
Synthetic Routes to 2-aryl-1H-pyrrolo[2,3-b]pyridin-4-amines: Cross-Coupling and Challenges in SEM-Deprotection

Abstract

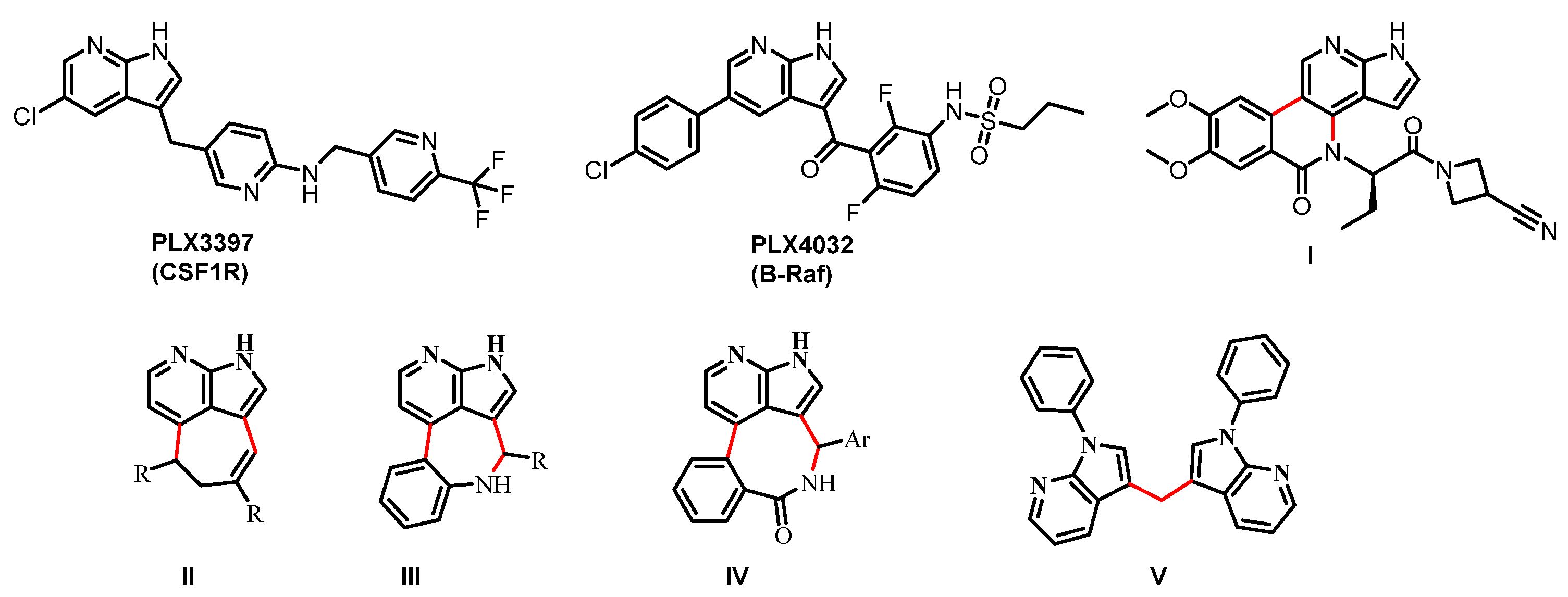
1. Introduction
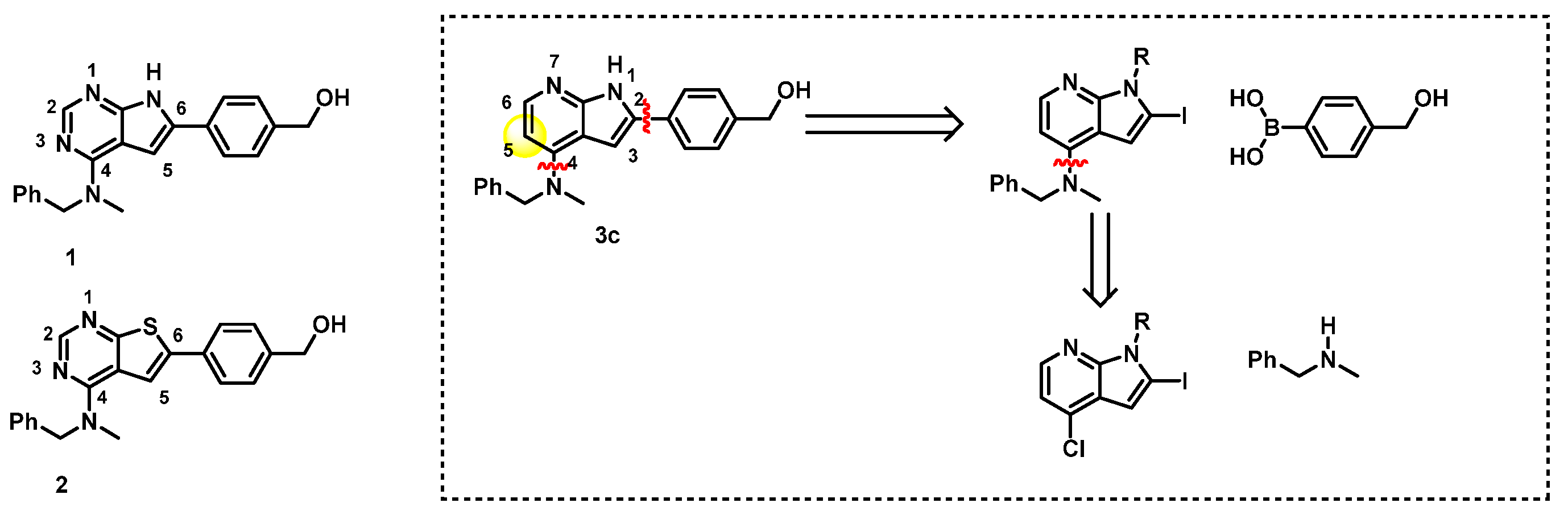
2. Results
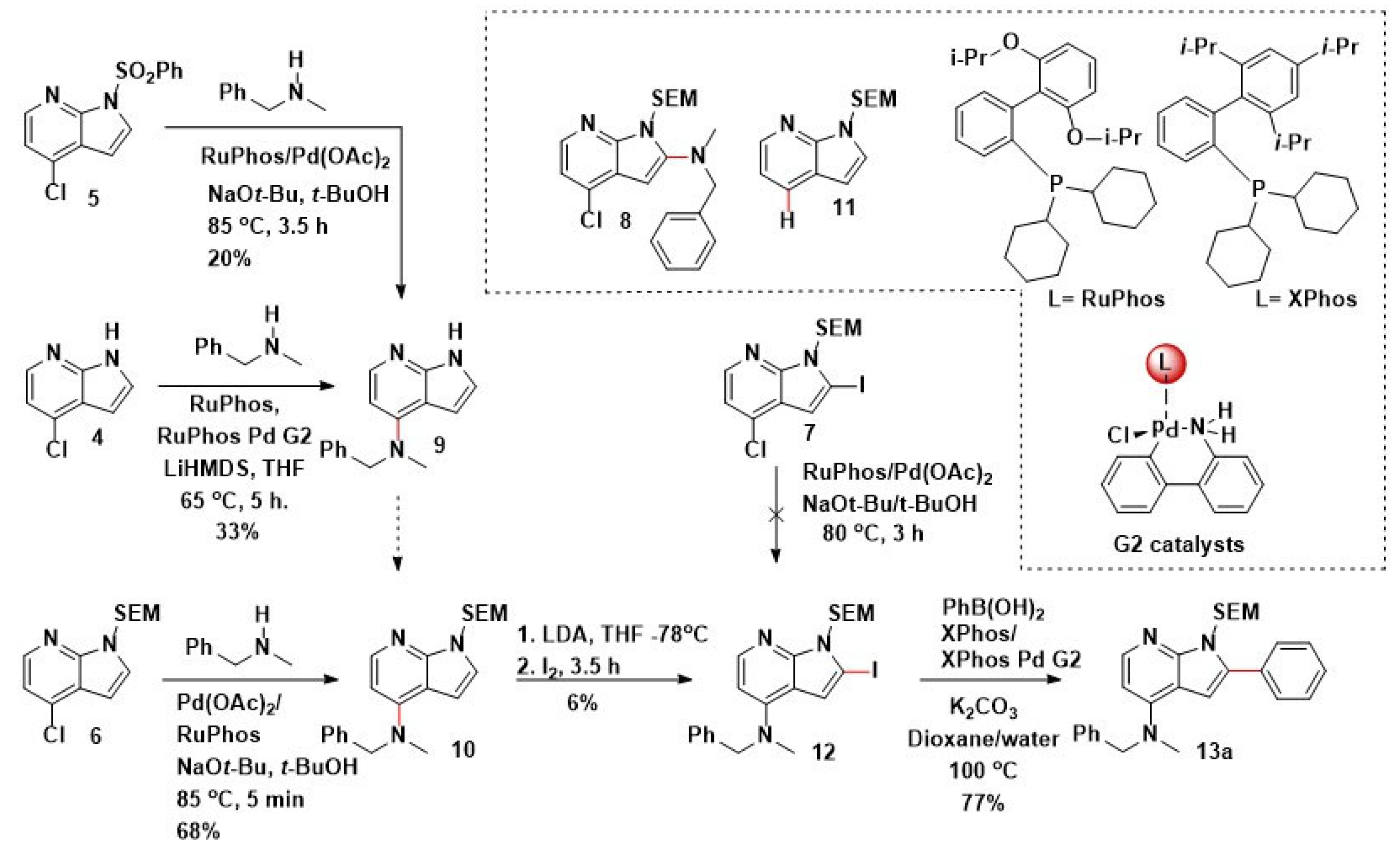
2.1. Route A: Amination Followed by Suzuki–Miyaura Cross-Coupling
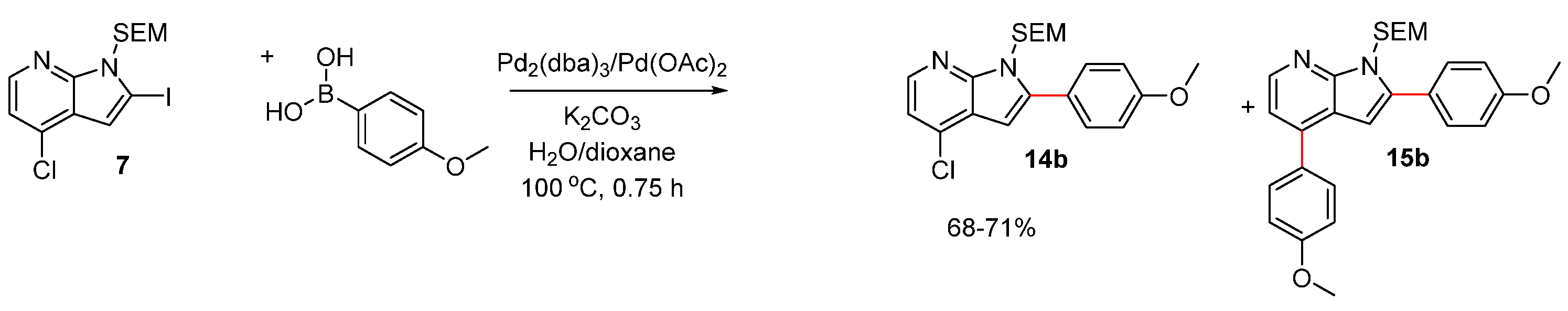
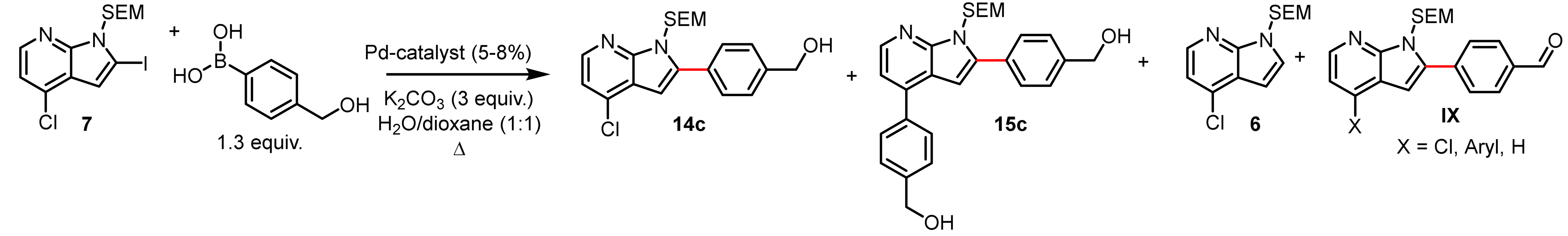
2.2. Route B: Suzuki–Miyaura Cross-Coupling Followed by Amination
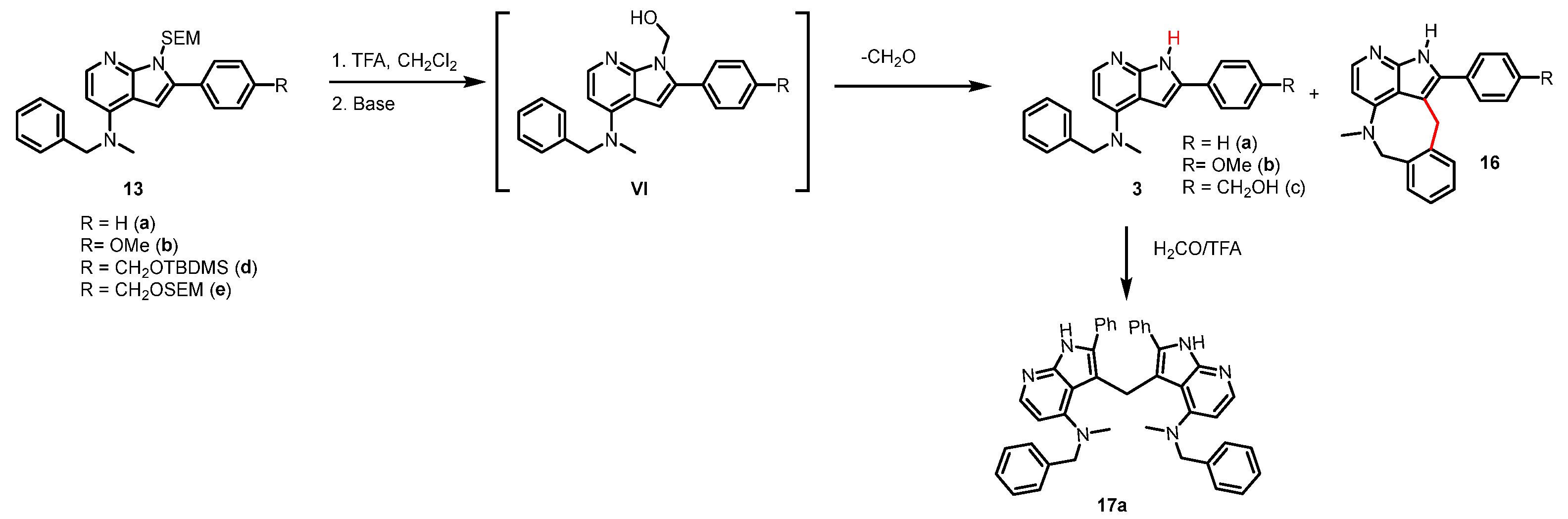
2.3. SEM-Deprotection
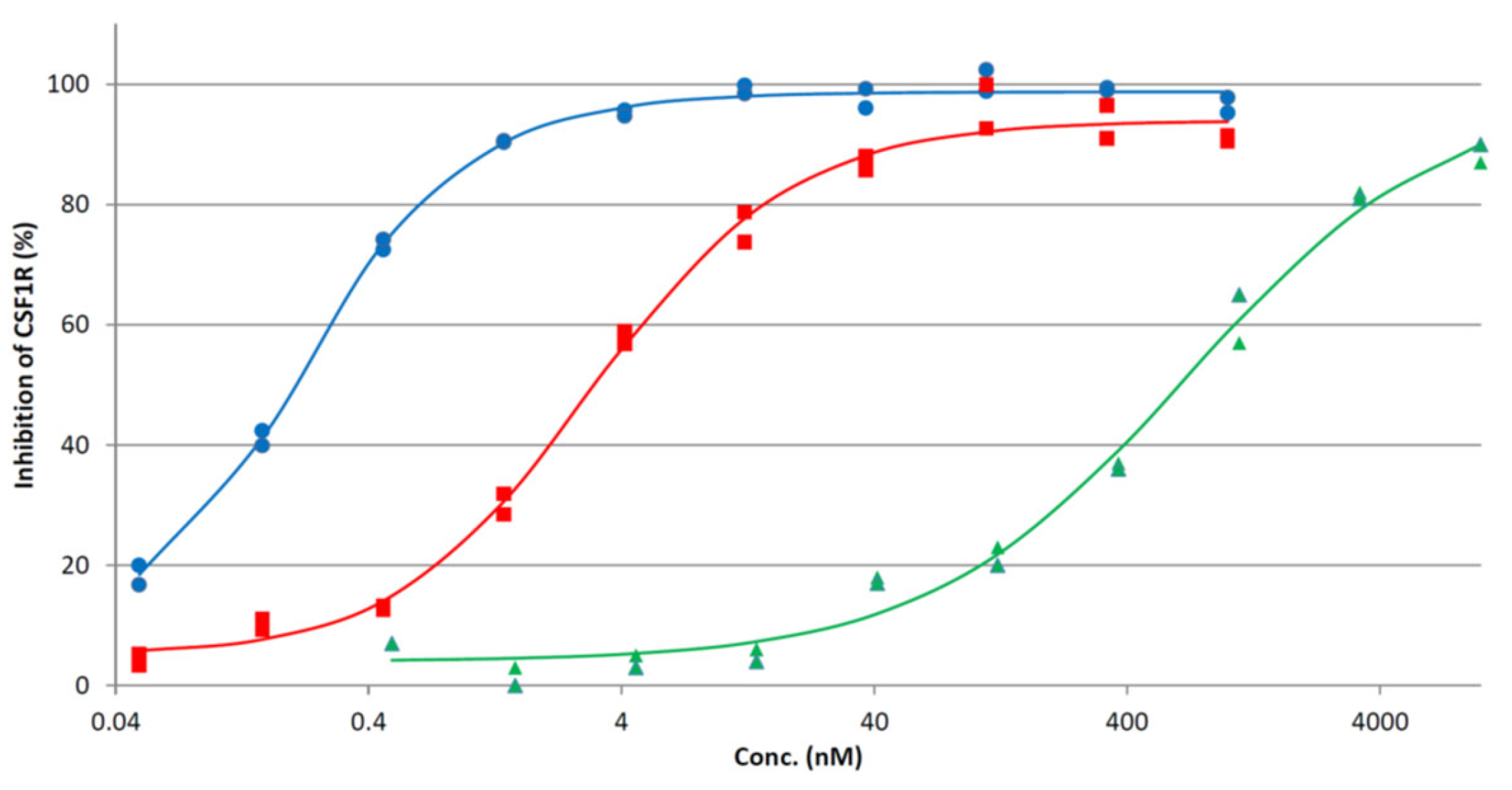
2.4. Enzymatic CSF1R Kinase Activity
3. Materials and Methods
3.1. Chemicals and Analysis
3.2. Synthesis
3.2.1. 4-Chloro-2-phenyl-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrrolo[2,3-b]pyridine (14a)
3.2.2. 4-Chloro-2-(4-methoxyphenyl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrrolo[2,3-b]pyridine (14b)
3.2.3. (4-(4-Chloro-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrrolo[2,3-b]pyridin-2-yl)phenyl)methanol (14c)
3.2.4. 2-(4-(((tert-Butyldimethylsilyl)oxy)methyl)phenyl)-4-chloro-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrrolo[2,3-b]pyridine (14d)
3.2.5. 4-Chloro-2-(4-(((2-(trimethylsilyl)ethoxy)methoxy)methyl)phenyl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrrolo[2,3-b]pyridine (14e)
3.2.6. N-Benzyl-N-methyl-2-phenyl-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrrolo[2,3-b]pyridin-4-amine (13a) via 12
3.2.7. N-Benzyl-N-methyl-2-phenyl-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrrolo[2,3-b]pyridin-4-amine (13a) via 14a
3.2.8. N-Benzyl-2-(4-methoxyphenyl)-N-methyl-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrrolo[2,3-b]-pyridin-4-amine (13b)
3.2.9. (4-(4-(Benzyl(methyl)amino)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrrolo[2,3-b]pyridin-2-yl)phenyl)methanol (13c)
3.2.10. N-Benzyl-2-(4-(((tert-butyldimethylsilyl)oxy)methyl)phenyl)-N-methyl-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrrolo[2,3-b]pyridin-4-amine (13d)
3.2.11. N-Benzyl-N-methyl-2-(4-(((2-(trimethylsilyl)ethoxy)methoxy)methyl)phenyl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrrolo[2,3-b]pyridin-4-amine (13e)
3.2.12. N-Benzyl-N-methyl-2-phenyl-1H-pyrrolo-[2,3-b]pyridin-4-amine (3a)
3.2.13. N-Benzyl-N-methyl-2-(4-methoxyphenyl-1H-pyrrolo-[2,3-b]pyridin-4-amine (3b)
3.2.14. (4-(4-(Benzyl(methyl)amino)-1H-pyrrolo[2,3-b]pyridin-2-yl)phenyl)methanol (3c)
3.2.15. 6-Methyl-1-phenyl-2,6,7,12-tetrahydro-2,3,6-triazabenzo[6,7]cycloocta[1,2,3-cd]indene (16a)
3.2.16. 1-(4-Methoxyphenyl)-6-methyl-2,6,7,12-tetrahydro-2,3,6-triazabenzo[6,7]cycloocta[1,2,3-cd]indene (16b)
3.2.17. (4-(6-Methyl-2,6,7,12-tetrahydro-2,3,6-triazabenzo[6,7]cycloocta[1,2,3-cd]inden-1-yl)phenyl)methanol (16c)
3.2.18. 3,3-Methylenebis(N-benzyl-N-methyl-2-phenyl-1H-pyrrolo[2,3-b]pyridin-4-amine (17a)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Smirnov, A.V.; English, D.S.; Rich, R.L.; Lane, J.; Teyton, L.; Schwabacher, A.W.; Luo, S.; Thornburg, R.W.; Petrich, J.W. Photophysics and Biological Applications of 7-Azaindole and Its Analogs. J. Phys. Chem. B 1997, 101, 2758–2769. [Google Scholar] [CrossRef]
- Herrera-Ochoa, D.; Llano, I.; Ripoll, C.; Cybulski, P.; Kreuzer, M.; Rocha, S.; García-Frutos, E.M.; Bravo, I.; Garzón-Ruiz, A. Protein aggregation monitoring in cells under oxidative stress: A novel fluorescent probe based on a 7-azaindole-BODIPY derivative. J. Mater. Chem. B 2024, 12, 7577–7590. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.-B.; Wang, S. Luminescence and reactivity of 7-azaindole derivatives and complexes. Chem. Soc. Rev. 2010, 39, 3142–3156. [Google Scholar] [CrossRef] [PubMed]
- Tap, W.D.; Wainberg, Z.A.; Anthony, S.P.; Ibrahim, P.N.; Zhang, C.; Healey, J.H.; Chmielowski, B.; Staddon, A.P.; Cohn, A.L.; Shapiro, G.I.; et al. Structure-guided blockade of CSF1R kinase in tenosynovial giant-cell tumor. N. Engl. J. Med. 2015, 373, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Bollag, G.; Hirth, P.; Tsai, J.; Zhang, J.; Ibrahim, P.N.; Cho, H.; Spevak, W.; Zhang, C.; Zhang, Y.; Habets, G.; et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 2010, 467, 596–599. [Google Scholar] [CrossRef]
- Farmer, L.J.; Ledeboer, M.W.; Hoock, T.; Arnost, M.J.; Bethiel, R.S.; Bennani, Y.L.; Black, J.J.; Brummel, C.L.; Chakilam, A.; Dorsch, W.A.; et al. Discovery of VX-509 (Decernotinib): A Potent and Selective Janus Kinase 3 Inhibitor for the Treatment of Autoimmune Diseases. J. Med. Chem. 2015, 58, 7195–7216. [Google Scholar] [CrossRef]
- Foote, K.M.; Nissink, J.W.M.; McGuire, T.; Turner, P.; Guichard, S.; Yates, J.W.T.; Lau, A.; Blades, K.; Heathcote, D.; Odedra, R.; et al. Discovery and Characterization of AZD6738, a Potent Inhibitor of Ataxia Telangiectasia Mutated and Rad3 Related (ATR) Kinase with Application as an Anticancer Agent. J. Med. Chem. 2018, 61, 9889–9907. [Google Scholar] [CrossRef]
- Juchum, M.; Guenther, M.; Doering, E.; Sievers-Engler, A.; Laemmerhofer, M.; Laufer, S. Trisubstituted Imidazoles with a Rigidized Hinge Binding Motif Act as Single Digit nM Inhibitors of Clinically Relevant EGFR L858R/T790M and L858R/T790M/C797S Mutants: An Example of Target Hopping. J. Med. Chem. 2017, 60, 4636–4656. [Google Scholar] [CrossRef]
- Barberis, C.; Moorcroft, N.; Arendt, C.; Levit, M.; Moreno-Mazza, S.; Batchelor, J.; Mechin, I.; Majid, T. Discovery of N-substituted 7-azaindoles as PIM1 kinase inhibitors—Part I. Bioorg. Med. Chem. Lett. 2017, 27, 4730–4734. [Google Scholar] [CrossRef]
- Wang, H.; Lawson, J.D.; Scorah, N.; Kamran, R.; Hixon, M.S.; Atienza, J.; Dougan, D.R.; Sabat, M. Design, synthesis and optimization of novel Alk5 (activin-like kinase 5) inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 4334–4339. [Google Scholar] [CrossRef]
- Price, D.J.; Drewry, D.H.; Schaller, L.T.; Thompson, B.D.; Reid, P.R.; Maloney, P.R.; Liang, X.; Banker, P.; Buckholz, R.G.; Selley, P.K.; et al. An orally available, brain-penetrant CAMKK2 inhibitor reduces food intake in rodent model. Bioorg. Med. Chem. Lett. 2018, 28, 1958–1963. [Google Scholar] [CrossRef] [PubMed]
- Barl, N.M.; Sansiaume-Dagousset, E.; Karaghiosoff, K.; Knochel, P. Full Functionalization of the 7-Azaindole Scaffold by Selective Metalation and Sulfoxide/Magnesium Exchange. Angew. Chem. Int. Ed. 2013, 52, 10093–10096. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.; David, E.; Toutov, A.A.; Snieckus, V. In Situ Anionic Shielding for Regioselective Metalation: Directed peri and Iterative Metalation Routes to Polyfunctionalized 7-Azaindoles. Angew. Chem. Int. Ed. 2012, 51, 2722–2726. [Google Scholar] [CrossRef] [PubMed]
- Elsayed, M.S.A.; Nielsen, J.J.; Park, S.; Park, J.; Liu, Q.; Kim, C.H.; Pommier, Y.; Agama, K.; Low, P.S.; Cushman, M. Application of Sequential Palladium Catalysis for the Discovery of Janus Kinase Inhibitors in the Benzo[c]pyrrolo[2,3-h][1,6]naphthyridin-5-one (BPN) Series. J. Med. Chem. 2018, 61, 10440–10462. [Google Scholar] [CrossRef]
- Angiolini, M.; Borgia, A.L.; Bandiera, T. Multistep synthesis of a new tricyclic azaindole-based scaffold. Tetrahedron Lett. 2009, 50, 5156–5158. [Google Scholar] [CrossRef]
- Wang, T.; Ledeboer, M.W.; Duffy, J.P.; Pierce, A.C.; Zuccola, H.J.; Block, E.; Shlyakter, D.; Hogan, J.K.; Bennani, Y.L. A novel chemotype of kinase inhibitors: Discovery of 3,4-ring fused 7-azaindoles and deazapurines as potent JAK2 inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 153–156. [Google Scholar] [CrossRef]
- Lee, S.H.; Kim, K.; Jeon, Y.U.; Kundu, A.; Dey, P.; Hwang, J.Y.; Mishra, N.K.; Kim, H.S.; Kim, I.S. Lewis acid-mediated cross-coupling reaction of 7-azaindoles and aldehydes: Cytotoxic evaluation of C3-linked bis-7-azaindoles. Tetrahedron Lett. 2019, 60, 150974. [Google Scholar] [CrossRef]
- Bjørnstad, F.; Havik, S.; Aarhus, T.I.; Mahdi, I.; Unger, A.; Habenberger, P.; Degenhart, C.; Eickhoff, J.; Klebl, B.M.; Sundby, E.; et al. Pyrrolopyrimidine based CSF1R inhibitors: Attempted departure from Flatland. Eur. J. Med. Chem. 2024, 265, 116053. [Google Scholar] [CrossRef]
- Aarhus, T.I.; Bjørnstad, F.; Wolowczyk, C.; Larsen, K.U.; Rognstad, L.; Leithaug, T.; Unger, A.; Habenberger, P.; Wolf, A.; Bjørkøy, G.; et al. Synthesis and Development of Highly Selective Pyrrolo[2,3-d]pyrimidine CSF1R Inhibitors Targeting the Autoinhibited Form. J. Med. Chem. 2023, 66, 6959–6980. [Google Scholar] [CrossRef]
- Aarhus, T.I.; Eickhoff, J.; Klebl, B.; Unger, A.; Boros, J.; Choidas, A.; Zischinsky, M.-L.; Wolowczyk, C.; Bjørkøy, G.; Sundby, E.; et al. A highly selective purine-based inhibitor of CSF1R potently inhibits osteoclast differentiation. Eur. J. Med. Chem. 2023, 255, 115344. [Google Scholar] [CrossRef]
- Caldwell, J.J.; Davies, T.G.; Donald, A.; McHardy, T.; Rowlands, M.G.; Aherne, G.W.; Hunter, L.K.; Taylor, K.; Ruddle, R.; Raynaud, F.I.; et al. Identification of 4-(4-Aminopiperidin-1-yl)-7H-pyrrolo[2,3-d]pyrimidines as Selective Inhibitors of Protein Kinase B through Fragment Elaboration. J. Med. Chem 2008, 51, 2147–2157. [Google Scholar] [CrossRef] [PubMed]
- Henderson, J.L.; McDermott, S.M.; Buchwald, S.L. Palladium-Catalyzed Amination of Unprotected Halo-7-azaindoles. Org. Lett 2010, 12, 4438–4441. [Google Scholar] [CrossRef] [PubMed]
- Henderson, J.L.; Buchwald, S.L. Efficient Pd-catalyzed amination reactions for heterocycle fuctionalization. Org. Lett 2010, 12, 4442–4445. [Google Scholar] [CrossRef] [PubMed]
- Bugge, S.; Kaspersen, S.J.; Sundby, E.; Hoff, B.H. Route selection in the synthesis of C-4 and C-6 substituted thienopyrimidines. Tetrahedron 2012, 68, 9226–9233. [Google Scholar] [CrossRef]
- Han, J.; Henriksen, S.; Nørsett, K.G.; Sundby, E.; Hoff, B.H. Balancing potency, metabolic stability and permeability in pyrrolopyrimidine-based EGFR inhibitors. Eur. J. Med. Chem. 2016, 124, 583–607. [Google Scholar] [CrossRef]
- Elavarasan, P.; Sathananthan, K.; Ponnusamy, S. Synthesis of 5-aryl-3,3′-bis-indolyl and bis-7-aza-indolyl methanone derivatives from 5-bromo-7-azaindoles via sequential methylenation using microwave irradiation, CAN oxidation, and Suzuki coupling reactions. RSC Adv. 2022, 12, 30712–30721. [Google Scholar] [CrossRef]
- Pollok, B.A.; Hamman, B.D.; Rodems, S.M.; Makings, L.R. Optical Probes and Assays. WO Patent 2000066766A1, 9 November 2000. [Google Scholar]
- Bell, I.M.; Gallicchio, S.N.; Wood, M.R.; Quigley, A.G.; Stump, C.A.; Zartman, C.B.; Fay, J.F.; Li, C.-C.; Lynch, J.J.; Moore, E.L.; et al. CGRP Receptor Antagonist. ACS Med. Chem. Lett. 2010, 1, 24–29. [Google Scholar] [CrossRef]
- Cheng, C.-C.; Chang, C.-P.; Yu, W.-S.; Hung, F.-T.; Liu, Y.-I.; Wu, G.-R.; Chou, P.-T. Comprehensive Studies on Dual Excitation Behavior of Double Proton versus Charge Transfer in 4-(N-Substituted amino)-1H-pyrrolo[2,3-b]pyridines. J. Phys. Chem. A 2003, 107, 1459–1471. [Google Scholar] [CrossRef]
- El-Gamal, M.I.; Abdel-Maksoud, M.S.; El-Din, M.M.G.; Yoo, K.H.; Baek, D.; Oh, C.-H. Cell-Based Biological Evaluation of a New Bisamide FMS Kinase Inhibitor Possessing Pyrrolo[3,2-c]pyridine Scaffold. Arch. Pharm. 2014, 347, 635–641. [Google Scholar] [CrossRef]
- Layek, M.; Gajare, V.; Kalita, D.; Islam, A.; Mukkanti, K.; Pal, M. A highly effective synthesis of 2-alkynyl-7-azaindoles: Pd/C-mediated alkynylation of heteroaryl halides in water. Tetrahedron 2009, 65, 4814–4819. [Google Scholar] [CrossRef]
- Nakajima, Y.; Tojo, T.; Morita, M.; Hatanaka, K.; Shirakami, S.; Tanaka, A.; Sasaki, H.; Nakai, K.; Mukoyoshi, K.; Hamaguchi, H.; et al. Synthesis and evaluation of 1H-pyrrolo[2,3-b]pyridine derivatives as novel immunomodulators targeting Janus kinase 3. Chem. Pharm. Bull. 2015, 63, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhi, B.; Baum, J.; Chen, Y.; Crockett, R.; Huang, L.; Eisenberg, S.; Ng, J.; Larsen, R.; Martinelli, M.; et al. A Practical Synthesis of 2-((1H-Pyrrolo[2,3-b]pyridine-4-yl)methylamino)-5-fluoronicotinic Acid. J. Org. Chem. 2006, 71, 4021–4023. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Jeon, H.B. Convenient N-formylation of amines in dimethylformamide with methyl benzoate under microwave irradiation. Bull. Korean Chem. Soc. 2010, 31, 1424–1426. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Pd mol% | Temp. [°C] | Scale [mg] | Time [h] | Product Distribution (mol %) 1 | ||||
| 14c | 15c 2 | Unk. 3 | 6 | IX 4 | ||||||
| 1 | Pd2(dba)3 | 7 | 100 | 100 | 1.75 | 92 | 4 | 4 | 0 | 0 |
| 2 | Pd2(dba)3 | 7 | 80 | 100 | 1.75 | 89 | 5 | 5 | 0 | 0 |
| 3 | Pd2(dba)3 | 7 | 60 | 50 | 2.5 | 87 | 6 | 7 | 0 | 0 |
| 4 | XPhos/ XPhos Pd G2 | 7 | 80 | 50 | 2.75 | 15 | 23 | 13 | 0 | 49 |
| 5 | Pd(OAc)2 | 5 | 80 | 50 | 2.75 | 68 | 7 | 17 | 2 | 6 |
| 6 | PEPPSITM-SIPr 5 | 5 | 80 | 50 | 0.3 | 44 | 17 | 13 | 9 | 18 |
| 7 | (Dppf)PdCl2 6 | 5 | 80 | 50 | 0.3 | 79 | 3 | 10 | 5 | 3 |
| 8 | Pd(PPh3)4 | 8 | 80 | 50 | 5 | 92 | 0 | 0 | 8 | 0 |
| 9 | Pd(PPh3)4 | 5 | 80 | 1233 | 9 | 95 | 0 | 0 | 5 | 0 |
| 10 | Pd(PPh3)4 | 5 | 90 | 2087 | 22 | 85 | 9 | 0 | 6 | 0 |
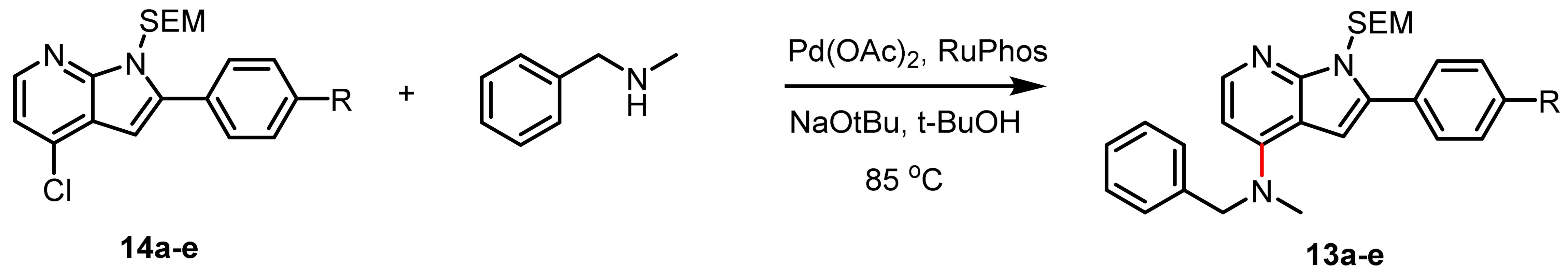 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Subs. | R | Scale (mg) 1 | Time (h) | Conv. (%) 2 | Yield (%) 3 | Product |
| 1 | 14a | H | 230 | 1 | >99 | 74 | 13a |
| 2 | 14b | OCH3 | 650 | 1 | >99 | 76 | 13b |
| 3 | 14c | CH2OH | 100 | 24 | ca 40 | - | 13c |
| 4 | 14d | CH2OTBDMS | 816 | 0.5 | >99 | 89 | 13d |
| 5 | 14e | CH2OSEM | 455 | 0.33 | >99 | 89 | 13e |
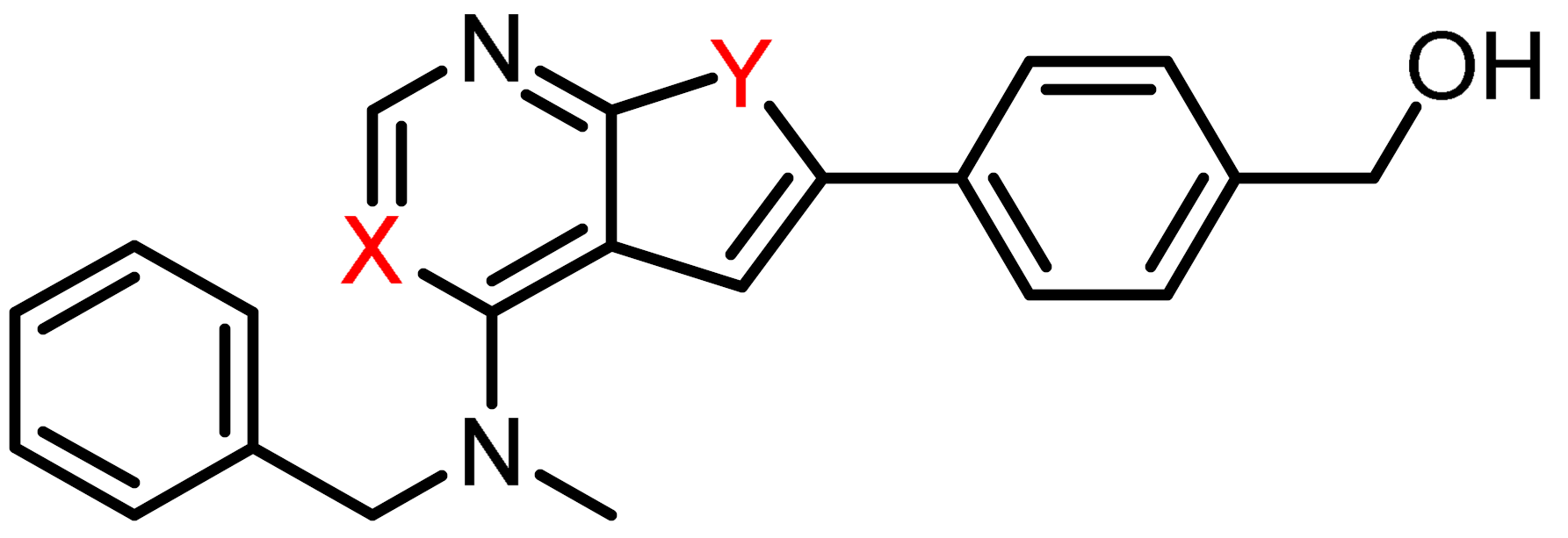 | ||||
|---|---|---|---|---|
| Inhibitor | X | Y | CSF1R IC50 1 | CSF1R IC50 2 |
| 3c | CH | N | 3.1 | 105 |
| 1 3 | N | N | 1.0 | 5 |
| 2 3 | N | S | 707 | 450 |
| PLX3397 3 | 5 | 20 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Merugu, S.R.; Selmer-Olsen, S.; Kaada, C.J.; Sundby, E.; Hoff, B.H. Synthetic Routes to 2-aryl-1H-pyrrolo[2,3-b]pyridin-4-amines: Cross-Coupling and Challenges in SEM-Deprotection. Molecules 2024, 29, 4743. https://doi.org/10.3390/molecules29194743
Merugu SR, Selmer-Olsen S, Kaada CJ, Sundby E, Hoff BH. Synthetic Routes to 2-aryl-1H-pyrrolo[2,3-b]pyridin-4-amines: Cross-Coupling and Challenges in SEM-Deprotection. Molecules. 2024; 29(19):4743. https://doi.org/10.3390/molecules29194743
Chicago/Turabian StyleMerugu, Srinivas Reddy, Sigrid Selmer-Olsen, Camilla Johansen Kaada, Eirik Sundby, and Bård Helge Hoff. 2024. "Synthetic Routes to 2-aryl-1H-pyrrolo[2,3-b]pyridin-4-amines: Cross-Coupling and Challenges in SEM-Deprotection" Molecules 29, no. 19: 4743. https://doi.org/10.3390/molecules29194743
APA StyleMerugu, S. R., Selmer-Olsen, S., Kaada, C. J., Sundby, E., & Hoff, B. H. (2024). Synthetic Routes to 2-aryl-1H-pyrrolo[2,3-b]pyridin-4-amines: Cross-Coupling and Challenges in SEM-Deprotection. Molecules, 29(19), 4743. https://doi.org/10.3390/molecules29194743