

Effects of Chalcogen Atoms on Excited-State Double-Proton Transfer Behavior for 3,6-bis(4,5-Dihydroxyoxazo-2-yl)benzene-1,2-diol Derivatives: A Computational Investigation

Abstract
1. Introduction
2. Results and Discussion
2.1. Geometrical Analyses
2.2. Hydrogen Bonding Strength
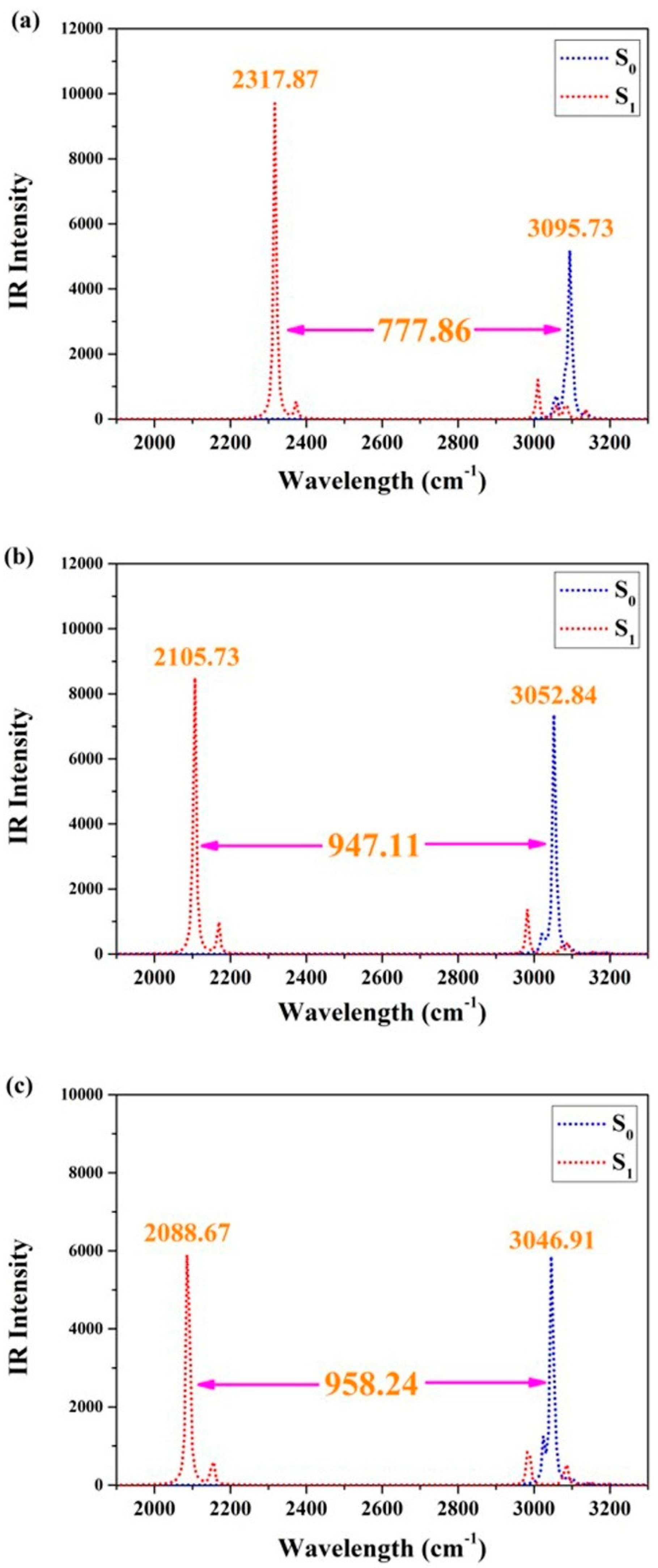
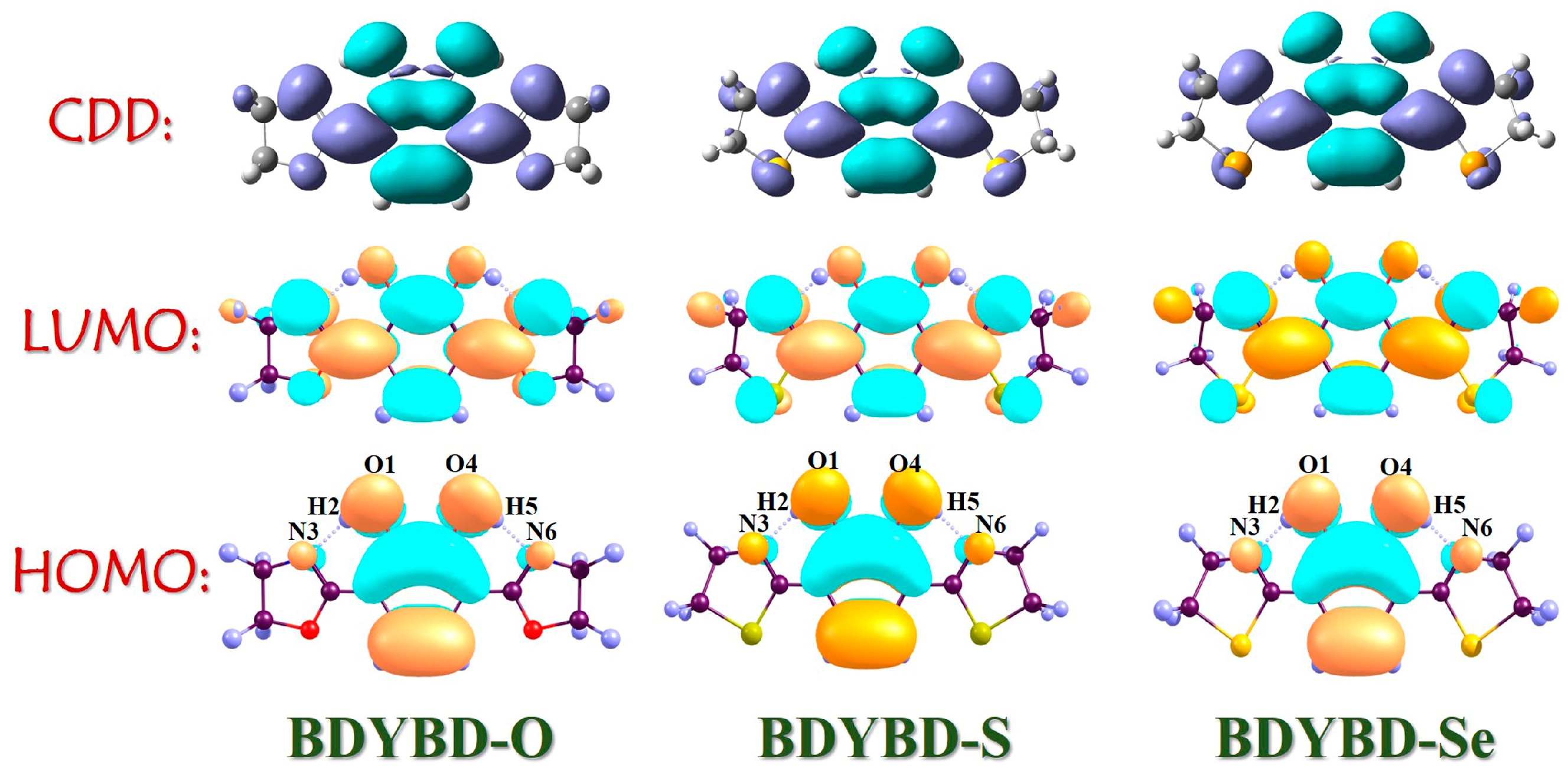
2.3. Vertical Excitation Properties
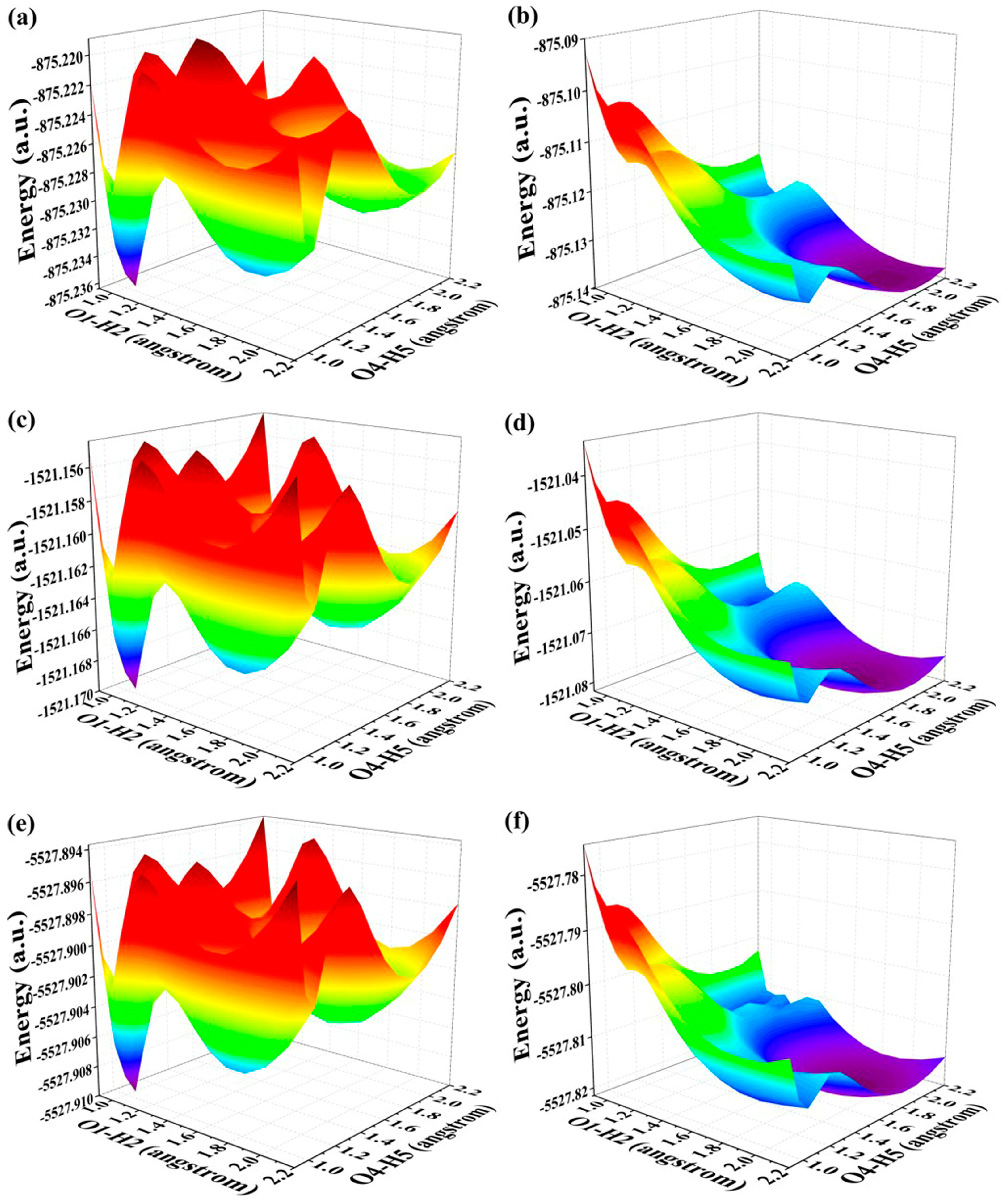
2.4. Mechanism Exploration
3. Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Desiraju, G. Hydrogen bridges in crystal engineering: Interactions without borders. Acc. Chem. Res. 2002, 35, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Rozas, I. On the nature of hydrogen bonds: An overview on computational studies and a word about patterns. Phys. Chem. Chem. Phys. 2007, 9, 2782–2790. [Google Scholar] [CrossRef]
- Wu, Y.; Houk, K.; Valentine, J.; Nam, W. Is intramolecular hydrogen-bonding important for bleomycin reactivity—A molecular mechanics study. Inorg. Chem. 1992, 31, 718–720. [Google Scholar] [CrossRef]
- Yang, Y.; Dou, D. Triply and quadruply hydrogen bonded systems: Design, structure and application. Prog. Chem. 2014, 26, 706–726. [Google Scholar]
- Hay, B.; Gutowski, M.; Dixon, D.; Garza, J.; Vargas, R.; Moyer, B. Structural criteria for the rational design of selective ligands: Convergent hydrogen bonding sites for the nitrate anion. J. Am. Chem. Soc. 2004, 126, 7925–7934. [Google Scholar] [CrossRef] [PubMed]
- Raymo, F.; Bartberger, M.; Houk, K.; Stoddart, J. The magnitude of [C-H···O] hydrogen bonding in molecular and supramolecular assemblies. J. Am. Chem. Soc. 2001, 123, 9264–9267. [Google Scholar] [CrossRef] [PubMed]
- Bradley, J.; Huib, J. On the role of water in intermolecular proton-transfer reactions. J. Am. Chem. Soc. 2007, 129, 13412–13420. [Google Scholar]
- Liu, Y.; Chu, T. Size effect of water cluster on the excited-state proton transfer in aqueous solvent. Chem. Phys. Lett. 2011, 505, 117–121. [Google Scholar] [CrossRef]
- Huynh, M.; Meyer, T. Proton-coupled electron transfer. Chem. Rev. 2007, 107, 5004–5064. [Google Scholar] [CrossRef] [PubMed]
- Hammes-Schiffer, S. Current theoretical challenges in proton-coupled electron transfer: Electron-proton nanadiabaticity, proton relays, and ultrafast dynamics. J. Phys. Chem. Lett. 2011, 2, 1410–1416. [Google Scholar] [CrossRef]
- Hamlin, T.A.; Poater, J.; Guerra, C.F.; Bickelhaupt, F.M. B-DNA model systems in non-terran bio-solvents: Implications for structure, stability and replication. Phys. Chem. Chem. Phys. 2017, 19, 16969–16978. [Google Scholar] [CrossRef] [PubMed]
- Poater, J.; Swart, M.; Guerra, C.F.; Bickelhaupt, F.M. Selectivity in DNA replication. Interplay of steric shape, hydrogen bonds, π-stacking and solvent effects. Chem. Commun. 2011, 47, 7326–7328. [Google Scholar] [CrossRef]
- Weller, A. Uber die fluoreszenz der salizylaure und verandter vibindungen. Naturwissenschafter 1955, 42, 175–176. [Google Scholar] [CrossRef]
- Kim, S.; Seo, J.; Jung, H.; Kim, J.; Park, S. White luminescence from polymer thin films containing excited-state intramolecular proton-transfer dyes. Adv. Mater. 2005, 17, 2077. [Google Scholar] [CrossRef]
- Shang, C.; Sun, C. Substituent effects on photophysical properties of ESIPT-based fluorophores bearing the 4-diethylaminosalicylaldehyde core. J. Mol. Liq. 2022, 367, 120477. [Google Scholar] [CrossRef]
- Guerra, C.F.; van der Wijst, T.; Poater, J.; Swart, M.; Bickelhaupt, F.M. Adenine versus guanine quartets in aqueous solution: Dispersion-corrected DFT study on the differences in π-stacking and hydrogen-bonding behavior. Theor. Chem. Acc. 2010, 125, 245–252. [Google Scholar] [CrossRef]
- Tomin, V.; Demchenko, A.; Chou, P. Thermodynamic vs. kinetic control of excited-state proton transfer reactions. J. Photochem. Photobiol. C 2015, 22, 1–18. [Google Scholar] [CrossRef]
- Sun, C.; Zhao, H.; Liu, X.; Yin, H.; Shi, Y. Tunable ESIPT reaction and antioxidant activities of 3-hydroxyflavone and its derivatives by altering atomic electronegativity. Org. Chem. Front. 2018, 5, 3435–3442. [Google Scholar] [CrossRef]
- Demchenko, A.; Tang, K.; Chou, P. Excited-state proton coupled charge transfer modulated by molecular structure and media polarization. Chem. Soc. Rev. 2013, 42, 1379–1408. [Google Scholar] [CrossRef]
- Zhou, P.; Han, K. Unraveling the detailed mechanism of excited-state proton transfer. Acc. Chem. Res. 2018, 51, 1681–1690. [Google Scholar] [CrossRef] [PubMed]
- Poater, J.; Swart, M.; Bickelhaupt, F.M.; Guerra, C.F. B-DNA structure and stability: The role of hydrogen bonding, π-π stacking interactions, twist-angle, and solvation. Org. Biomol. Chem. 2014, 12, 4691–4700. [Google Scholar] [CrossRef]
- Durko-Maciag, M.; Ulrich, G.; Jacquemin, D.; Myliwiec, J.; Massue, J. Solid-state emitters presenting a modular excited-state proton transfer (ESIPT) process: Recent advances in dual-state emission and lasing applications. Phys. Chem. Chem. Phys. 2023, 25, 15085–15098. [Google Scholar] [CrossRef]
- Mohan, M.; Satyanarayan, M.; Trivedi, D. Photophysics of proton transfer in hydrazides: A combined theoretical and experimental analysis towards OLED device application. New J. Chem. 2019, 43, 10413. [Google Scholar] [CrossRef]
- Almacellas, D.; Guerra, C.F.; Poater, J. Strengthened cooperativity of DNA-based cyclic hydrogen-bonded rosettes by subtle functionalization. Org. Biomol. Chem. 2023, 21, 8403–8412. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Han, K. ESIPT-based AIE luminogens: Design strategies, applications, and mechanisms. Aggregate 2022, 3, 160. [Google Scholar] [CrossRef]
- Zhao, J.; Liu, C. Computational insights into excited state intramolecular double proton transfer behavior associated with atomic electronegativity for bis(2’-benzothiazolyl)hydroquinone. Molecules 2023, 28, 5951. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, H.; Fan, L.; Li, F.; Song, P. Unveiling and regulating the solvent-polarity-associated excited state intramolecular double proton transfer for 1-bis(benzothiazolyl)naphthalene-diol fluorophore. Spectrochim. Acta Part A 2023, 299, 122831. [Google Scholar] [CrossRef]
- Peng, C.; Shen, J.; Chen, Y.; Wu, P.; Hung, W.; Hu, W.; Chou, P. Optically triggered stepwise double-proton transfer in an intramolecular proton relay: A case study of 1,8-dihydroxy-2-naphthaldehyde. J. Am. Chem. Soc. 2015, 137, 14349–14357. [Google Scholar] [CrossRef]
- Hatakeyama, T.; Shiren, K.; Nakajima, K.; Nomura, S.; Nakatsuka, S.; Kinoshita, K.; Ni, J.; Ono, Y.; Ikuta, T. Ultrapure blue thermally activated delayed fluorescence molecules: Efficient HOMO-LUMO separation by the multiple resonance effect. Adv. Mater. 2016, 28, 2777–2781. [Google Scholar] [CrossRef]
- Matsui, K.; Oda, S.; Yoshiura, K.; Nakajima, K.; Yasuda, N.; Hatakeyama, T. One-shot multiple borylation toward BN-doped nanographenes. J. Am. Chem. Soc. 2018, 140, 1195–1198. [Google Scholar] [CrossRef]
- Song, L.; Meng, X.; Zhao, J.; Han, H.; Zheng, D. Effects of azole rings with different chalcogen atoms on ESIPT behavior for benzochalcogenazolyl-substituted hydroxyfluorenes. Spectrochim. Acta Part A 2022, 264, 120296. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Zhan, L.; Cao, X.; Lv, X.; Gong, S.; Chen, Z.; Zhou, C.; Huang, Z.; Ni, F.; Zou, Y.; et al. Simple acridan-based multi-resonance structures enable highly efficient narrowband green TADF electroluminescence. Adv. Opt. Mater. 2021, 9, 2100825. [Google Scholar] [CrossRef]
- Meng, F.; Chen, Y.; Chen, C.; Chou, P. Syntheses and excited-state intramolecular proton transfer of 3-hydroxythioflavone and its sulfone analogue. ChemPhotoChem 2018, 2, 475–480. [Google Scholar] [CrossRef]
- Enchev, V.; Markova, N.; Stoyanova, M.; Petrov, P.; Rogozherov, M.; Kuchukova, N.; Timtcheva, I.; Monev, V.; Angelova, S.; Spassova, M. Excited state proton transfer in 3,6-bis(4,5-dihydroxyoxazo-2-yl)benzene-1,2-diol. Chem. Phys. Lett. 2013, 563, 43–49. [Google Scholar] [CrossRef]
- Orlando, C.; Wirth, J.; Heath, D. Red-luminescent and near-infrared-luminescent benzazole derivatives. Chem. Commun. 1971, 23, 1551. [Google Scholar] [CrossRef]
- Zhao, G.; Han, K. Time-dependent density functional theory study on hydrogen-bonded intramolecular charge-transfer excited state of 4-dimethylamino-benzonitrile in methanol. J. Comput. Chem. 2008, 29, 2010–2017. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, Y.; Jiang, K.; Shi, D.; Sun, J. Excited-state N-H···S hydrogen bond between indole and dimethyl sulfide: Time-dependent density functional theory study. Phys. Chem. Chem. Phys. 2011, 13, 15299–15304. [Google Scholar] [CrossRef]
- Zhao, G.; Han, K. Site-specific solvation of the photoexcited protochlorophyllide a in methanol: Formation of the hydrogen-bonded intermediate state induced by hydrogen-bond strengthening. Biophys. J. 2008, 94, 38–46. [Google Scholar] [CrossRef]
- Zhao, J.; Chen, J.; Song, P.; Liu, J.; Ma, F. The charge transfer phenomenon in benzene-pyrene-sulfoxide/methanol system: Role of the intermolecular hydrogen bond in excited states. J. Clust. Sci. 2015, 26, 1463–1472. [Google Scholar] [CrossRef]
- Zhao, G.; Han, K. Hydrogen bonding in the electronic excited state. Acc. Chem. Res. 2012, 45, 404–413. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.; Essen, H. The characterization of atomic interactions. J. Chem. Phys. 1984, 80, 1943–1960. [Google Scholar] [CrossRef]
- Emamian, S.; Lu, T.; Kruse, H.; Emamian, H. Exploring nature and predicting strength of hydrogen bonds: A correlation analysis between atoms-in-molecules descriptors, binding energies, and energy components of symmetry-adapted perturbation theory. J. Comput. Chem. 2019, 40, 2868–2881. [Google Scholar] [CrossRef]
- Miertus, S.; Scrocco, E.; Tomasi, J. Electrostatic interaction of a solute with a continuum. A direct utilization of ab initio molecular potentials for the prevision of solvent effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- McDonald, L.; Liu, B.; Taraboletti, A.; Whiddon, K.; Shriver, L.; Konopka, M.; Liu, Q.; Pang, Y. Fluorescent flavonoids for endoplasimic reticulum cell imaging. J. Mater. Chem. B 2016, 4, 7902–7908. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Ma, Y.; Yang, Y.; Liu, S.; Liu, Y.; Song, Y. Excited state intramolecular proton transfer mechanism of o-hydroxynaphthyl phenanthroimidazole. Chin. Phys. B 2018, 27, 023103. [Google Scholar] [CrossRef]
- Jana, S.; Dalapati, S.; Guchhait, N. Functional group induced excited state intramolecular proton transfer process in 4-amino-2-methylsulfanyl-pyrimidine-5-carboxylic acid ethyl ester: A combined spectroscopic and density functional theory study. Photochem. Photobiol. Sci. 2013, 12, 1636–1648. [Google Scholar] [CrossRef]
- Xiao, S.; Lou, Z.; Ji, D.; Zhao, J. Understanding solvent polarity dependent excited state behavior and ESIPT mechanism for 2-benzo[b]thiphen-3-yl-3-hydroxy-6-methoxy-chroman-4-one compound. Chem. Phys. Lett. 2021, 769, 138409. [Google Scholar] [CrossRef]
- Abeywickrama, C.; Wijesinghe, K.; Stahelin, R.; Pang, Y. Bright red-emitting pyrene derivatives with a large Stokes shift for nucleus staining. Chem. Commun. 2017, 53, 5886–5889. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, Y.; Liu, Y.; Jiang, K. Structure-lock induced phosphorescence lifetime enhancing of (9H-carbazol-9-yl)(phenyl)methanone: An organic phosphorescent materials. J. Lumin. 2020, 227, 117587. [Google Scholar] [CrossRef]
- Ros, P.; Schuit, G. A molecular orbital calculations on copper chloride complexes. Theor. Chem. Acc. 1966, 4, 1–12. [Google Scholar] [CrossRef]
- Zhao, J.; Jin, B.; Tang, Z. Solvent-polarity-dependent conformation and ESIPT behaviors for 2-(benzimidazole-2-yl)-3-hydroxychromone: A novel dynamical mechanism. Phys. Chem. Chem. Phys. 2022, 24, 27660–27669. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Song, P.; Feng, L.; Wang, X.; Tang, Z. Theoretical insights into atomic-electronegativity-regulated ESIPT behavior for B-bph-fla-OH fluorophore. J. Mol. Liq. 2023, 380, 121763. [Google Scholar] [CrossRef]
- Li, G.; Han, K. The sensing mechanism studies of the fluorescent probes with electronically excited state calculations. WIREs Comput. Mol. Sci. 2018, 8, 1351. [Google Scholar] [CrossRef]
- Schlegel, H. Optimization of equilibrium geometries and transition structures. J. Comput. Chem. 1982, 3, 214–218. [Google Scholar] [CrossRef]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Petersson, G.; Nakatsuji, H.; et al. Gaussian 16; Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Miehlich, B.; Savin, A.; Stoll, H.; Preuss, H. Results obtained with the correlation energy density functionals of Becke and Lee, Yang and Parr. Chem. Phys. Lett. 1989, 157, 200–206. [Google Scholar] [CrossRef]
- Becke, A. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Feller, D. The role of databases in support computational chemistry calculations. J. Comput. Chem. 1996, 17, 1571–1586. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effects of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Cammi, R.; Tomasi, J. Remarks on the use of the apparent surface charges (ASC) methods in solvation problems: Iterative versus matrix-inversion procedures and renormalization of the apparent charges. J. Comput. Chem. 1995, 16, 1449–1458. [Google Scholar] [CrossRef]
- Cances, E.; Mennucci, B.; Tomasi, J. A new integral equation formalism for the polarizable continuum model: Theoretical background and applications to isotropic and anisotropic dielectrics. J. Chem. Phys. 1997, 107, 3032–3041. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BDYBD-O | BDYBD-S | BDYBD-Se | ||||
|---|---|---|---|---|---|---|
| S0 | S1 | S0 | S1 | S0 | S1 | |
| O1–H2 | 0.999 | 1.042 | 1.001 | 1.056 | 1.001 | 1.057 |
| H2···N3 | 1.709 | 1.576 | 1.687 | 1.528 | 1.684 | 1.526 |
| Δ(O1H2N3) | 147.51 | 151.17 | 147.82 | 152.06 | 147.86 | 152.03 |
| S0 | S1 | ∆ρ (S1–S0) | ∆E (S1–S0) | |||
|---|---|---|---|---|---|---|
| ρ | EHB | ρ | EHB | ρ | EHB | |
| BDYBD-O | 0.05211 | −10.882 | 0.07187 | −15.290 | 0.01976 | 4.408 |
| BDYBD-S | 0.05581 | −11.708 | 0.08223 | −17.602 | 0.02642 | 5.894 |
| BDYBD-Se | 0.05602 | −11.755 | 0.08279 | −17.726 | 0.02677 | 5.971 |
| Transition | λ | f | Composition | CI (%) | |
|---|---|---|---|---|---|
| BDYBD-O | S0 → S1 | 336.45 | 0.5885 | H → L | 97.24 |
| S0 → S2 | 279.76 | 0.0622 | H-1 → L | 96.61 | |
| S0 → S3 | 251.44 | 0.0402 | H-2 → L | 97.48 | |
| BDYBD-S | S0 → S1 | 361.97 | 0.7153 | H → L | 97.68 |
| S0 → S2 | 311.36 | 0.0506 | H-1 → L | 92.07 | |
| S0 → S3 | 290.62 | 0.0446 | H-2 → L | 98.28 | |
| BDYBD-Se | S0 → S1 | 364.53 | 0.5271 | H → L | 97.78 |
| S0 → S2 | 338.62 | 0.0527 | H-1 → L | 96.12 | |
| S0 → S3 | 280.46 | 0.0356 | H-2 → L | 98.15 |
| BDYBD-O | BDYBD-S | BDYBD-Se | |
|---|---|---|---|
| I → III | 3.0508 | 2.7893 | 2.2153 |
| I → II | 1.6354 | 1.2846 | 1.1421 |
| II → III | 2.7355 | 2.5954 | 2.244 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, D.; Liu, C.; Zhang, M.; Zhao, J. Effects of Chalcogen Atoms on Excited-State Double-Proton Transfer Behavior for 3,6-bis(4,5-Dihydroxyoxazo-2-yl)benzene-1,2-diol Derivatives: A Computational Investigation. Molecules 2024, 29, 461. https://doi.org/10.3390/molecules29020461
Yang D, Liu C, Zhang M, Zhao J. Effects of Chalcogen Atoms on Excited-State Double-Proton Transfer Behavior for 3,6-bis(4,5-Dihydroxyoxazo-2-yl)benzene-1,2-diol Derivatives: A Computational Investigation. Molecules. 2024; 29(2):461. https://doi.org/10.3390/molecules29020461
Chicago/Turabian StyleYang, Dapeng, Chang Liu, Meiyi Zhang, and Jinfeng Zhao. 2024. "Effects of Chalcogen Atoms on Excited-State Double-Proton Transfer Behavior for 3,6-bis(4,5-Dihydroxyoxazo-2-yl)benzene-1,2-diol Derivatives: A Computational Investigation" Molecules 29, no. 2: 461. https://doi.org/10.3390/molecules29020461
APA StyleYang, D., Liu, C., Zhang, M., & Zhao, J. (2024). Effects of Chalcogen Atoms on Excited-State Double-Proton Transfer Behavior for 3,6-bis(4,5-Dihydroxyoxazo-2-yl)benzene-1,2-diol Derivatives: A Computational Investigation. Molecules, 29(2), 461. https://doi.org/10.3390/molecules29020461