functional theory in COVID-19 drug modeling. Drug Discov. Today 2022, 27, 1411–1419. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Xie, W.; Xue, X.; Yang, K.; Ma, J.; Liang, W.; Zhao, Q.; Zhou, Z.; Pei, D.; Ziebuhr, J. Design of wide-spectrum inhibitors targeting coronavirus main proteases. PLoS Biol. 2005, 3, e324. [Google Scholar]
- Anand, K.; Ziebuhr, J.; Wadhwani, P.; Mesters, J.R.; Hilgenfeld, R. Coronavirus main proteinase (3CLpro) structure: Basis for design of anti-SARS drugs. Science 2003, 300, 1763–1767. [Google Scholar] [CrossRef]
- Jaramillo, P.; Domingo, L.R.; Chamorro, E.; Pérez, P. A further exploration of a nucleophilicity index based on the gas-phase ionization potentials. J. Mol. Struct. THEOCHEM 2008, 865, 68–72. [Google Scholar] [CrossRef]
- Tariq, S.; Mutahir, S.; Khan, M.A.; Mutahir, Z.; Hussain, S.; Ashraf, M.; Bao, X.; Zhou, B.; Stark, C.B.W.; Khan, I.U. Synthesis, in Vitro Cholinesterase Inhibition, Molecular Docking, DFT, and ADME Studies of Novel 1,3,4-Oxadiazole-2-Thiol Derivatives. Chem. Biodivers. 2022, 19, e202200157. [Google Scholar] [CrossRef]
- Celik, S. DFT investigations and molecular docking as potent inhibitors of SARS-CoV-2 main protease of 4-phenylpyrimidine. J. Mol. Struct. 2023, 1277, 134895. [Google Scholar] [CrossRef]
- Lakshminarayanan, S.; Jeyasingh, V.; Murugesan, K.; Selvapalam, N.; Dass, G. Molecular electrostatic potential (MEP) surface analysis of chemo sensors: An extra supporting hand for strength, selectivity & non-traditional interactions. J. Photochem. Photobiol. 2021, 6, 100022. [Google Scholar]
- Liu, K.; Lu, X.; Shi, H.; Xu, X.; Kong, R.; Chang, S. nCoVDock2: A docking server to predict the binding modes between COVID-19 targets and its potential ligands. Nucleic Acids Res. 2023, 51, W365–W371. [Google Scholar] [CrossRef] [PubMed]
- Alghamdi, A.; Abouzied, A.S.; Alamri, A.; Anwar, S.; Ansari, M.; Khadra, I.; Zaki, Y.H.; Gomha, S.M. Synthesis, molecular docking, and dynamic simulation targeting main protease (Mpro) of new, Thiazole clubbed pyridine scaffolds as potential COVID-19 inhibitors. Curr. Issues Mol. Biol. 2023, 45, 1422–1442. [Google Scholar] [CrossRef]
- Hatada, R.; Okuwaki, K.; Mochizuki, Y.; Handa, Y.; Fukuzawa, K.; Komeiji, Y.; Okiyama, Y.; Tanaka, S. Fragment molecular orbital based interaction analyses on COVID-19 main protease—Inhibitor N3 complex (PDB ID: 6LU7). J. Chem. Inf. Model. 2020, 60, 3593–3602. [Google Scholar] [CrossRef]
- Khan, M.A.; Mutahir, S.; Jabar, G.; Wenwei, Z.; Tariq, M.A.; Almehizia, A.A.A.; Mustafa, M. DFT, Molecular Docking, ADME, and Cardiotoxicity studies of Persuasive Thiazoles as Potential Inhibitors of the Main Protease of SARS-CoV-2. Chem. Biodivers. 2024, e202401775. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, F.S.; Amanlou, M. Simeprevir, potential candidate to repurpose for coronavirus infection: Virtual screening and molecular docking study. Life Sci. 2020, 258, 118205. [Google Scholar] [CrossRef]
- Ma, S.-K.E.; Wong, W.C.; Leung, C.-W.R.; Lai, S.-T.T.; Lo, Y.-C.J.; Wong, K.-H.H.; Chan, M.-C.; Que, T.-L.; Chow, K.-W.M.; Yuen, M.-C.; et al. Review of vector-borne diseases in Hong Kong. Travel Med. Infect. Dis. 2011, 9, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Carvelli, J.; Demaria, O.; Vély, F.; Batista, L.; Benmansour, N.C.; Fares, J.; Carpentier, S.; Thibult, M.-L.; Morel, A.; André, P. Identification of immune checkpoints in COVID-19. 2020. [Google Scholar] [CrossRef]
- Liu, C.; Zhou, Q.; Li, Y.; Garner, L.V.; Watkins, S.P.; Carter, L.J.; Smoot, J.; Gregg, A.C.; Daniels, A.D.; Jervey, S. Research and development on therapeutic agents and vaccines for COVID-19 and related human coronavirus diseases. ACS Cent. Sci. 2020, 6, 315–331. [Google Scholar] [CrossRef]
- Sivasankarapillai, V.S.; Pillai, A.M.; Rahdar, A.; Sobha, A.P.; Das, S.S.; Mitropoulos, A.C.; Mokarrar, M.H.; Kyzas, G.Z.J.N. On facing the SARS-CoV-2 (COVID-19) with combination of nanomaterials and medicine: Possible strategies and first challenges. Nanomaterials 2020, 10, 852. [Google Scholar] [CrossRef] [PubMed]
- Owis, A.I.; El-Hawary, M.S.; El Amir, D.; Aly, O.M.; Abdelmohsen, U.R.; Kamel, M.S.J.R.A. Molecular docking reveals the potential of Salvadora persica flavonoids to inhibit COVID-19 virus main protease. RSC Adv. 2020, 10, 19570–19575. [Google Scholar] [CrossRef]
- Mutahir, S.; Khan, M.A.; Naglah, A.M.; Al-Omar, M.A.; Almehizia, A.A.; Huwaimel, B.; Abouzied, A.S.; Alharbi, A.S.; Refat, M.S. Structural Characterization and Molecular Docking Screening of Most Potent 1, 2, 4-Triazine Sulfonamide Derivatives as Anti-Cancer Agents. Crystals 2023, 13, 767. [Google Scholar] [CrossRef]
- Arshad, R.; Khan, M.A.; Mutahir, S.; Hussain, S.; Al-Hazmi, G.H.; Refat, M.S. DFT, Molecular Docking and ADME Studies of Thiazolidinones as Tyrosinase Inhibitors. Polycycl. Aromat. Compd. 2022, 43, 6750–6765. [Google Scholar] [CrossRef]
- Bahl, A.S.; Verma, V.K.; Bhatia, J.; Arya, D.S. Integrating in silico and in vivo approach for investigating the role of polyherbal oil in prevention and treatment of COVID-19 infection. Chem. Biol. Interact. 2022, 367, 110179. [Google Scholar] [CrossRef] [PubMed]
- Garg, S.; Roy, A. In silico analysis of selected alkaloids against main protease (Mpro) of SARS-CoV-2. Chem. Biol. Interact. 2020, 332, 109309. [Google Scholar] [CrossRef]
- Shao, H.P.; Wang, T.H.; Zhai, H.L.; Bi, K.X.; Zhao, B.Q. Discovery of inhibitors against SARS-CoV-2 main protease using fragment-based drug design. Chem. Biol. Interact. 2023, 371, 110352. [Google Scholar] [CrossRef]
- Nazeer, U.; Rasool, N.; Mujahid, A.; Mansha, A.; Zubair, M.; Kosar, N.; Mahmood, T.; Raza Shah, A.; Shah, S.A.A.; Zakaria, Z.A. Selective arylation of 2-bromo-4-chlorophenyl-2-bromobutanoate via a Pd-catalyzed Suzuki cross-coupling reaction and its electronic and non-linear optical (NLO) properties via DFT studies. Molecules 2020, 25, 3521. [Google Scholar] [CrossRef] [PubMed]
- Aliveisi, R.; Taherpour, A.; Yavari, I. A DFT Study of Electronic Structures and Relative Stabilities of Isomeric n, m-Diazaphenanthrenes. Polycycl. Aromat. Compd. 2019, 39, 462–469. [Google Scholar] [CrossRef]
- Mebi, C.A. DFT study on structure, electronic properties, and reactivity of cis-isomers of [(NC5H4S)2 Fe(CO)2]. J. Chem. Sci. 2011, 123, 727–731. [Google Scholar] [CrossRef]
- Orio, M.; Pantazis, D.A.; Neese, F. Density functional theory. Photosynth. Res. 2009, 102, 443–453. [Google Scholar] [CrossRef]
- Mutahir, S.; Khan, M.A.; Almehizia, A.A.; Abouzied, A.S.; Khalifa, N.E.; Naglah, A.M.; Deng, H.; Refat, M.S.; Khojali, W.M.; Huwaimel, B. Design, Synthesis, Characterization and Computational Studies of Mannich Bases Oxadiazole Derivatives as New Class of Jack Bean Urease Inhibitors. Chem. Biodivers. 2023, 20, e202300241. [Google Scholar] [CrossRef]
- Fadlalla, M.; Ahmed, M.; Ali, M.; Elshiekh, A.A.; Yousef, B.A. Molecular docking as a potential approach in repurposing drugs against COVID-19: A systematic review and novel pharmacophore models. Curr. Pharmacol. Rep. 2022, 8, 212–226. [Google Scholar] [CrossRef]
- Mutahir, S.; Khan, M.A.; Khan, I.U.; Yar, M.; Ashraf, M.; Tariq, S.; Ye, R.-L.; Zhou, B.-J. Organocatalyzed and mechanochemical solvent-free synthesis of novel and functionalized bis-biphenyl substituted thiazolidinones as potent tyrosinase inhibitors: SAR and molecular modeling studies. Eur. J. Med. Chem. 2017, 134, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V.J.S.r. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Bikiewicz, A.; Banach, M.; von Haehling, S.; Maciejewski, M.; Bielecka-Dabrowa, A.J.E.H.F. Adjuvant breast cancer treatments cardiotoxicity and modern methods of detection and prevention of cardiac complications. ESC Heart Fail. 2021, 8, 2397–2418. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).