
Identification of GM1-Ganglioside Secondary Accumulation in Fibroblasts from Neuropathic Gaucher Patients and Effect of a Trivalent Trihydroxypiperidine Iminosugar Compound on Its Storage Reduction
 , ,
, ,  , , , , , , , and
, , , , , , , and
Abstract
:1. Introduction
2. Results
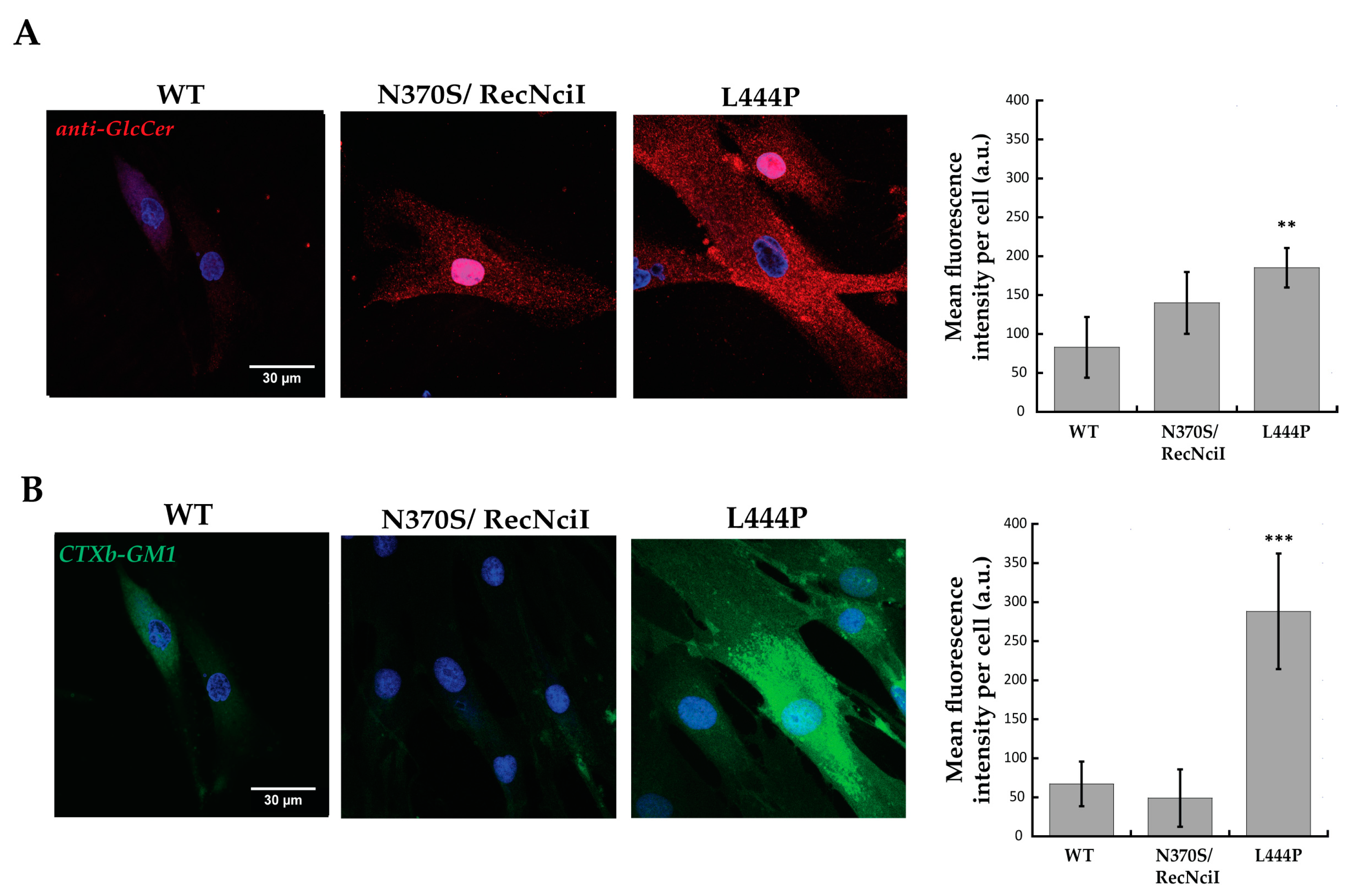
2.1. Increased GlcCer and GM1 Contents in L444P Fibroblasts Highlighted by Confocal-Microscopy and Flow-Cytometry Analyses
2.2. Decreased Accumulation of GM1 in L444P Fibroblasts Treated with CV82
2.3. Unchanged β-Gal Enzyme Activity in WT and Mutated Fibroblasts from Juvenile GM1 Patients Treated with CV82
2.4. Increased Accumulation of GlcCer and GM1 in R131C Fibroblasts Treated with CV82
3. Discussion
4. Materials and Methods
4.1. Cell Cultures
4.2. Inhibitory Activity towards Human Lysosomal β-Galactosidase (β-Gal)
4.3. Pharmacological Chaperoning Activity
4.4. Confocal-Microscopy Analysis
4.5. Flow-Cytometry Analysis
4.6. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Castillon, G.; Chang, S.-C.; Moride, Y. Global Incidence and Prevalence of Gaucher Disease: A Targeted Literature Review. J. Clin. Med. 2022, 12, 85. [Google Scholar] [CrossRef]
- Wang, M.; Li, F.; Zhang, J.; Lu, C.; Kong, W. Global Epidemiology of Gaucher Disease: An Updated Systematic Review and Meta-Analysis. J. Pediatr. Hematol./Oncol. 2023, 45, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Dvir, H.; Harel, M.; McCarthy, A.A.; Toker, L.; Silman, I.; Futerman, A.H.; Sussman, J.L. X-ray Structure of Human Acid-Beta-Glucosidase, the Defective Enzyme in Gaucher Disease. EMBO Rep. 2003, 4, 704–709. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, G.A. Phenotype, Diagnosis, and Treatment of Gaucher’s Disease. Lancet 2008, 372, 1263–1271. [Google Scholar] [CrossRef]
- Zimran, A.; Elstein, D. Management of Gaucher Disease: Enzyme Replacement Therapy. Pediatr. Endocrinol. Rev. 2014, 12 (Suppl. S1), 82–87. [Google Scholar]
- Platt, F.M.; Jeyakumar, M.; Andersson, U.; Priestman, D.A.; Dwek, R.A.; Butters, T.D.; Cox, T.M.; Lachmann, R.H.; Hollak, C.; Aerts, J.M.; et al. Inhibition of Substrate Synthesis as a Strategy for Glycolipid Lysosomal Storage Disease Therapy. J. Inherit. Metab. Dis. 2001, 24, 275–290. [Google Scholar] [CrossRef]
- Weinreb, N.J.; Goker-Alpan, O.; Kishnani, P.S.; Longo, N.; Burrow, T.A.; Bernat, J.A.; Gupta, P.; Henderson, N.; Pedro, H.; Prada, C.E.; et al. The Diagnosis and Management of Gaucher Disease in Pediatric Patients: Where Do We Go from Here? Mol. Genet. Metab. 2022, 136, 4–21. [Google Scholar] [CrossRef]
- Boyd, R.E.; Lee, G.; Rybczynski, P.; Benjamin, E.R.; Khanna, R.; Wustman, B.A.; Valenzano, K.J. Pharmacological Chaperones as Therapeutics for Lysosomal Storage Diseases. J. Med. Chem. 2013, 56, 2705–2725. [Google Scholar] [CrossRef] [PubMed]
- Pereira, D.M.; Valentão, P.; Andrade, P.B. Tuning Protein Folding in Lysosomal Storage Diseases: The Chemistry behind Pharmacological Chaperones. Chem. Sci. 2018, 9, 1740–1752. [Google Scholar] [CrossRef]
- Prichard, K.; Campkin, D.; O’Brien, N.; Kato, A.; Fleet, G.W.J.; Simone, M.I. Biological Activities of 3,4,5-trihydroxypiperidines and Their N- and O-derivatives. Chem. Biol. Drug Des. 2018, 92, 1171–1197. [Google Scholar] [CrossRef]
- Sánchez-Fernández, E.M.; García Fernández, J.M.; Mellet, C.O. Glycomimetic-Based Pharmacological Chaperones for Lysosomal Storage Disorders: Lessons from Gaucher, GM1-Gangliosidosis and Fabry Diseases. Chem. Commun. 2016, 52, 5497–5515. [Google Scholar] [CrossRef]
- Martínez-Bailén, M.; Clemente, F.; Matassini, C.; Cardona, F. GCase Enhancers: A Potential Therapeutic Option for Gaucher Disease and Other Neurological Disorders. Pharmaceuticals 2022, 15, 823. [Google Scholar] [CrossRef] [PubMed]
- McCafferty, E.H.; Scott, L.J. Migalastat: A Review in Fabry Disease. Drugs 2019, 79, 543–554. [Google Scholar] [CrossRef]
- Nilsson, O.; Svennerholm, L. Accumulation of Glucosylceramide and Glucosylsphingosine (Psychosine) in Cerebrum and Cerebellum in Infantile and Juvenile Gaucher Disease. J. Neurochem. 1982, 39, 709–718. [Google Scholar] [CrossRef]
- Gornati, R.; Berra, B.; Montorfano, G.; Martini, C.; Ciana, G.; Ferrari, P.; Romano, M.; Bembi, B. Glycolipid Analysis of Different Tissues and Cerebrospinal Fluid in Type II Gaucher Disease. J. Inherit. Metab. Dis. 2002, 25, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Fuller, M.; Rozaklis, T.; Lovejoy, M.; Zarrinkalam, K.; Hopwood, J.J.; Meikle, P.J. Glucosylceramide Accumulation Is Not Confined to the Lysosome in Fibroblasts from Patients with Gaucher Disease. Mol. Genet. Metab. 2008, 93, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Vanni, C.; Clemente, F.; Paoli, P.; Morrone, A.; Matassini, C.; Goti, A.; Cardona, F. 3,4,5-Trihydroxypiperidine Based Multivalent Glucocerebrosidase (GCase) Enhancers. ChemBioChem 2022, 23, e202200077. [Google Scholar] [CrossRef]
- Migdalska-Richards, A.; Schapira, A.H.V. The Relationship between Glucocerebrosidase Mutations and Parkinson Disease. J. Neurochem. 2016, 139 (Suppl. S1), 77–90. [Google Scholar] [CrossRef]
- Turnbull, W.B.; Precious, B.L.; Homans, S.W. Dissecting the Cholera Toxin−Ganglioside GM1 Interaction by Isothermal Titration Calorimetry. J. Am. Chem. Soc. 2004, 126, 1047–1054. [Google Scholar] [CrossRef]
- Tonin, R.; Caciotti, A.; Procopio, E.; Fischetto, R.; Deodato, F.; Mancardi, M.M.; Di Rocco, M.; Ardissone, A.; Salviati, A.; Marangi, A.; et al. Pre-Diagnosing and Managing Patients with GM1 Gangliosidosis and Related Disorders by the Evaluation of GM1 Ganglioside Content. Sci. Rep. 2019, 9, 17684. [Google Scholar] [CrossRef]
- Capitini, C.; Feo, F.; Caciotti, A.; Tonin, R.; Lulli, M.; Coviello, D.; Guerrini, R.; Calamai, M.; Morrone, A. Fluorescent In Situ Staining and Flow Cytometric Procedures as New Pre-Diagnostic Tests for Sialidosis, GM1 Gangliosidosis and Niemann–Pick Type C. Biomedicines 2022, 10, 1962. [Google Scholar] [CrossRef] [PubMed]
- Clemente, F.; Martínez-Bailén, M.; Matassini, C.; Morrone, A.; Falliano, S.; Caciotti, A.; Paoli, P.; Goti, A.; Cardona, F. Synthesis of a New β-Galactosidase Inhibitor Displaying Pharmacological Chaperone Properties for GM1 Gangliosidosis. Molecules 2022, 27, 4008. [Google Scholar] [CrossRef] [PubMed]
- Goebl, A.; Ferrier, R.A.; Ferreira, P.; Pinto-Rojas, A.; Matshes, E.; Choy, F.Y.M. Gaucher Disease with Prenatal Onset and Perinatal Death Due to Compound Heterozygosity for the Missense R131C and Null Rec Nci I GBA Mutations. Pediatr. Dev. Pathol. 2011, 14, 240–243. [Google Scholar] [CrossRef]
- Bendikov-Bar, I.; Horowitz, M. Gaucher Disease Paradigm: From ERAD to Comorbidity. Hum. Mutat. 2012, 33, 1398–1407. [Google Scholar] [CrossRef] [PubMed]
- Reczek, D.; Schwake, M.; Schröder, J.; Hughes, H.; Blanz, J.; Jin, X.; Brondyk, W.; Van Patten, S.; Edmunds, T.; Saftig, P. LIMP-2 Is a Receptor for Lysosomal Mannose-6-Phosphate-Independent Targeting of β-Glucocerebrosidase. Cell 2007, 131, 770–783. [Google Scholar] [CrossRef]
- Tsuji, S.; Choudary, P.V.; Martin, B.M.; Stubblefield, B.K.; Mayor, J.A.; Barranger, J.A.; Ginns, E.I. A Mutation in the Human Glucocerebrosidase Gene in Neuronopathic Gaucher’s Disease. N. Engl. J. Med. 1987, 316, 570–575. [Google Scholar] [CrossRef]
- Theophilus, B.; Latham, T.; Grabowski, G.A.; Smith, F.I. Gaucher Disease: Molecular Heterogeneity and Phenotype-Genotype Correlations. Am. J. Hum. Genet. 1989, 45, 212–225. [Google Scholar]
- Sinclair, G.; Choy, F.Y.; Humphries, L. A Novel Complex Allele and Two New Point Mutations in Type 2 (Acute Neuronopathic) Gaucher Disease. Blood Cells Mol. Dis. 1998, 24, 420–427. [Google Scholar] [CrossRef]
- Babajani, G.; Tropak, M.B.; Mahuran, D.J.; Kermode, A.R. Pharmacological Chaperones Facilitate the Post-ER Transport of Recombinant N370S Mutant β-Glucocerebrosidase in Plant Cells: Evidence That N370S Is a Folding Mutant. Mol. Genet. Metab. 2012, 106, 323–329. [Google Scholar] [CrossRef]
- Flores-Leon, M.; Outeiro, T.F. More than Meets the Eye in Parkinson’s Disease and Other Synucleinopathies: From Proteinopathy to Lipidopathy. Acta Neuropathol. 2023, 146, 369–385. [Google Scholar] [CrossRef]
- Hahn, C.N.; del Pilar Martin, M.; Schröder, M.; Vanier, M.T.; Hara, Y.; Suzuki, K.; Suzuki, K.; d’Azzo, A. Generalized CNS Disease and Massive GM1-Ganglioside Accumulation in Mice Defective in Lysosomal Acid Beta-Galactosidase. Hum. Mol. Genet. 1997, 6, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Nicoli, E.-R.; Annunziata, I.; d’Azzo, A.; Platt, F.M.; Tifft, C.J.; Stepien, K.M. GM1 Gangliosidosis-A Mini-Review. Front. Genet. 2021, 12, 734878. [Google Scholar] [CrossRef] [PubMed]
- Breiden, B.; Sandhoff, K. Mechanism of Secondary Ganglioside and Lipid Accumulation in Lysosomal Disease. Int. J. Mol. Sci. 2020, 21, 2566. [Google Scholar] [CrossRef] [PubMed]
- Walkley, S.U.; Vanier, M.T. Secondary Lipid Accumulation in Lysosomal Disease. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2009, 1793, 726–736. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GD-Derived Fibroblasts | Mutated Gene | Nucleotide Changes | Amino Acid Changes | β-Glucocerebrosidase Activity in Fibroblast (n.v.: 120–400 nmol/mg/h) |
|---|---|---|---|---|
| N370S | GBA NM_000157.1 | c.1226A>G/ RecNcil | p.N370S/ RecNcil | 44.4 |
| L444P | GBA NM_000157.1 | c.1448T>C/ c.1448T>C | p.L444P/ p.L444P | 11.8 |
| R131C | GBA NM_000157.1 | c.508C>T/ c.508C>T | p.R131C/ p.R131C | 1.7 |
 | GCase Inhibition (%) (a) | IC50 (µM) (a) | Mutated GCase Activity Rescue (a) | |
| N370S/RecNcil | L444P/L444P | |||
| 100 | 7 ± 1 competitive inhibition (Ki = 3.1 ± 0.2 μM) | 1.21 at 10 μM | 1.07 at 10 μM | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ceni, C.; Clemente, F.; Mangiavacchi, F.; Matassini, C.; Tonin, R.; Caciotti, A.; Feo, F.; Coviello, D.; Morrone, A.; Cardona, F.; et al. Identification of GM1-Ganglioside Secondary Accumulation in Fibroblasts from Neuropathic Gaucher Patients and Effect of a Trivalent Trihydroxypiperidine Iminosugar Compound on Its Storage Reduction. Molecules 2024, 29, 453. https://doi.org/10.3390/molecules29020453
Ceni C, Clemente F, Mangiavacchi F, Matassini C, Tonin R, Caciotti A, Feo F, Coviello D, Morrone A, Cardona F, et al. Identification of GM1-Ganglioside Secondary Accumulation in Fibroblasts from Neuropathic Gaucher Patients and Effect of a Trivalent Trihydroxypiperidine Iminosugar Compound on Its Storage Reduction. Molecules. 2024; 29(2):453. https://doi.org/10.3390/molecules29020453
Chicago/Turabian StyleCeni, Costanza, Francesca Clemente, Francesca Mangiavacchi, Camilla Matassini, Rodolfo Tonin, Anna Caciotti, Federica Feo, Domenico Coviello, Amelia Morrone, Francesca Cardona, and et al. 2024. "Identification of GM1-Ganglioside Secondary Accumulation in Fibroblasts from Neuropathic Gaucher Patients and Effect of a Trivalent Trihydroxypiperidine Iminosugar Compound on Its Storage Reduction" Molecules 29, no. 2: 453. https://doi.org/10.3390/molecules29020453