
High-Density Energetic Materials with Low Mechanical Sensitivity and Twinning Derived from Nitroimidazole Fused Ring

Abstract
1. Introduction
2. Results and Discussion
2.1. Synthesis
2.2. Spectral Analyses
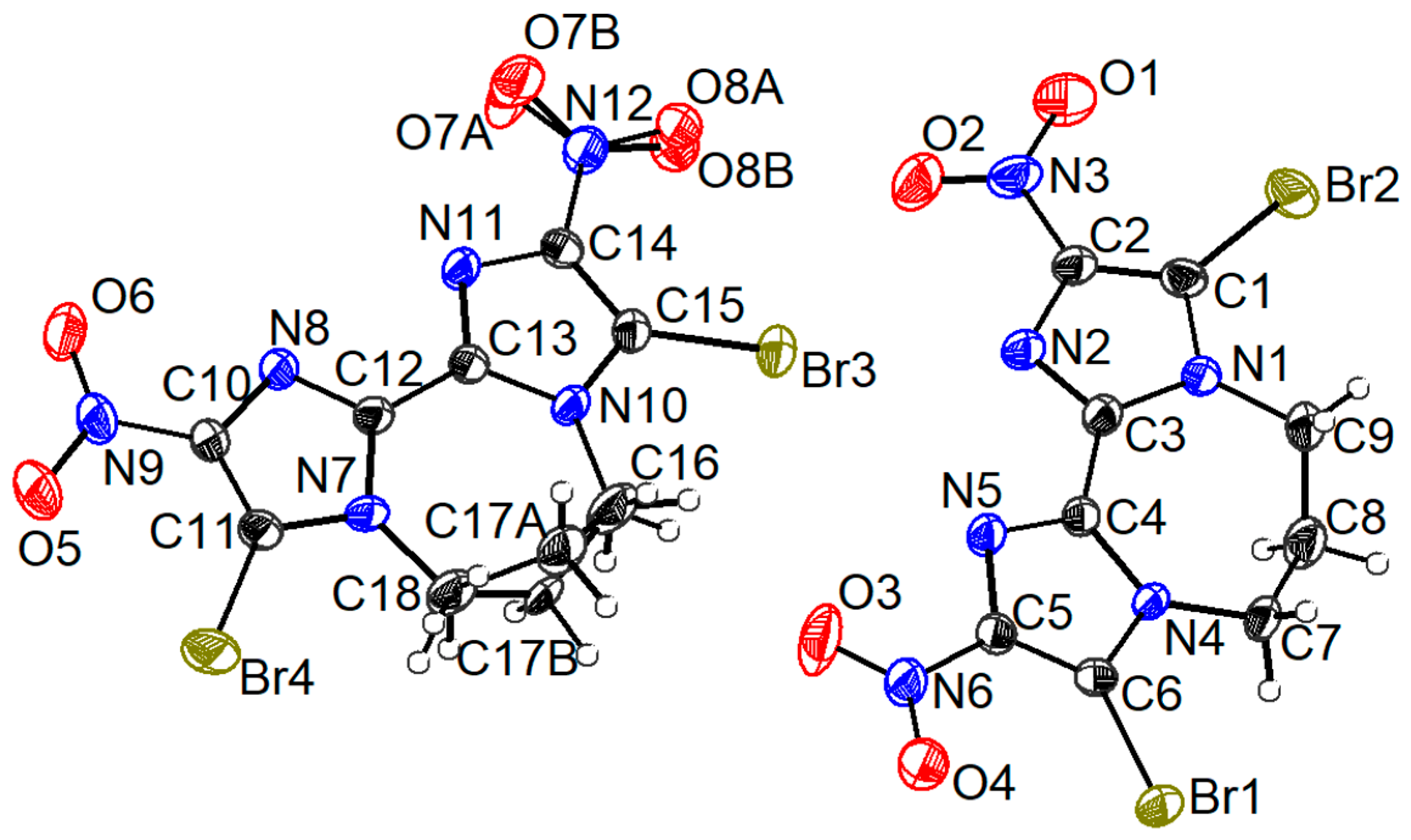
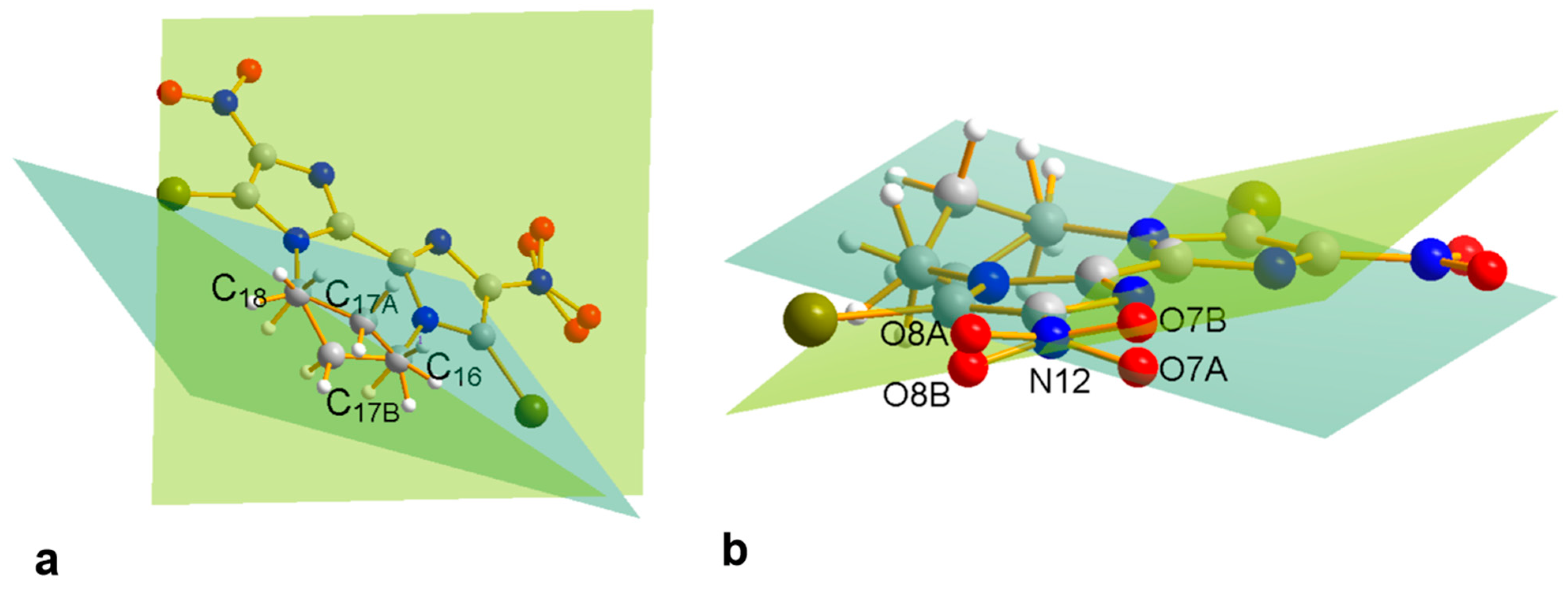
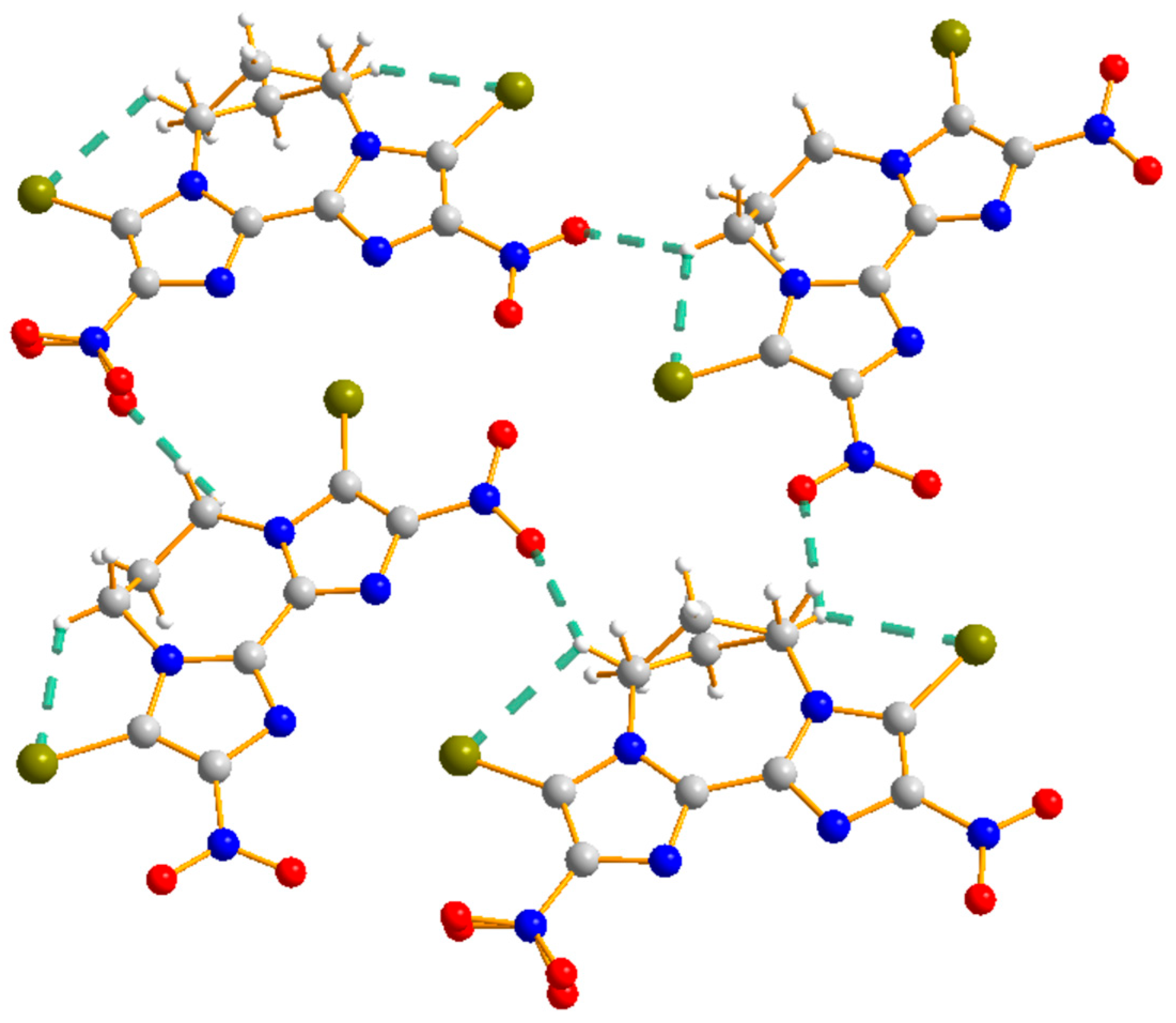
2.3. Crystallographic Analyses
2.4. Analyses of the Electronic Properties
2.4.1. Non-Covalent Interaction Analysis
2.4.2. Hirshfeld Surface Analysis
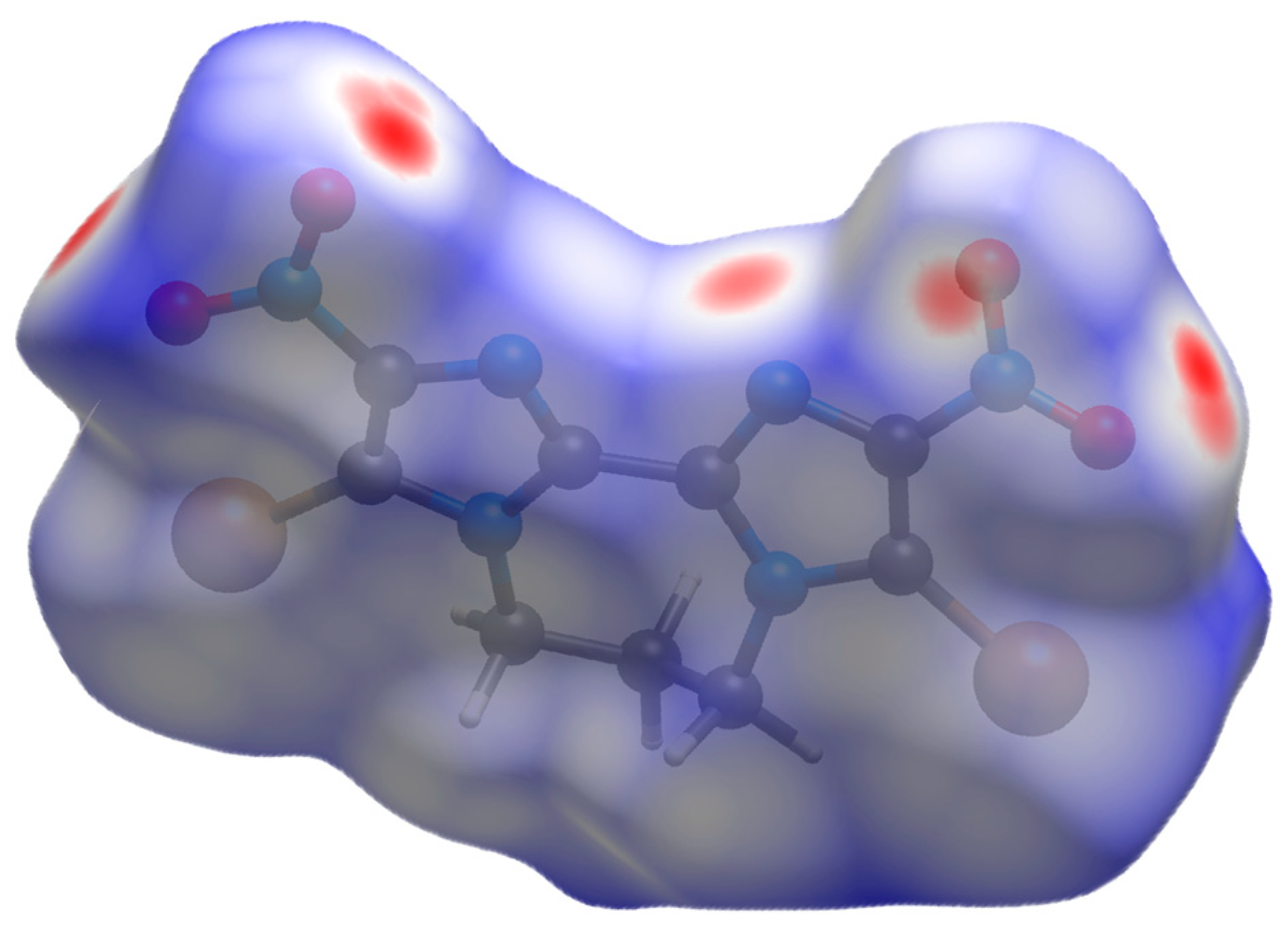
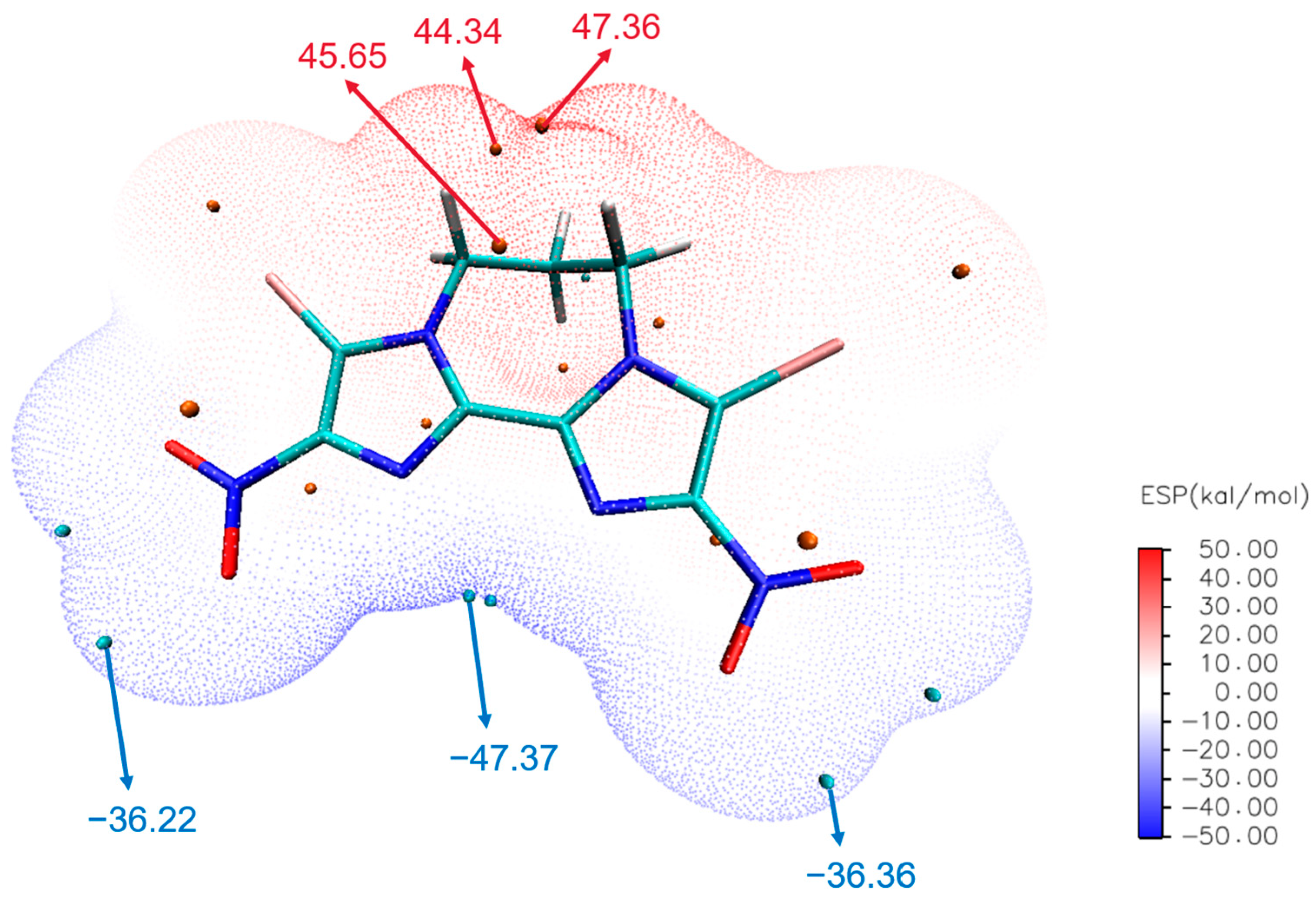
2.4.3. Electrostatic Potential Analysis
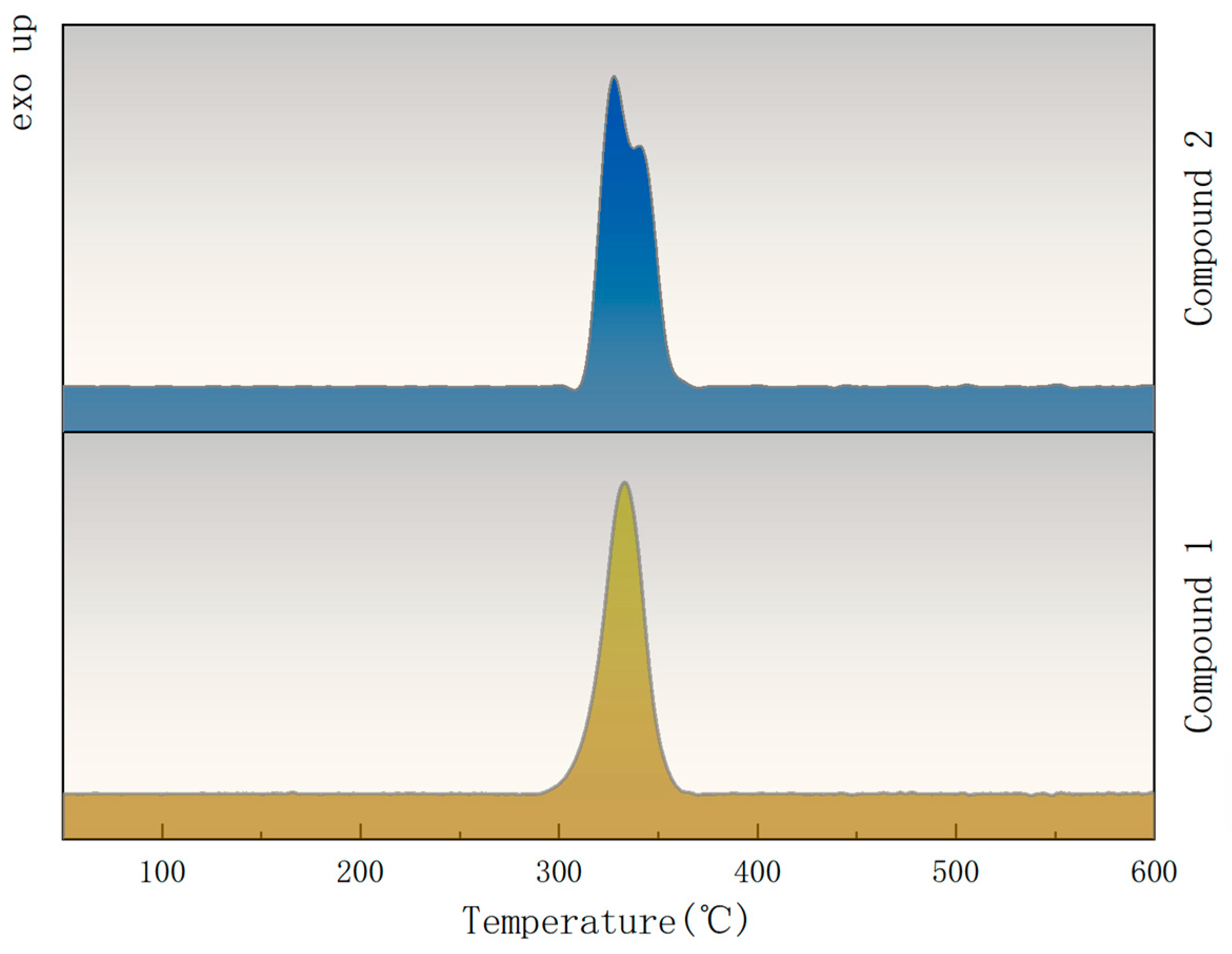
2.5. Physicochemical and Explosive Properties
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Xu, Y.; Shen, C.; Lin, Q.; Wang, P.; Jiang, C.; Lu, M. 1-Nitro-2-trinitromethyl substituted imidazoles: A new family of high performance energetic materials. J. Mater. Chem. A 2016, 4, 17791–17800. [Google Scholar] [CrossRef]
- Zhang, Z.Q.; Qian, W.; Lu, H.; Yang, W.; Zhang, C.; Fan, G.; Ma, Q. Polymorphism in a Nonsensitive-High-Energy Material: Discovery of a New Polymorph and Crystal Structure of 4,4′,5,5′-Tetranitro-1H,1′H-[2,2′-biimidazole]-1,1′-diamine. Cryst. Growth Des. 2020, 20, 8005–8014. [Google Scholar] [CrossRef]
- Gao, H.; Zhang, Q.; Shreeve, J.M. Fused Heterocycle-based Energetic Materials. J. Mater. Chem. A 2020, 8, 4193–4216. [Google Scholar] [CrossRef]
- Yin, P.; He, C.; Shreeve, J.M. Fused Heterocycle-based Energetic Salts: Alliance of Pyrazole and 1,2,3-Triazole. J. Mater. Chem. A 2016, 4, 1514–1519. [Google Scholar] [CrossRef]
- Veljković, I.S.; Radovanović, J.I.; Veljković, D.Ž. How aromatic system size affects the sensitivities of highly energetic molecules? RSC Adv. 2021, 11, 31933–31940. [Google Scholar] [CrossRef] [PubMed]
- Denffer, M.; Klapötke, T.M.; Sabaté, C.M. Hydrates of 5-Amino-1H-tetrazolium Halogenide Salts Starting Materials for the Synthesis of Energetic Compounds. Z. Anorg. Allg. Chem. 2008, 634, 2575–2582. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, Z.; Zhang, H.; Zhang, Z.; Wu, H.; Jin, M.; Wu, C.; Yang, D.; Yin, Y. Lattice-Mismatch-Induced Twinning for Seeded Growth of Anisotropic Nanostructures. ACS Nano 2015, 9, 3307–3313. [Google Scholar] [CrossRef]
- López-Güell, K.; Forrer, N.; Cartoixà, X.; Zardo, I.; Rurali, R. Phonon Transport in GaAs and InAs Twinning Superlattices. J. Phys. Chem. C 2022, 126, 16851–16858. [Google Scholar] [CrossRef]
- Sasaki, T.; Sakamoto, S.; Takamizawa, S. Twinning-Based Organosuperelasticity and Chirality in a Single Crystal of an Achiral Donor–Acceptor Type Schiff Base Induced by Charge-Transfer Interactions. Cryst. Growth Des. 2020, 20, 8079–8083. [Google Scholar] [CrossRef]
- Ouyang, S.; Yang, Y.Q.; Han, M.; Xia, Z.H.; Huang, B.; Luo, X.; Zhao, G.M.; Chen, Y.X. Structure of A-C Type Intervariant Interface in Nonmodulated Martensite in a Ni–Mn–Ga Alloy. ACS Appl. Mater. Interfaces 2016, 8, 16985–16996. [Google Scholar] [CrossRef]
- Dai, Z.R.; Sun, S.; Wang, Z.L. Phase Transformation, Coalescence, and Twinning of Monodisperse FePt Nanocrystals. Nano Lett. 2001, 1, 443–447. [Google Scholar] [CrossRef]
- Lan, Z.P.; Lai, X.; Roberts, K.; Klapper, H. X-ray Topographic and Polarized Optical Microscopy Studies of Inversion Twinning in Sodium Chlorate Single Crystals Grown in the Presence of Sodium Dithionate Impurities. Cryst. Growth Des. 2014, 14, 6084–6092. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Quantitative Analysis of Molecular Surface Based on Improved Marching Tetrahedra Algorithm. J. Mol. Graph. Model. 2012, 38, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lu, T. Efficient Evaluation of Electrostatic Potential with Computerized Optimized Code. Phys. Chem. Chem. Phys. 2021, 23, 20323. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Đunovića, A.B.; Veljković, D.Ž. Halogen Bonds as a Tool in the Design of High Energetic Materials: Evidence from Crystal Structures and Quantum Chemical Calculations. CrystEngComm 2021, 23, 6915–6922. [Google Scholar] [CrossRef]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilakab, D.; Spackman, M.A. CrystalExplorer: A Program for Hirshfeld Surface Analysis, Visualization and Quantitative Analysis of Molecular Crystals. J. Appl. Crystallogr. 2021, 54, 1006–1011. [Google Scholar] [CrossRef]
- Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer 17; University of Western Australia: Perth, Australia, 2017. [Google Scholar]
- Lu, T.; Manzetti, S. Wave Function and Reactivity Study of Benzo[a]Pyrene Diol Epoxide and Its Enantiomeric Forms. Struct. Chem. 2014, 25, 1521–1533. [Google Scholar] [CrossRef]
- Murray, J.S.; Concha, M.C.; Politzer, P. Links between Surface Electrostatic Potentials of Energetic Molecules, Impact Sensitivities and C–NO2/N–NO2 Bond Dissociation Energies. Mol. Phys. 2009, 107, 89–97. [Google Scholar] [CrossRef]
- Zhang, J.H.; Shreeve, J.M. Nitroaminofurazans with Azo and Azoxy Linkages: A Comparative Study of Structural, Electronic, Physicochemical, and Energetic Properties. J. Phys. Chem. C 2015, 119, 12887–12895. [Google Scholar] [CrossRef]
- Klapötke, T.M. Energetic Materials Encyclopedia, 1st ed.; Walter De Gruyter GmbH: Berlin, Germany, 2021; pp. (DADP) 359–363, (TNT) 1894–1986. [Google Scholar]
- Muravyev, N.V.; Meerov, D.B.; Monogarov, K.A.; Melnikov, I.N.; Kosareva, E.K.; Fershtat, L.L.; Sheremetev, A.B.; Dalinger, I.L.; Fomenkov, I.V.; Pivkina, A.N. Sensitivity of Energetic Materials: Evidence of Thermodynamic Factor on a Large Array of CHNOFCl Compounds. Chem. Eng. J. 2021, 421, 129804. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.1; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Suceska, M. EXPLO5 6.01; Brodarski Institute: Zagreb, Croatia, 2013. [Google Scholar]
- Perera, M.D.; Sinha, A.S.; Aakeröy, C.B. Enhancing Chemical Stability of Tetranitro Biimidazole-based Energetic Materials through Co-crystallization. Can. J. Chem. 2020, 98, 358–364. [Google Scholar] [CrossRef]
- Bruker. APEX3 v2019.1; Bruker Nano, Inc.: Madison, WI, USA, 2019. [Google Scholar]
- Sheldrick, G.M. SHELXT—Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. The Influence of Polarization Functions on Molecular Orbital Hydrogenation Energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Ochterski, J.W.; Petersson, G.A.; Montgomery, J.A. A Complete Basis Set Model Chemistry. V. Extensions to Six or More Heavy Atoms. J. Chem. Phys. 1996, 104, 2598–2619. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 2 |
|---|---|
| Temperature (K) | 296 |
| Crystal system | Monoclinic |
| Space group | P21/c |
| Unit cell dimensions a orientation | 14.4361 (11) |
| b orientation | 12.6417 (9) |
| c orientation | 13.9228 (10) |
| α orientation | 90 |
| β orientation | 95.278 (2) |
| γ orientation | 90 |
| Volume | 2530.1 (3) |
| Z | 8 |
| Density | 2.216 |
| Max. transmission | 0.7456 |
| Min. transmission | 0.4363 |
| Goodness-of-fit on F2 | 1.033 |
| Final R indices (I > 2sigma (I)) | R1 = 0.0321 |
| wR2 = 0.0762 | |
| R indices (all data) | R1 = 0.0517 |
| wR2 = 0.0827 | |
| CCDC Number | 2295688 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Lv, M.; Zhang, G.; Dong, Z.; Ye, Z. High-Density Energetic Materials with Low Mechanical Sensitivity and Twinning Derived from Nitroimidazole Fused Ring. Molecules 2024, 29, 353. https://doi.org/10.3390/molecules29020353
Liu Y, Lv M, Zhang G, Dong Z, Ye Z. High-Density Energetic Materials with Low Mechanical Sensitivity and Twinning Derived from Nitroimidazole Fused Ring. Molecules. 2024; 29(2):353. https://doi.org/10.3390/molecules29020353
Chicago/Turabian StyleLiu, Yaxin, Meifang Lv, Guofeng Zhang, Zhen Dong, and Zhiwen Ye. 2024. "High-Density Energetic Materials with Low Mechanical Sensitivity and Twinning Derived from Nitroimidazole Fused Ring" Molecules 29, no. 2: 353. https://doi.org/10.3390/molecules29020353
APA StyleLiu, Y., Lv, M., Zhang, G., Dong, Z., & Ye, Z. (2024). High-Density Energetic Materials with Low Mechanical Sensitivity and Twinning Derived from Nitroimidazole Fused Ring. Molecules, 29(2), 353. https://doi.org/10.3390/molecules29020353