
Developing and Evaluating the Greenness of a Reliable, All-in-One Thin-Film Microextraction Protocol for Determining Fentanyl, Methadone, and Zolpidem in Plasma, Urine, and Oral Fluid

Abstract

1. Introduction
2. Results and Discussion
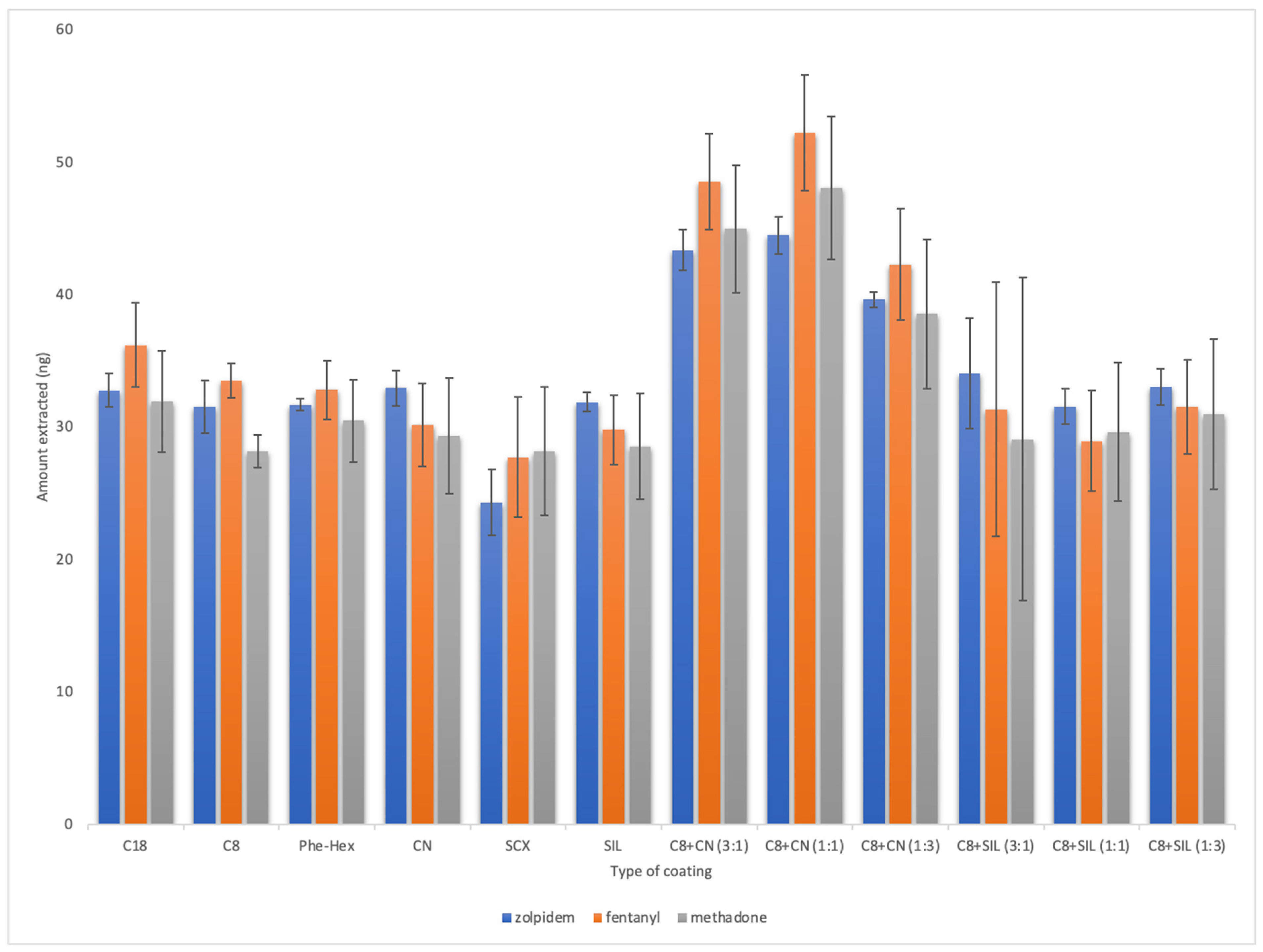
2.1. Method Development
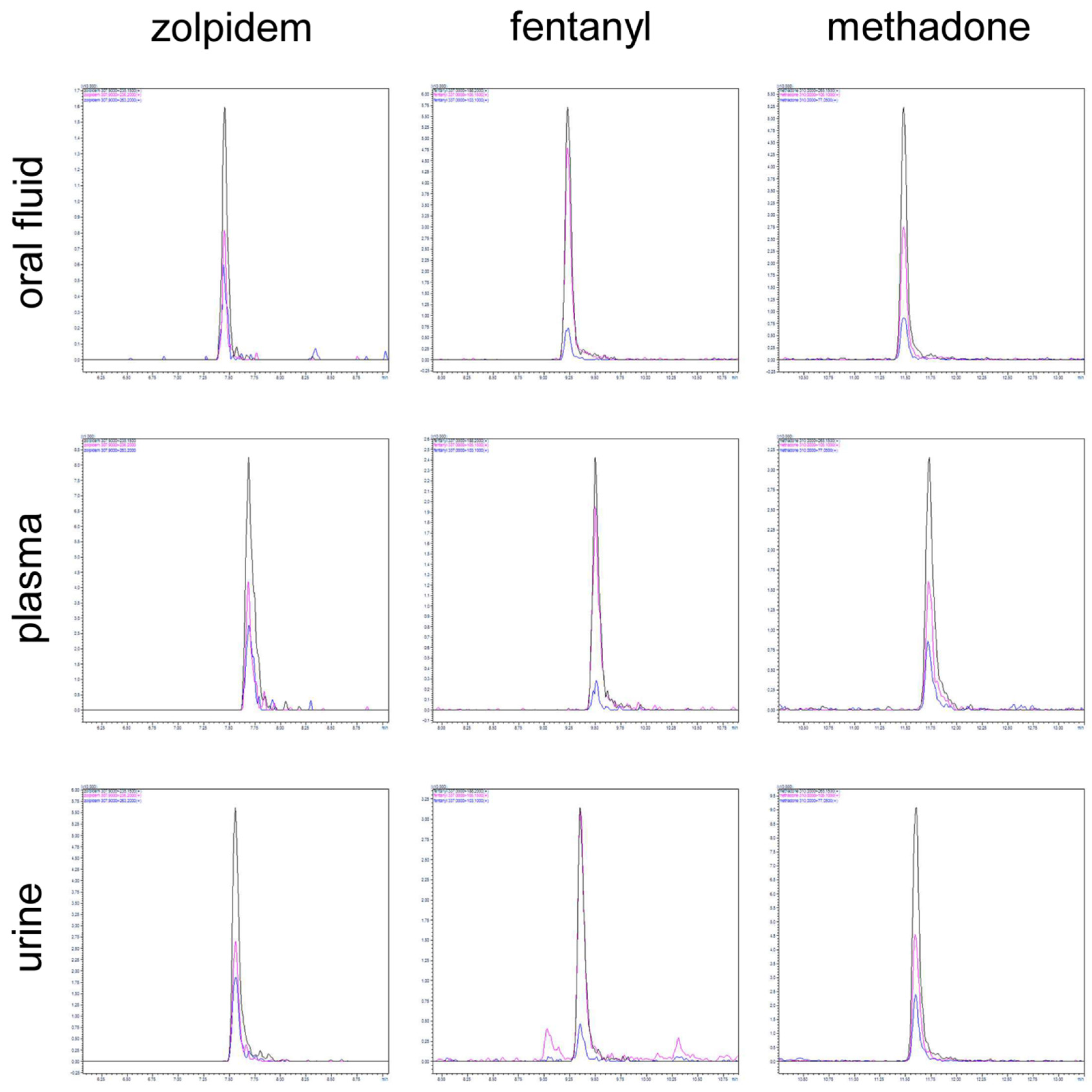
2.2. Method Validation
2.3. In Silico Assessment of the TFME-LC-MS/MS Protocol’s Greenness
3. Materials and Methods
3.1. Human Matrices
3.2. Chemicals
3.3. Working/Standard Solutions and Calibrators
3.4. Preparation of the Thin-Film Microextraction Blades
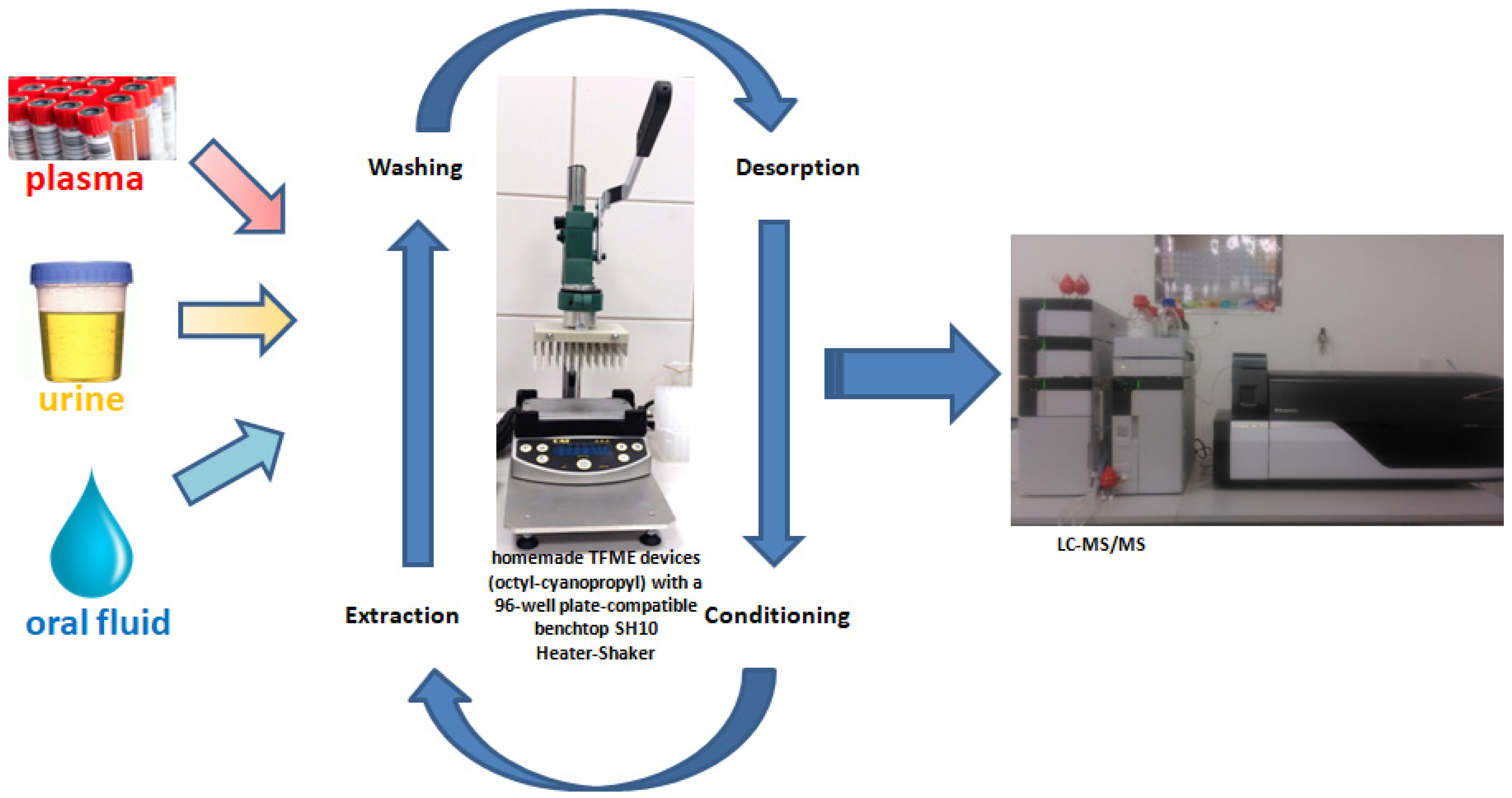
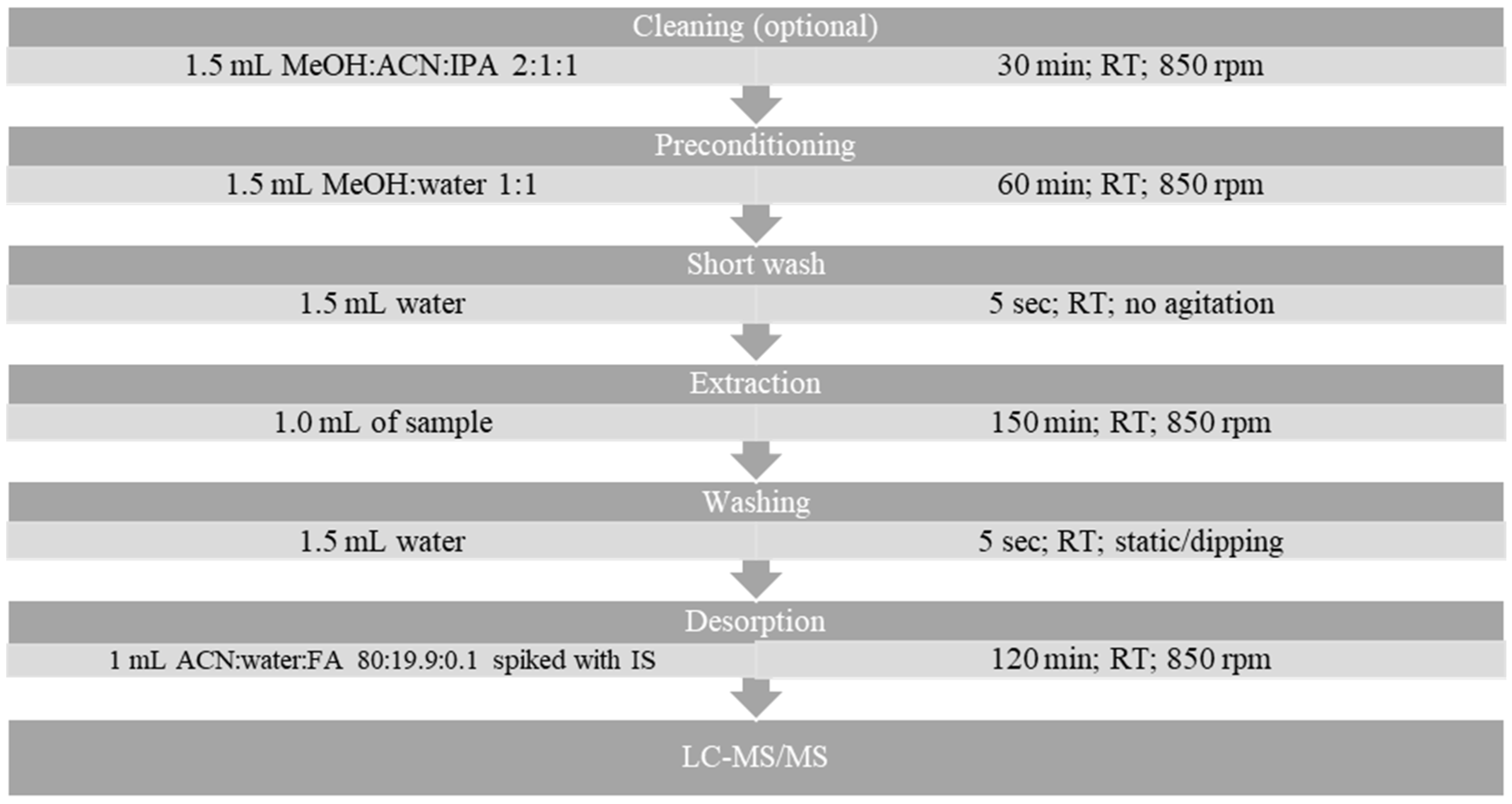
3.5. Sample Preparation and Extraction Protocol
3.6. LC-MS/MS Method
3.7. Validation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Health Topic. Available online: https://www.who.int/news-room/fact-sheets/detail/opioid-overdose (accessed on 16 November 2023).
- Centers for Disease Control and Prevention. Drug Overdose. Available online: https://www.cdc.gov/drugoverdose/index.html (accessed on 16 November 2023).
- National Academies of Sciences, Engineering, and Medicine; Health and Medicine Division, Board on Health Sciences Policy; Committee on Medication-Assisted Treatment for Opioid Use Disorder. The Effectiveness of Medication-Based Treatment for Opioid Use Disorder. In Medications for Opioid Use Disorder Save Lives; Mancher, M., Leshner, A.I., Eds.; The National Academies Press: Washington, DC, USA, 2019; pp. 33–62. [Google Scholar]
- Strang, J. (Ed.) Drug Misuse and Dependence: UK Guidelines on Clinical Management; Global and Public Health 2017, Independent Expert Working Group; Taylor & Francis: London, UK, 2017; pp. 97–99, Chapter 4.4.7—Methadone Dosing. [Google Scholar]
- FDA Drug Safety Communication: FDA Warns about Serious Risks and Death when Combining Opioid Pain or Cough Medicines with Benzodiazepines; Requires Its Strongest Warning. Available online: https://www.fda.gov/drugs/drug-safety-and-availability/fda-drug-safety-communication-fda-warns-about-serious-risks-and-death-when-combining-opioid-pain-or (accessed on 16 November 2023).
- Brandt, J.; Leong, C. Benzodiazepines and Z-Drugs: An Updated Review of Major Adverse Outcomes Reported on in Epidemiologic Research. Drugs R D. 2017, 17, 493–507. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.R.; Chand, R.; Kumar, S.; Mittal, N.; Srinivasamn, S.; Rajabzadeh, A.M. Recent biosensing advances in the rapid detection of illicit drugs. TrAC Trends Anal. Chem. 2020, 131, 116006. [Google Scholar] [CrossRef]
- Joosten, F.; Parrilla, M.; van Nuijs, A.L.N.; Ozoemena, K.I.; De Wael, K. Electrochemical detection of illicit drugs in oral fluid: Potential for forensic drug testing. Electrochim. Acta 2022, 436, 141309. [Google Scholar] [CrossRef]
- Farley, M.; Tran, H.; Towler, S.; Gevorkyan, J.; Pearring, S.; Rodda, L.N. A Single Method for 127 Recommended and Additional DUID Drugs in Blood and Urine by LC-MS-MS. J. Anal. Toxicol. 2022, 46, 658–669. [Google Scholar] [CrossRef]
- Trana, A.D.; Mannocchi, G.; Pirani, F.; Maida, N.; Gottardi, M.; Pichini, S.; Busardò, F.P. A Comprehensive HPLC-MS-MS Screening Method for 77 New Psychoactive Substances, 24 Classic Drugs and 18 Related Metabolites in Blood, Urine and Oral Fluid. J. Anal. Toxicol. 2020, 44, 769–783. [Google Scholar] [CrossRef] [PubMed]
- Kyriakou, C.; Marchei, E.; Scaravelli, G.; García-Algar, O.; Supervía, A.; Graziano, S. Identification and quantification of psychoactive drugs in whole blood using dried blood spot (DBS) by ultra-performance liquid chromatography tandem mass spectrometry. J. Pharm. Biomed. Anal. 2016, 128, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Pablo, A.; Breaud, A.R.; Clarke, W. Automated analysis of dried urine spot (DUS) samples for toxicology screening. Clin. Biochem. 2020, 75, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Scanferla, D.T.P.; Lini, R.S.; Marchioni, C.; Mossini, S.A.G. Drugs of abuse: A narrative review of recent trends in biological sample preparation and chromatographic techniques. Forensic Chem. 2022, 30, 100442. [Google Scholar] [CrossRef]
- D’Ovidio, C.; Bonelli, M.; Rosato, E.; Tartaglia, A.; Ulusoy, H.İ.; Samanidou, V.; Furton, K.G.; Kabir, A.; Ali, I.; Savini, F.; et al. Novel Applications of Microextraction Techniques Focused on Biological and Forensic Analyses. Separations 2022, 9, 18. [Google Scholar] [CrossRef]
- Carasek, E.; Scur, R.; Bernardi, G. High-throughput analytical methods employing microextraction techniques: Towards fast analyses lasting a few seconds. Adv. Sample Prep. 2023, 8, 100095. [Google Scholar] [CrossRef]
- Silveira, G.O.; Pego, A.M.F.; Pereira, E.; Silva, J.; Yonamine, M. Green sample preparations for the bioanalysis of drugs of abuse in complex matrices. Bioanalysis 2019, 11, 295–312. [Google Scholar] [CrossRef] [PubMed]
- Mandrioli, R.; Mercolini, L.; Protti, M. Blood and Plasma Volumetric Absorptive Microsampling (VAMS) Coupled to LC-MS/MS for the Forensic Assessment of Cocaine Consumption. Molecules 2020, 25, 1046. [Google Scholar] [CrossRef]
- Mestad, I.O.; Gjelstad, A.; Pedersen-Bjergaard, S.; Øiestad, E.L. Green and sustainable drug analysis—Combining microsampling and microextraction of drugs of abuse. Sust. Chem. Pharm. 2021, 24, 100517. [Google Scholar]
- Ask, K.S.; Øiestad, E.L.; Pedersen-Bjergaard, S.; Gjelstad, A. Dried blood spots and parallel artificial liquid membrane extraction-A simple combination of microsampling and microextraction. Anal. Chim. Acta. 2018, 1009, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Garcés, N.; Bojko, B.; Hein, D.; Pawliszyn, J. Solid phase microextraction devices prepared on plastic support as potential single-use samplers for bioanalytical applications. Anal. Chem. 2015, 87, 9722–9730. [Google Scholar] [CrossRef] [PubMed]
- Boyacı, E.; Gorynski, K.; Rodriguez-Lafuente, A.; Bojko, B.; Pawliszyn, J. Introduction of solid-phase microextraction as a high-throughput sample preparation tool in laboratory analysis of prohibited substances. Anal. Chim. Acta 2014, 809, 69–81. [Google Scholar] [CrossRef]
- Bessonneau, V.; Boyaci, E.; Maciazek-Jurczyk, M.; Pawliszyn, J. In vivo solid phase microextraction sampling of human saliva for non-invasive and on-site monitoring. Anal. Chim. Acta 2015, 856, 35–45. [Google Scholar] [CrossRef]
- Vasiljevic, T.; Gómez-Ríos, G.A.; Li, F.; Liang, P.; Pawliszyn, J. High-throughput quantification of drugs of abuse in biofluids via 96-solid-phase microextraction-transmission mode and direct analysis in real time mass spectrometry. Rapid Commun. Mass Spectrom. 2019, 33, 1423–1433. [Google Scholar] [CrossRef]
- Vasiljevic, T.; Gómez-Ríos, G.A.; Pawliszyn, J. Single-Use Poly(etheretherketone) Solid-Phase Microextraction-Transmission Mode Devices for Rapid Screening and Quantitation of Drugs of Abuse in Oral Fluid and Urine via Direct Analysis in Real-Time Tandem Mass Spectrometry. Anal. Chem. 2018, 90, 952–960. [Google Scholar] [CrossRef]
- Płotka-Wasylka, J. A new tool for the evaluation of the analytical procedure: Green Analytical Procedure Index. Talanta 2018, 181, 204–209. [Google Scholar] [CrossRef]
- Pena-Pereira, F.; Wojnowski, W.; Tobiszewski, M. AGREE-Analytical GREEnness Metric Approach and Software. Anal. Chem. 2020, 92, 10076–10082. [Google Scholar] [CrossRef]
- Plotka-Wasylka, J.; Wojnowski, W. Complementary green analytical procedure index (ComplexGAPI) and software. Green Chem. 2021, 23, 8657–8665. [Google Scholar] [CrossRef]
- Wojnowski, W.; Tobiszewski, M.; Pena-Pereira, F.; Psillakis, E. AGREEprep—Analytical greenness metric for sample preparation. TrAC Trends Anal. Chem. 2022, 149, 116553. [Google Scholar] [CrossRef]
- Sobczak, Ł.; Kołodziej, D.; Goryński, K. Modifying current thin-film microextraction (TFME) solutions for analyzing prohibited substances: Evaluating new coatings using liquid chromatography. J. Pharm. Anal. 2022, 12, 470–480. [Google Scholar] [CrossRef] [PubMed]
- Kołodziej, D.; Sobczak, Ł.; Goryński, K. PANDA Microextraction (PolyAmide Non-coated Device for Adsorption-based Microextraction) and Novel 3D Printed TFME (Thin-Film Microextraction) Supports. Anal. Chem. 2022, 94, 2764–2771. [Google Scholar] [CrossRef] [PubMed]
- Bioanalytical Method Validation Guidance for Industry. Food and Drug Administration. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/bioanalytical-method-validation-guidance-industry (accessed on 16 November 2023).
- Mula, J.; Chiara, F.; Manca, A.; Palermiti, A.; Maiese, D.; Cusato, J.; Simiele, M.; Giuseppe De Rosa, F.; Di Perri, G.; De Nicolò, A.; et al. Analytical validation of a novel UHPLC-MS/MS method for 19 antibiotics quantification in plasma: Implementation in a LC-MS/MS Kit. Biomed. Pharmac. 2023, 163, 114790. [Google Scholar] [CrossRef] [PubMed]
- Kahl, K.W.; Seither, J.Z.; Reidy, L.J. LC-MS-MS vs ELISA: Validation of a Comprehensive Urine Toxicology Screen by LC-MS-MS and a Comparison of 100 Forensic Specimens. J. Anal. Toxicol. 2019, 43, 734–745. [Google Scholar] [CrossRef] [PubMed]
- Matuszewski, B.K.; Constanzer, M.L.; Chavez-Eng, C.M. Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC-MS/MS. Anal. Chem. 2003, 75, 3019–3030. [Google Scholar] [CrossRef] [PubMed]
- De Nicolò, A.; Cantù, M.; D’Avolio, A. Matrix effect management in liquid chromatography mass spectrometry: The internal standard normalized matrix effect. Bioanalysis 2017, 9, 1093–1105. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.N.; Pawliszyn, J. Effect of Binding Components in Complex Sample Matrices on Recovery in Direct Immersion Solid-Phase Microextraction: Friends or Foe? Anal. Chem. 2018, 90, 2430–2433. [Google Scholar] [CrossRef]
- Reyes-Garcés, N.; Bojko, B.; Pawliszyn, J. High throughput quantification of prohibited substances in plasma using thin film solid phase microextraction. J. Chromatogr. A 2014, 1374, 40–49. [Google Scholar] [CrossRef]
- Galuszka, A.; Migaszewski, Z.M.; Konieczka, P.; Namiesnik, J. Analytical Eco-Scale for assessing the greenness of analytical procedures. TrAC Trends Anal. Chem. 2012, 37, 61–72. [Google Scholar] [CrossRef]
- Manousi, N.; Wojnowski, W.; Płotka-Wasylka, J.; Samanidou, V. Blue applicability grade index (BAGI) and software: A new tool for the evaluation of method practicality. Green Chem. 2023, 25, 7598–7604. [Google Scholar] [CrossRef]
- Reyes-Garcés, N.; Gionfriddo, E.; Gómez-Ríos, G.A.; Alam, M.N.; Boyacı, E.; Bojko, B.; Singh, V.; Grandy, J.; Pawliszyn, J. Advances in Solid Phase Microextraction and Perspective on Future Directions. Anal. Chem. 2018, 90, 302–360. [Google Scholar] [CrossRef]
- Zhou, W.; Wieczorek, M.N.; Jiang, R.W.; Pawliszyn, J. Comparison of different approaches for direct coupling of solid-phase microextraction to mass spectrometry for drugs of abuse analysis in plasma. J. Pharm. Anal. 2023, 13, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Mirnaghi, F.S.; Chen, Y.; Sidisky, L.M.; Pawliszyn, J. Optimization of the coating procedure for a high-throughput 96-blade solid phase microextraction system coupled with LC-MS/MS for analysis of complex samples. Anal. Chem. 2011, 83, 6018–6025. [Google Scholar] [CrossRef]
- Goryński, K.; Bojko, B.; Kluger, M.; Jerath, A.; Wąsowicz, M.; Pawliszyn, J. Development of SPME method for concomitant sample preparation of rocuronium bromide and tranexamic acid in plasma. J. Pharm. Biomed. Anal. 2014, 92, 183–192. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Matrix | Drugs of Abuse | Linearity Range ng mL−1 | R2 | Slope (m) | Intercept (q) | LOD ng mL−1 | LOQ ng mL−1 | |
|---|---|---|---|---|---|---|---|---|
| Urine | Zolpidem | 3–100 | 0.971 | 62,645 | 28,338 | 11.44 | 1 | 3 |
| Fentanyl | 1–100 | 0.991 | 97,278 | 70,241 | 8.00 | 0.4 | 1 | |
| Methadone | 3–100 | 0.998 | 126,872 | −8901 | 5.44 | 1 | 3 | |
| Plasma | Zolpidem | 3–100 | 0.996 | 19,998 | −17,490 | 12.13 | 1 | 3 |
| Fentanyl | 1–100 | 0.996 | 104,063 | −9263 | 6.67 | 0.4 | 1 | |
| Methadone | 1–100 | 0.998 | 169,015 | −59,173 | 6.78 | 0.4 | 1 | |
| Oral fluid | Zolpidem | 1–100 | 0.995 | 46,848 | −4588 | 9.13 | 0.3 | 1 |
| Fentanyl | 3–100 | 0.995 | 74,614 | −51,954 | 5.01 | 1 | 3 | |
| Methadone | 3–100 | 0.998 | 75,469 | −16,221 | 3.89 | 1 | 3 |
| Matrix | Drugs of Abuse | Matrix Effect (%) | Extraction Efficiency (%) | Precision Intra-Day (%) QC-LOQ | Precision Inter-Day (%) QC-LOQ | Accuracy (%) | Stability | |||
|---|---|---|---|---|---|---|---|---|---|---|
| RT (%) | F/T (%) | FR (%) | AS (%) | |||||||
| Urine | Zolpidem | 97.4 | 42.8 | 7.4 | 6.2 | 89 | 9 | 7 | 11 | 2 |
| Fentanyl | 96.0 | 62.7 | 4.9 | 5.5 | 99 | 10 | 11 | 4 | 3 | |
| Methadone | 96.0 | 76.2 | 4.0 | 8.8 | 104 | 9 | 5 | 3 | 0 | |
| Plasma | Zolpidem | 93.6 | 34.3 | 8.3 | 7.8 | 102 | −13 | −11 | 15 | −9 |
| Fentanyl | 93.3 | 34.5 | 4.3 | 8.1 | 104 | 5 | 4 | 13 | −1 | |
| Methadone | 92.2 | 62.4 | 4.7 | 11.1 | 99 | 5 | 2 | 15 | −2 | |
| Oral fluid | Zolpidem | 85.5 | 26.7 | 9.6 | 11.8 | 103 | −15 | 7 | 7 | 15 |
| Fentanyl | 87.4 | 38.0 | 4.8 | 12.0 | 101 | −5 | 3 | 3 | −9 | |
| Methadone | 87.7 | 36.7 | 2.7 | 12.0 | 102 | −9 | −11 | 9 | −5 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goryński, K.; Sobczak, Ł.; Kołodziej, D. Developing and Evaluating the Greenness of a Reliable, All-in-One Thin-Film Microextraction Protocol for Determining Fentanyl, Methadone, and Zolpidem in Plasma, Urine, and Oral Fluid. Molecules 2024, 29, 335. https://doi.org/10.3390/molecules29020335
Goryński K, Sobczak Ł, Kołodziej D. Developing and Evaluating the Greenness of a Reliable, All-in-One Thin-Film Microextraction Protocol for Determining Fentanyl, Methadone, and Zolpidem in Plasma, Urine, and Oral Fluid. Molecules. 2024; 29(2):335. https://doi.org/10.3390/molecules29020335
Chicago/Turabian StyleGoryński, Krzysztof, Łukasz Sobczak, and Dominika Kołodziej. 2024. "Developing and Evaluating the Greenness of a Reliable, All-in-One Thin-Film Microextraction Protocol for Determining Fentanyl, Methadone, and Zolpidem in Plasma, Urine, and Oral Fluid" Molecules 29, no. 2: 335. https://doi.org/10.3390/molecules29020335
APA StyleGoryński, K., Sobczak, Ł., & Kołodziej, D. (2024). Developing and Evaluating the Greenness of a Reliable, All-in-One Thin-Film Microextraction Protocol for Determining Fentanyl, Methadone, and Zolpidem in Plasma, Urine, and Oral Fluid. Molecules, 29(2), 335. https://doi.org/10.3390/molecules29020335